

Биологические мембраны: Журнал мембранной и клеточной биологии, 2022, T. 39, № 1, стр. 75-82
Индукция митохондриальной циклоспорин-зависимой поры ацилкарнитинами. Влияние концентрации и длины углеродной цепи
Н. И. Федотчева a, Е. В. Гришина a, В. В. Дынник a, *
a Институт теоретической и экспериментальной биофизики РАН
142290 Пущино, Московская обл., Россия
* E-mail: Dynnik@rambler.ru
Поступила в редакцию 29.04.2021
После доработки 31.05.2021
Принята к публикации 02.06.2021
- EDN: UFATSH
- DOI: 10.31857/S0233475522010066
Аннотация
Известно, что концентрации длинноцепочечных жирных кислот (LCFA) и их карнитиновых производных (LCАC) при ожирении и диабете сильно увеличиваются, а в условиях ишемии-реперфузии могут превышать десятки мкМ в очаге поражения клеток. Наиболее токсичными считаются LCАC, активированные производные LCFA. В данной работе исследовано влияние D,L-ацилкарнитинов с различной длиной углеродной цепи на индукцию митохондриальной циклоспорин А (CsA)-зависимой поры (mPTP). В экспериментах на изолированных митохондриях печени крыс тестировали гексаноилкарнитин (HC, С6:0), лауроилкарнитин (LC, C12:0), миристоилкарнитин (MC, C14:0) и пальмитоилкарнитин (PC, C16:0); регистрировали скорость дыхания митохондрий, митохондриальный потенциал (ΔΨ) и набухание митохондрий при окислении глутамата и пирувата в присутствии ацилкарнитинов в различных концентрациях. Показано, что все исследованные ацилкарнитины вызывают ингибирование дыхания митохондрий, активированного ADP-гексокиназной системой, а также индуцируют набухание митохондрий. Величины наблюдаемых эффектов обратно пропорциональны длине углеродной цепи ацилкарнитинов. Критические концентрации PC, МС, LC и НС, при которых происходит снижение ΔΨ, составляют 100, 110, 500 мкМ и 3 мМ соответственно. Индукция набухания митохондрий происходит при пороговых концентрациях PC, МС и LC 10, 25 и 200 мкМ соответственно. CsA, EGTA и ADP увеличивают пороговые концентрации PC и МС, необходимые для индукции mPTP, и не эффективны при более высоких концентрациях ацилкарнитинов. Таким образом, в ряду карнитиновых производных насыщенных жирных кислот с длиной углеродной цепи от 6 до 16 PC и МС являются наиболее токсичными агентами, способными вызывать индукцию mPTP.
ВВЕДЕНИЕ
Концентрации длинноцепочечных жирных кислот (LCFA) и их карнитиновых производных (LCАC) в плазме крови используются как маркеры ряда врожденных метаболических и сердечно-сосудистых заболеваний, а также диабета 2 типа (T2D) и стеатогепатита и др. [1, 2]. При ожирении и T2D, распространение которых в мире носит характер пандемии, риск возникновения инсульта/инфаркта увеличивается в несколько раз. Увеличение такого риска может быть обусловлено токсическим действием LCFA и LCAC, вызывающим нарушение Са2+-гомеостаза и гибель клеток по типу некроза или апоптоза.
Концентрации LCFA и LCАC в крови пациентов с ожирением и T2D в 2–4 раза превышают контрольные величины [1], а в условиях ишемии-реперфузии в очаге поражения клеток могут превышать десятки мкМ [3–5]. LCАC – основная активированная форма LCFA, которая переносится кровью, транспортируется в митохондрии и окисляется в виде Ацил-КоА. Длинноцепочечные Ацил-КоА являются ингибиторами NAD-зависимых дегидрогеназ, включая ферменты цикла Кребса и глутаматдегидрогеназу [6], а также транслокатора адениннуклеотидов (ANT) и других митохондриальных транспортеров [7–11].
Хорошо известно, что LCFA вызывают разобщение окислительного фосфорилирования митохондрий [12], стимулируют индукцию избытком Са2+ циклоспорин А (CsA)-чувствительной митохондриальной поры (mPTP) [13–15]. Принято считать, что индукция mPTP играет важную роль в гибели клеток по типу некроза, апоптоза или некроптоза [13–16]. Кальций является основным индуктором этой поры. Избыточное накопление Са2+ в митохондриях приводит к обратимому или необратимому открытию поры, деполяризации и деэнергизации митохондрий и индукции mPTP.
Исследования последних лет показывают, что LCАC и соответствующие Ацил-КоА накапливаются в постишемических митоходриях в больших концентрациях [9]. Очевидно, что длинноцепочечные Ацил-КоА могут быть основными токсинами, вызывающими подавление энергетики митохондрий и индукцию mPTP, вследствие ингибирования ими ключевых дегидрогеназ митохондрий и различных транспортеров [6–11]. Однако к настоящему времени механизмы токсического действия LCFA, LCАC, и Ацил-КоА на регуляцию Ca2+-гомеостаза, энергетику митохондрий и индукцию mPTP в клетках разных типов изучены недостаточно. Мало исследован также вопрос о сравнительном действии ацилкарнитинов с различной длиной углеродной цепи на индукцию CsA-зависимой поры, который и составляет предмет данной работы.
МАТЕРИАЛЫ И МЕТОДЫ
Условия выделения митохондрий и проведения экспериментов описаны ранее [17]. Все процедуры на животных выполнялись в соответствии с директивой ЕС 86/609/EEC и были одобрены комитетом по этике Института теоретической и экспериментальной биофизики РАН. Самцов крыс линии Вистар (возраст 6–8 недель) содержали в одинаковых условиях в кондиционированных и проветриваемых помещениях при температуре 20–22°C (свет/темнота – 12 ч/12 ч). Митохондрии печени выделяли с использованием стандартных методик дифференциального центрифугирования в среде, содержащей 300 мМ сахарозы, 1 мМ EGTA и 10 мМ трис-HCl (рН 7.4). Митохондриальные препараты дважды промывали средой выделения, не содержащей EGTA, ресуспендировали в среде того же состава и хранили на льду. Инкубационная среда для митохондрий содержала: 125 мМ KCl, 3 мМ KH2PO4, 10 мМ HEPES (pH 7.4), 1 мМ MgCl2. Содержание митохондриального белка определяли методом Лоури с бычьим сывороточным альбумином в качестве стандарта. Скорость потребления кислорода митохондриями определяли полярографическим методом с помощью закрытого кислородного электрода Кларка в термостатируемой ячейке объемом 1 мл, при 27°C и постоянном перемешивании. Разность электрических потенциалов (ΔΨ) на внутренней мембране митохондрий определяли по распределению липофильного катиона тетрафенилфосфония (ТРР+), концентрацию которого во внешней среде [ТРР+]out регистрировали с помощью ТРР+-селективного электрода. Среда инкубации содержала 125 мМ KCl, 3 мМ KH2PO4, 1 мМ MgSO4, 10 мМ HEPES (рН 7.4), 2мкМ ТРР+. Стационарную скорость дыхания в состоянии V3 при добавлении 750 мкМ ADP обеспечивали внесением в среду инкубации 5 мМ глюкозы и 0.5 ед. гексокиназы. В работе использовали реактивы фирм Sigma-Aldrich (США), Tocris (Великобритания).
Набухание митохондрий определяли спектральным методом при длине волны 540 нм с использованием спектрофотометра Ocean Optic USB-4000 (США). Митохондрии (0.3–0.4 мг/мл) инкубировали в среде, содержащей 125 мМ KCl, 1.5 мМ KH2PO4, 10 мМ HEPES (pH 7.4), в качестве субстратов окисления использовали глутамат (10 мМ) с пируватом (1 мМ) или сукцинат (5 мМ).
Статистический анализ экспериментальных данных проводили с t-критерия Стьюдента для сравнения пар величин с помощью программы SigmaPlot 11. Данные представлены как среднее значение ± S.E.M. Количество повторенных независимых экспериментов n = 4‒6. За уровень значимости принято p < 0.05.
РЕЗУЛЬТАТЫ
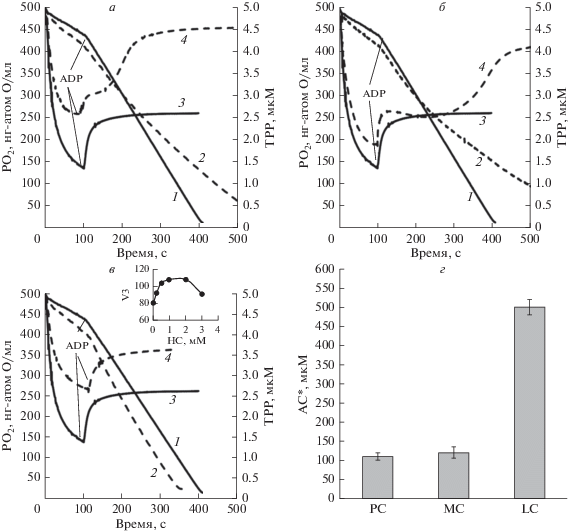
Влияние ацилкарнитинов на дыхание митохондрий и митохондриальный потенциал. Рис. 1 характеризует влияние ацилкарнитинов с различной длиной углеродной цепи (С6, С12–С16) на дыхание митохондрий и митохондриальный потенциал ΔΨ при активации энергетики митохондрий ADP-гексокиназной системой. Преинкубация митохондрий с 100 мкМ D,L-пальмитоилкарнитина (PC, С16:0) вызывает ингибирование дыхания и быструю диссипацию ΔΨ, которые наблюдаются после добавки ADP и активации дыхания митохондрий (скорость дыхания V3) (рис. 1а). Диссипация ΔΨ и деэнергизация митохондрий, так же как и набухание митохондрий (см. ниже), свидетельствуют об открытии mPTP при избытке PC. Средняя пороговая концентрация РС (для D,L-формы) составляет 110 мкМ (рис. 1г). При меньших концентрациях PC ([PC] < 60‒70 мкМ) сброс потенциала и открытие поры развиваются медленно. Сходный эффект вызывает миристаилкарнитин (МС, C14:0), для которого пороговая концентрация составляет 120 мкМ (рис. 1г). В сравнении с РС и МС, лауроилкарнитин (LC, C12:0) вызывает ингибирование дыхания и сброс ΔΨ только при концентрациях, превышающих 500 мкМ (рис. 1г). Короткоцепочечный гексаноилкарнитин (HC, С6:0) активирует дыхание и не вызывает сброс ΔΨ даже при концентрации в 2 мМ (рис. 1в). Слабое ингибирование дыхания митохондрий наблюдается только при больших концентрациях HC, превышающих 3 мМ (рис. 1в, вставка). Это ингибирование снимается при добавках малата или сукцината в среду инкубации (не показано), что указывает на возможное ингибирование дегидрогеназ цикла Кребса короткоцепочечными Ацил-КоА. Ранее нами было показано, что подобный эффект обратимого ингибирования энергетики митохондрий может наблюдаться даже при избытке пирувата [14].
Рис. 1.
Влияние ацилкарнитинов с различной длиной углеродной цепи на дыхание и мембранный потенциал митохондрий. а – Дыхание (1, 2) и мембранный потенциал (3, 4) митохондрий регистрировали в присутствии 1мМ ADP, глюкозы и гексокиназы в контроле (1, 3) и при преинкубации митохондрий с 110 мкМ пальмитоилкарнитина (PC, 2, 4). б – Дыхание (1, 2) и мембранный потенциал (3, 4) митохондрий регистрировали в присутствии 1мМ ADP, глюкозы и гексокиназы в контроле (1, 3) и при преинкубации митохондрий с 450 мкМ лаурoилкарнитина (LC, 2, 4). в – Дыхание (1, 2) и мембранный потенциал (3, 4) митохондрий регистрировали в присутствии 1мМ ADP, глюкозы и гексокиназы в контроле (1, 3) и при преинкубации митохондрий с 2.0 мМ гексаноилкарнитина (HC, 2, 4). г – Пороговые концентрации ацилкарнитинов (АС*), при которых происходит сброс мембранного потенциала (выход ТРР из митохондрий). MC – миристоилкарнитин. Все ацилкарнитины в D,L-форме. V3 – скорость дыхания митохондрий (нг-атом O/мл/мин).
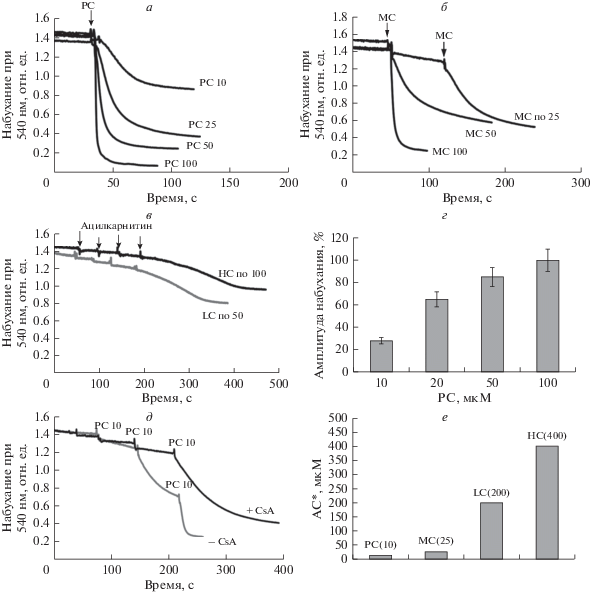
Влияние ацилкарнитинов на набухание митохондрий. На рис. 2 представлены данные о влиянии концентраций ацилкарнитинов (С6, С12–С16) на набухание митохондрий. Видно, что РС (рис. 2а) и МС (рис. 2б) обладают сходным токсическим действием. При разовых добавках в 25 и 50 мкМ соответственно, РС и МС индуцируют 50–60% набухание митохондрий, а при концентрациях 100 мкМ оба ацилкарнитина индуцируют быстрое 100% набухание митохондрий. Зависимость амплитуды набухания от концентрации РС показана на рис. 2г. В сравнении с действием длинноцепочечных ацилкарнитинов, эффекты карнитинов с более короткой длиной углеродной цепи LC (C12:0) и НС (С6:0) выражены слабо. Видно, что 25–30% набухание митохондрий развивается только при достижении концентраций LC и НС, превышающих 200 и 400 мкМ соответственно (рис. 2в).
Рис. 2.
Набухание митохондрий, индуцированное ацилкарнитинами с различной длиной углеродной цепи. а – Набухание митохондрий, индуцированное пальмитоилкарнитином в разных концентрациях (PC). б – Набухание митохондрий, индуцированное миристоилкарнитином (MC) в разных концентрациях. в – Набухание митохондрий при последовательных добавках лауроилкарнитина (LC) и гексаноилкарнитина (HC). г – зависимость амплитуды набухания митохондрий от концентрации PC при окислении глутамата с пируватом; д – индуцированное PC набухание митохондрий при окислении сукцината и влияние циклоспорина А; е – пороговые концентрации (значения указаны в скобках) ацилкарнитинов (АС*), индуцирующие набухание митохондрий. В качестве субстратов окисления были использованы глутамат 10 мМ + пируват 1 мМ и сукцинат (5 мМ), концентрация митохондриального белка 0.3–0.4 мг/мл, среда инкубации 120 мМ КСl, 10 мМ HEPES, 1.5 мМ фосфата, рН 7.4.
Влияние последовательных малых добавок РС и МС на набухание митохондрий показано на рис. 3. Однократные добавки 10 мкМ РС и 25 мкМ МС не вызывают заметный эффект, тогда как последующие добавки индуцируют быстрое набухание митохондрий (контроль, рис. 3а, 3б, нижние кривые). Набухание митохондрий при однократных добавках 10 мкМ РС (рис. 3а) и 25 мкМ МС (рис. 3б) эффективно предотвращается CsA и EGTA (рис. 3а, 3б, верхние кривые), тогда как вторая добавка РС и третья добавка МС вызывают 100% набухание митохондрий, что указывает на ограниченный защитный эффект CsA и EGTA, наблюдающийся при пороговых концентрациях LCAC.
Рис. 3.
Влияние циклоспорина А и EGTA на индуцированное ацилкарнитинами набухание митохондрий. Набухание митохондрий, индуцированное последовательными добавками пальмитоилкарнитина (РС), 10 мкМ каждая (а), и миристоилкарнитином (МС), 25 мкМ каждая добавка (б), в присутствии циклоспорина А (2 мкМ) и EGTA (1 мМ); в скобках указаны скорости набухания (ΔD/мин). Ингибирование скорости (в) и амплитуды (г) индуцированного 20 мкМ РС и 50 мкМ МС набухания митохондрий циклоспорином А и EGTA. * – за 100% принята максимальная амплитуда набухания, наблюдаемая в присутствии 100 мкМ РС. Субстраты окисления глутамат (10 мМ) и пируват (1 мМ).
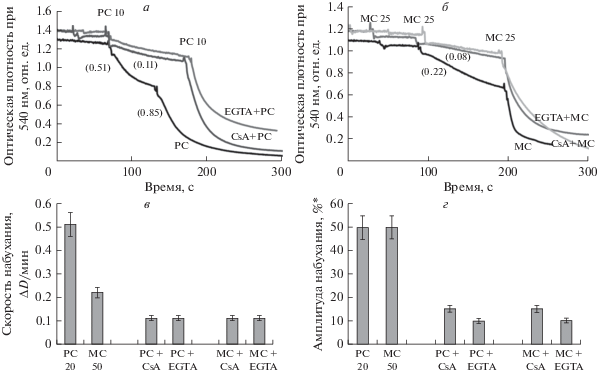
Влияние CsA и EGTA на скорости и амплитуды набухания митохондрий при действии 20 мкМ РС и 50 мкМ МС представлена на рис. 3в, 3г соответственно. Из данных, представленных на этих рисунках следует, что только при концентрациях РС и МС близких к пороговым величинам 20 мкМ и 50 мкМ, CsA и EGTA оказывают характерный для CsA-зависимой поры (mPTP) защитный эффект. Защитный эффект отсутствует при более высоких концентрациях РС и МС (рис. 3а, 3б).
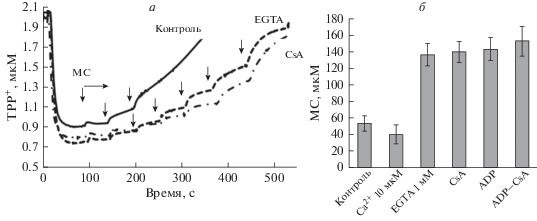
Влияние миристоилкарнитина на диссипацию митохондриального потенциала. Ограниченный защитный эффект CsA и EGTA. Из данных приведенных на рис. 4а видно, что в контроле (верхняя кривая) третья добавка 20 мкМ МС вызывает снижение митохондриального потенциала ΔΨ. Преинкубация митоходрий с CsA или EGTA препятствует индукции поры при таких условиях, тогда как последующие добавки МС вызывают концентрационно-зависимую диссипацию ΔΨ, указывая на ограниченный защитный эффект этих ингибиторов. Известно, что ADP, Mg2+, ингибиторы ANT (карбоксиаттрактилозид, бонгкрековая кислота) и др. также ингибируют индукцию mPTP, вызванную ионами кальция [16]. При индукции поры РС или МС также наблюдается защитный эффект этих соединений. Из рис. 3б следует, что пороговая концентрация МС (МС*), вызывающая диссипацию ΔΨ увеличивается более чем в 2 раза в присутствии CsA, EGTA, ADP (ADP + Mg2+ + гексокиназа) или при сочетанном действии CsA и EGTA, тогда как наличие Ca2+ в среде инкубации усиливает действие МС, снижая пороговую величину МС*. Взаимное синергичное действие РС и Ca2+при индукции mPTP нами было показано ранее на митохондриях печени и сердца [18, 19].
Рис. 4.
Влияние циклоспорина А и EGTA на индуцированное ацилкарнитинами снижение мембранного потенциала митохондрий. Изменения мембранного потенциала, индуцированные последовательными добавками миристоилкарнитина (МС), 20 мкМ каждая добавка в присутствии 2 мкМ CsA и 1 мМ EGTA (а); влияние Са2+ и ингибиторов митохондриальной поры (EGTA 1 мМ, CsA 2 мкМ и ADP 0.7 мМ) на пороговые концентрации миристоилкарнитина (б).
ОБСУЖДЕНИЕ
Полученные результаты позволяют сделать вывод о том, что длинноцепочечные LCАC (МС и РС) можно рассматривать, как наиболее токсичные агенты в ряду производных насыщенных жирных кислот, вызывающих индукцию CsA-зависимой поры.
Хорошо известно, что CsA, адениновые нуклеотиды (ATP, ADP), ингибиторы ANT и Mg2+ препятствуют индукции mPTP кальцием [16], а эффект неорганического фосфата (Рi) дозозависим [20, 21]. Механизмы регуляции и структурные элементы этого белкового мегакомплекса остаются предметом споров в течение нескольких десятков лет [13–16]. В настоящее время считается, что этот мегаканал формируется с участием ATP-синтазы (F1F0), ANT, переносчика фосфата Рi (PiC) и ключевого регулятора поры циклофилина D (CypD) [16]. CypD и F1F0 являются мишенями CsA [16, 22].
В наших экспериментах CsA и ADP вызывают увеличение пороговой концентрации МС (МС*), необходимой для диссипации ΔΨ (рис. 4б). Однако, в отличие от классической модели индукции открытия поры избытком Ca2+, при индукции поры LCAC защитный эффект CsA ограничен концентрациями РС или МС, близкими к пороговым. Из рис. 3 видно, что СsA эффективен только при малых концентрациях РС и МС. Уже вторая добавка 10 мкМ РС или 25 мкМ МС вызывает быстрое набухание митохондрий. Эти данные могут свидетельствовать о наличии разных мишеней в мегакомплексе поры, на которые может быть направлено действие Ca2+ и LCАC и соответствующих ацил-КоА.
CypD рассматривается в качестве ключевой мишени CsA при индукции mPTP кальцием [16, 22]. Одной из наиболее вероятных мишеней действия LCAC, а также соответствующих ацил-КоА, при индукции mPTP может быть ANT. Известно, что цитоплазматические и митохондриальные длинноцепочечные ацил-КоА, связываясь с ANT, вызывают ингибирование этого переносчика аденилатов [9–11]. Поэтому, известная гипотеза об участии LCFA и их производных в стабилизации ANT в цитоплазматической или митохондриальной конформациях и роли таких конформационных переходов в регуляции mPTP [23] требует дальнейшего изучения.
Известно, что LCFA могут вызывать образование липидной поры [24–26]. Длинноцепочечные насыщенные жирные кислоты: пальмитиновая (С16:0), стеариновая (C18:0) и эйкозаноевая (C20:0) кислоты, имеющие, в сравнении с другими LCFA, высокое сродство к Са2+, могут образовывать в мембранах разного типа липидную пору. Образование такой обратимой липидной поры (комплекса жирной кислоты и Са2+) предотвращается в присутствии EGTA [25, 26]. Теоретически можно было бы допустить, что деацетилирование пальмитоил-КоА и миристоил-КоА в митохондриях в условиях наших экспериментов может приводить к локальному накоплению соответствующих жирных кислот и индукции липидной поры. Однако индукция поры с участием МС в наших условиях наблюдается в присутствии EGTA и CsA (рис. 4б), что свидетельствует об участии LCAC в регуляции классической CsA-зависимой mPTP.
Конфликт интересов. Авторы заявляют, что у них нет конфликта интересов.
Источники финансирования. Работа выполнена в рамках Государственного контракта 075-00381-21-00 Института теоретической и экспериментальной биофизики РАН (ИТЭБ РАН).
Соответствие принципам этики. Исследование проводилось в соответствии с этическими принципами, сформулированными в Хельсинкской декларации по использованию лабораторных животных. Все процедуры на животных были одобрены комитетом по этике ИТЭБ РАН (N28 / 2021, 9 февраля 2021 г.).
Список литературы
Koves T.R., Ussher J.R., Noland R.C., Slentz D., Mosedale M., Ilkayeva O., Bain J., Stevens R., Dyck J.R., Newgard C.B., Lopaschuk G.D., Muoio D.M. 2008. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 7 (1), 45–56. https://doi.org/10.1016/j.cmet.2007.10.013
Fromenty B., Robin M.A., Igoudjil A., Mansouri A., Pessayre D. 2004. The ins and outs of mitochondrial dysfunction in NASH. Diabetes Metab. 30 (2), 121–138. https://doi.org/10.1016/s1262-3636(07)70098-8
Ford D.A., Han X., Horner C.C., Gross W. 1996. Gross accumulation of unsaturated acylcarnitine molecular species during acute myocardial ischemia: Metabolic compartmentalization of products of fatty acyl chain elongation in the acylcarnitine pool. Biochemistry. 35 (24), 7903–7909. https://doi.org/10.1021/bi960552n
Lesnefsky E.J., Moghaddas S., Tandler B., Kerner J., Hoppel C.L. 2001. Mitochondrial dysfunction in cardiac disease: Ischemia–reperfusion, aging, and heart failure. J. Mol. Cell. Cardiol. 33 (6), 1065–1089. https://doi.org/10.1006/jmcc.2001.1378
Liepinsh E., Makrecka-Kuka M., Volska K., Kuka J., Makarova E., Antone U., Sevostjanovs E., Vilskersts R., Strods A., Tars K., Dambrova M. 2016. Long-chain acylcarnitines determine ischaemia/reperfusion-induced damage in heart mitochondria. Biochem. J. 473 (9), 1191–1202. https://doi.org/10.1042/BCJ20160164
Erfle J.D., Sauer F. 1969. The inhibitory effects of acyl-coenzyme A esters on the pyruvate and alpha-oxoglutarate dehydrogenase complexes. Biochim. Biophys. Acta. 178 (3), 441–452. https://doi.org/10.1016/0005-2744(69)90213-7
Lai J.C., Cooper A.J. 1991. Neurotoxicity of ammonia and fatty acids: Differential inhibition of mitochondrial dehydrogenases by ammonia and fatty acyl coenzyme A derivatives. Neurochem. Res. 16 (7), 795–803.
Farrell H.M.Jr., Wickham E.D., Reeves H.C. 1995. Effects of long-chain acyl-coenzyme A’s on the activity of the soluble form of nicotinamide adenine dinucleotide phosphate-specific isocitrate dehydrogenase from lactating bovine mammary gland. Arch. Biochem. Biophys. 321 (1), 199–208. https://doi.org/10.1006/abbi.1995.1386
Paulson D.J., Shug A.L. 1984. Inhibition of the adenine nucleotide translocator by matrix-localized palmityl-CoA in rat heart mitochondria. Biochim. Biophys. Acta. 766 (1), 70–76. https://doi.org/10.1016/0005-2728(84)90218-4.10
Schoënfeld P., Bohnensack R. 1997. Fatty acid-promoted mitochondrial permeability transition by membrane depolarization and binding to the ADP/ATP carrier. FEBS Lett. 420, 167–170.
Ciapaite J., Van Eikenhorst D., Bakker S., Diamant M., Heine R.J., Wagner M.J., Westerhoff H.V., Krab K. 2005. Modular kinetic analysis of the adenine nucleotide translocator-mediated effects of palmitoyl-CoA on the oxidative phosphorylation in isolated rat liver mitochondria. Diabetes. 54 (4), 944–951. https://doi.org/10.2337/diabetes.54.4.944
Wojtczak L., Wieckowski M.R. 1999. The mechanisms of fatty acid-induced proton permeability of the inner mitochondrial membrane. J. Bioenerg. Biomembr. 31, 447–455.
Hunter D.R., Haworth R.A. 1979. The Ca2+-induced membrane transition in mitochondria. III. Transitional Ca2+ release. Arch. Biochem. Biophys. 195 (2), 468–477.
Crompton M., Ellinger H., Costi A. 1988. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem. J. 255, 357–360.
Bernardi P., Broekemeir K.M., Pfeiffer D.R. 1994. Recent progress on regulation of the mitochondrial permeability transition pore; a cyclosporin-sensitive pore in the inner mitochondrial membrane. J. Bioenerg. Biomembr. 26 (5), 509–517. https://doi.org/10.1007/BF00762735
Kwong J.Q., Molkentin J.D. 2015. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 21 (2), 206–214. https://doi.org/10.1016/j.cmet.2014.12.001
Гришина Е.В., Галимова М.Х., Джафаров Р.Х., Сергеев А.С., Федотчева Н.И., Дынник В.В. 2015. Индукция циклоспорин-чувствительной митохондриальной поры субстратами, образующими ацетил-СоА, в норме и при диабете 2 типа. Биол. мембраны. 32 (5–6), 319–327. https://doi.org/10.7868/S0233475515050047
Berezhnov A.V., Fedotova E.I., Nenov M.N., Kasymov V.A., Pimenov O.Y., Dynnik V.V. 2020. Dissecting cellular mechanisms of long-chain acylcarnitines-driven cardiotoxicity: Disturbance of calcium homeostasis, activation of Ca2+-dependent phospholipases, and mitochondrial energetics collapse. Int. J. Mol. Sci. 21 (20), 7461. https://doi.org/10.3390/ijms21207461
Dynnik V.V., Grishina E.V., Fedotcheva N.I. 2020. The mitochondrial NO-synthase/guanylate cyclase/protein kinase G signaling system underpins the dual effects of nitric oxide on mitochondrial respiration and opening of the permeability transition pore. FEBS J. 287 (8), 1525–1536.https://doi.org/10.1111/febs.15090
Chalmers S., Nicholls D.G. 2003. The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J. Biol. Chem. 278 (2), 19062–19070.
Mishra J., Ariea J., Davani L., Gayathri K., Natarajan K., Kwok W.M., David F., Stowe J., Camara A.K.S. 2019. Cyclosporin A increases mitochondrial buffering of calcium: An additional mechanism in delaying mitochondrial permeability transition pore opening. Cells. 8, 1052. https://doi.org/10.3390/cells8091052
Fayaz S.M., Raj Y.V., Krishnamurthy R.G. 2015. CypD: The key to the death door. CNS Neurol. Disord. Drug Targets. 14, 654–663.
Zoratti M., Szabo I. 1995. Mitochondrial permeability pore. Biochim. Biophys. Acta. 1241, 139–176.
Sultan A., Sokolove P.M. 2001. Palmitic acid opens a novel cyclosporin A-insensitive pore in the inner mitochondrial membrane. Arch. Biochem. Biophys. 386 (1), 37–51. https://doi.org/10.1006/abbi.2000.2194
Mironova G.D., Gateau-Roesch O., Levrat C., Gritsenko E., Pavlov E., Lazareva A.V., Limarenko E., Rey C., Louisot P., Saris N.E. 2001. Palmitic and stearic acids bind Ca2+ with high affinity and form nonspecific channels in black-lipid membranes. Possible relation to Ca2+-activated mitochondrial pores. J. Bioenerg. Biomembr. 33 (4), 319–331. https://doi.org/10.1023/a:1010659323937
Mironova G.D., Pavlov E.V. 2021. Mitochondrial cyclosporine A-independent palmitate/Ca2+-induced permeability transition pore (PA-mPT Pore) and its role in mitochondrial function and protection against calcium overload and glutamate toxicity. Cells. 10, 125. https://doi.org/10.3390/cells10010125
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии