

Биологические мембраны: Журнал мембранной и клеточной биологии, 2022, T. 39, № 3, стр. 163-171
Гемостаз и тромбоз. Пространственная организация биохимических процессов на микроуровне
М. А. Пантелеев a, b, c, *, А. М. Шибеко a, b, Д. Ю. Нечипуренко a, b, c, Е. А. Береснева a, Н. А. Подоплелова a, b, А. Н. Свешникова a, b, c
a Центр теоретических проблем физико-химической фармакологии РАН
109029 Москва, Россия
b Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии
им. Дмитрия Рогачева Минздрава России
117198 Москва, Россия
c Московский государственный университет им. М.В. Ломоносова
119991 Москва, Россия
* E-mail: mapanteleev@yandex.ru
Поступила в редакцию 10.12.2021
После доработки 20.01.2022
Принята к публикации 20.01.2022
- EDN: NCXCRU
- DOI: 10.31857/S0233475522030094
Аннотация
Системы свертывания крови и фибринолиза представляют собой ферментативные каскады в плазме крови, управляющие процессами формирования и растворения фибринового сгустка соответственно. Однако критические процессы в обеих системах происходят не в жидкой фазе, а на специализированных “скаффолдах”: двух- или трехмерных матрицах, обеспечивающих особые условия для протекания биохимических реакций. В настоящий момент можно выделить следующие принципиальные категории скаффолдов: а) обогащенные фосфатидилсерином фосфолипидные мембраны, предоставляемые прокоагулянтной субпопуляцией активированных тромбоцитов, а также поврежденным эндотелием, мембранами апоптотических телец в атеросклеротической бляшке, липопротеидами и микровезикулами плазмы, б) комплекс фибрина и белков внеклеточного матрикса, ассоциированный с тромбоцитами и являющийся ведущим скаффолдом для про- и анти-фибринолитических процессов, в) полимеры фосфатов, включая тромбоцитарные полифосфаты и внеклеточные ловушки нейтрофилов. Для некоторых из этих скаффолдов существуют предположения об их физиологической значимости и физическом смысле, в то время как роль других представляется загадочной или, как минимум, патофизиологической. Здесь мы рассмотрим существующие представления о ролях и механизмах участия этих скаффолдов в гемостазе и тромбозе.
ВВЕДЕНИЕ
Системы свертывания крови и фибринолиза представляют собой характерные примеры ферментативных систем внеклеточной регуляции [1]. В основе и той и другой лежат протеолитические ферменты, активирующие друг друга в каскадах, содержащих многочисленные обратные связи (рис. 1) [2, 3]. Результатом работы каскада свертывания крови является фибриновый сгусток, предотвращающий потерю крови; задачей системы фибринолиза является растворение этого фибрина. Задачи обеих систем являются принципиально пространственными, и сложная структура каскадов свертывания и фибринолиза, по-видимому, связана с наличием в них модулей, отвечающих за отдельные функциональные подзадачи [4–6]. Так, положительная обратная связь активации фактора XI тромбином наделяет каскад свертывания автоволновыми свойствами [7], активация фактора свертывания V тромбином и формирование протромбиназы играет определяющую роль в пороге по активации [4, 8], а активация фактора свертывания VII важна для порога по скорости потока и распознавания геометрии места повреждения [8, 9].
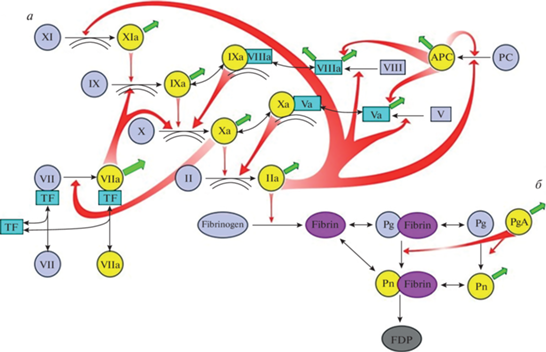
Рис. 1.
Каскады свертывания (а) и фибринолиза (б). Обозначения: реакции превращения факторов свертывания в активные формы показаны односторонними тонкими черными стрелками. При этом фигурные красные стрелки показывают, под действием каких именно ферментов происходит активация. Реакции потери активности в результате ингибирования показаны тонкими зелеными стрелками (для простоты стрелки изображены как просто “уход”, т.е. не показано, с какими именно ингибиторами происходит связывание). Обратимые реакции формирования комплексов показаны двусторонними тонкими черными стрелками. Белки свертывания обозначены либо названиями, либо римскими цифрами, либо аббревиатурами (TF – тканевой фактор, PC – протеин С, APC – активированный протеин С, Pg – плазминоген, Pn – плазмин, FDP – продукты деградации фибрина). Чтобы избежать перегруженности, на схеме не показаны: связывание тромбина с тромбомодулином, активация и секреция тромбоцитов, контактная активация свертывания.
На протяжении многих лет признается, что главные реакции каскада свертывания крови идут на поверхности фосфолипидных мембран [10], предположительно предоставляемых в первую очередь прокоагулянтной субпопуляцией активированных тромбоцитов [11]. Это включает активацию фактора Х внешней теназой [12] и внутренней теназой [13, 14], активацию протромбина протромбиназой, инактивацию фактора Va активированным протеином С и другие. Точно так же процесс фибринолиза по определению тесно связан с фибрином. В этих областях науки продолжает таиться много загадок – в части механизмов связывания белков свертывания с мембраной [15–17], путей сборки мембранных комплексов и доставки субстрата [12, 13, 18], видов прокоагулянтных поверхностей в организме [11, 19]. Однако исследования последнего десятилетия привели к более масштабному пересмотру картины: как будет описано ниже, взаимодействие белков свертывания и фибринолиза с “нерастворимой фазой” оказалось куда более многообразным и масштабным, чем представлялось еще не так давно [5, 11, 15, 17, 20–28].
В силу этого мы хотели бы предложить более дерзкий пересмотр существующих представлений и обсудить, как выглядят биохимические реакции, проходящие в “нерастворимой фазе”, в свете этих открытий. Мы постараемся показать, что в гемостазе и тромбозе можно выделить три блока таких “скаффолдов”, имеющих более сложный и гибкий смысл, чем предполагалось ранее. Под скаффолдами здесь и далее мы будем подразумевать двух- и трехмерные молекулярные структуры, служащие плацдармом для разворачивающихся биохимических процессов и управляющие их скоростями.
1. Прокоагулянтные мембраны – ключевой скаффолд для прокоагулянтных реакций свертывания, имеющий, по-видимому, два физических смысла: а) локальное концентрирование белков, позволяющее преодолеть диффузионный предел скорости химической реакции и проводить быстрые реакции при низких концентрациях факторов свертывания, б) управление пространственным распределением факторов свертывания путем создания “неподвижной фазы”, где белки защищены от ингибиторов, потока и дополнительно стабилизированы мультимеризацией.
2. Комплекс фибрина и ассоциированных с ним молекул внеклеточного матрикса и адгезионных молекул (фибронектин, тромбоспондин, фактор Виллебранда), который неразрывно связан с тромбоцитарным агрегатом через рецепторы-интегрины тромбоцитов. Этот комплекс влияет на свертывание (через фактор VIII, ассоциированный с фактором Виллебранда; путем прямого влияния на связывание фактора VIII; также путем адсорбции и протекции тромбина), управляет антифибринолитическими реакциями, находящимися между свертыванием и фибринолизом (фактор XIII активируется тромбином в фибрин-зависимых реакциях), является мишенью антифибринолитических модификаций (фактор XIIIa, TAFI), а также служит основой для всего каскада фибринолиза (ускорение активации плазминогена тканевым активатором плазминогена, защита плазмина от сверхбыстрого ингибирования в плазме крови). Сейчас считается, что пространственное распределение фибрина в тромбах далеко не гомогенное [29, 30]. В богатых тромбоцитами тромбах фактор Виллебранда оказывается критически важным элементом архитектуры, берущим на себя механическую функцию и препятствующим фибринолизу [20, 26].
3. Полимеры фосфатов в разном виде, главными из которых являются: а) полифосфаты тромбоцитов, являющиеся важными кандидатами в активаторы контактного пути в артериальном тромбе и модуляцию огромного количества других реакций; б) внеклеточные ловушки нейтрофилов, играющие огромную роль в венозном тромбозе; в) ДНК предположительно лейкоцитарного происхождения (возможно, частично также из внеклеточных ловушек нейтрофилов [22]), стабилизирующая тромбы и препятствующая их лизису [26, 27].
Разберем эти скаффолды и связанные с ними механизмы один за другим. В данном обзоре мы не будем подробно описывать молекулярные и биохимические особенности этих реакций, а сосредоточимся на попытке принципиального понимания их функционирования.
ПРОКОАГУЛЯНТНЫЕ МЕМБРАНЫ: ТРОМБОЦИТЫ И НЕ ТОЛЬКО
Возможная величина константы скорости любой ферментативной реакции ограничена сверху частотой столкновений между молекулами, которая в свою очередь определяется скоростью диффузии. Для белков в воде при комнатной температуре [31] этот диффузионный предел определяет верхнюю границу каталитической эффективности как 109 M–1 с–1. Характерные концентрации ферментов свертывания крови не превышают десятков нМ; для некоторых из них нормой являются концентрации порядка пМ. Если у нас есть фермент с концентрацией 1 пМ (10–12 М), то он сможет катализировать лишь 0.1% своего субстрата в секунду. На практике же большинство реальных ферментов имеет константы не больше 107 M–1 с–1, и тогда результат оказывается еще более ничтожным. Очевидно, что для системы свертывания, которая отвечает за критические защитные функции, скорость срабатывания в десятки часов неприемлема. С другой стороны, производить заметно более высокие концентрации ферментов, которые имеют лишь сигнальное значение и не производят полезного продукта, кажется невыгодным для организма. Однако если перевести эти многочисленные ферменты в небольшой замкнутый объем или, даже лучше, посадить на небольшую мембрану (рис. 2а), то эффективность реакций резко увеличится. Это позволяет предположить, что широкое использование мембранных реакций в свертывании крови, равно как и во внутриклеточной сигнализации, обусловлено потребностью сочетать высокую скорость процессов с низкими концентрациями управляющих ферментов.
Рис. 2.
а – Мембранные реакции, б – микрофотография агрегата тромбоцитов, содержащего прокоагулянтные тромбоциты; в – микрофотография отдельного прокоагулянтного тромбоцита и его “шапки” в различных каналах [11]; г – вытеснение прокоагулянтных тромбоцитов из тромба (слева – комбинированное изображение, полученное методом наложения дифференциально-интерференционного контраста (ДИК) и эпифлуоресцентного изображения в канале аннексина V, cправа – эпифлуоресцентное изображение в канале аннексина V [29], желтый контур соответствует границе тромба, выделенной в канале ДИК).
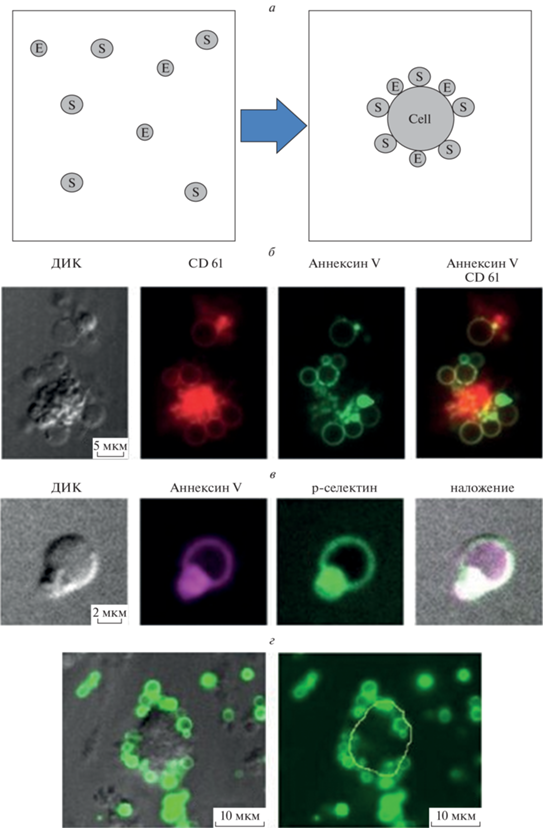
Несмотря на то, что эта идея кажется прозрачной и очевидной, проверить ее на практике непросто. Исходная идея регуляторной роли мембраны, ускоряющей свертывание за счет локального концентрирования или “редукции числа измерений”, появилась еще в 1980-е и была способна объяснить экспериментальные наблюдения, в том числе такие как увеличение наблюдаемой константы Михаэлиса для протромбиназы при увеличении концентрации липидных везикул [32]. Действительно, если цель мембраны – сконцентрировать факторы, то увеличение площади мембраны ведет к их разбавлению. В согласии с этим, мы сейчас знаем, что лишь часть тромбоцитов переходит в состояние прокоагулянтных для ускорения реакций свертывания [33–39], и возможное объяснение может быть в том, что слишком много тромбоцитов для этого будет вредно (рис. 2б). Более того, мы знаем, что белки свертывания на прокоагулянтных тромбоцитах концентрируются в специальной структуре – “шапке” (рис. 2в) [15], что может дополнительно ускорить реакции.
С другой стороны, уже почти 20 лет назад было показано, что растворимый фосфатидилсерин может поддерживать формирование как минимум одного комплекса (протромбиназы) не хуже, чем мембрана [40]. Это не очень хорошо согласуется с идеей “редукции числа измерений” или с идеей преодоления диффузионного барьера. Кроме того, диффузионные ограничения, описанные выше, недостаточно сильные, чтобы ферменты на максимуме возможностей не могли их преодолеть.
Альтернативная гипотеза, которая приходит на ум: мембраны могут быть важны для пространственной организации процесса. Около двадцати лет назад была предложена концепция “клеточной модели гемостаза” [41], которая делала упор на мембранных реакциях как особых компартментах, где белки свертывания защищены от ингибирования. Перемещение между компартментами управляется диффузией (нужно выйти из-под защиты мембраны и продиффундировать к соседнему тромбоциту или от поврежденного субэндотелия к тромбоциту), и действительно, в согласии с этим рост сгустка управляется диффузией фактора IXa, который гораздо медленнее ингибируется в плазме, чем фактор Xa [6, 42]. Интересно, что на тромбоцитах, помимо фосфатидилсерина, существуют иные способы связывания факторов свертывания (рецепторы, такие как гликопротеин Ib-V-IX, для тромбина и фактора XI, не имеющих доменов для связывания мембраны, а также возможность связывания фактора VIII тем же гликопротеином Ib-V-IX через фактор фон Виллебранда), которые, судя по всему, не ускоряют реакций с их участием, но защищают от ингибиторов [43].
Наличие связывания с мембраной может также менять наблюдаемые коэффициенты диффузии факторов свертывания в тромбоцитарных агрегатах [44], что может позволить управлять пространственной организацией процесса. Далее, мы знаем, что быстрые потоки могут нарушать работу химических систем, и свертывание крови, в частности, может “выключаться”, даже когда скорости потока заметно меньше сосудистых [9]. Тогда “привязка” факторов к мембранам позволит защитить их от потока. В согласии с этим, для ряда факторов сообщается формирование гомо- и гетеродимеров на мембране [16, 45], которые более прочно с ней связаны и защищены от смыва потоком. Еще один возможный аргумент в пользу предположения о роли мембран в пространственной организации тромбов – вытеснение прокоагулянтных тромбоцитов на периферию тромба, где происходит формирование фибринового слоя [29]. Таким образом, локализация фибрина в определенных областях тромба может быть достигнута за счет соотвествующего перераспределения прокоагулянтных мембран. Однако вытеснение прокоагулянтных тромбоцитов на периферию тромбов, их последующее взаимодействие с другими клетками и попадание в кровоток может приводить и к патофизиологическим процессам: так, формирование патологических агрегатов нейтрофилов с прокоагулянтными тромбоцитами и их фрагментами может являться одним из ключевых механизмов тромбирования легких при ишемии кишечника [46].
Необходимо признать, что хотя обе гипотезы о физическом смысле мембранных реакций кажутся интуитивно понятными и подтверждены косвенными наблюдениями, в настоящий момент нет ни прямых доказательств их справедливости, ни четких предсказаний необходимых экспериментов для того, чтобы их проверить.
Дополнительная сложность связана с тем, что источник прокоагулянтной активности в гемостазе и тромбозе не понятен до конца. Хотя прокоагулянтные тромбоциты кажутся подходящими кандидатами на эту роль, вклад непрокоагулянтных тромбоцитов, микровезикул плазмы крови, липопротеинов, эритроцитов, поврежденного эндотелия может быть значительным как в физиологических, так и в патологических условиях [11, 19, 42].
ФИБРИЛЛЯРНЫЕ СКАФФОЛДЫ
Фибрин
Традиционно фибрин рассматривается в первую очередь как пассивная составляющая тромба, хотя его способность влиять на свертывание разными путями известна давно (недаром исторически его называли “антитромбин I” за способность связывать тромбин). Сейчас становится понятно, что фибрин плотно и специфически связывает многочисленные молекулы и клетки, формируя мощную основу для протекания разнообразных биохимических процессов. Особенно ярко это выражается в “шапках” прокоагулянтых тромбоцитов, где сеть фибрина ассоциирует многочисленные молекулы альфа-гранул [47]. Взаимодействие фибрина с фактором Виллебранда и тромбоспондином, взаимодействие тромбоцитов с ними всеми и коллагеном, а также атака системой фибринолиза (и ее продолжением – системой металлопротеиназ матрикса) и фибрина, и коллагена позволяет рассматривать совокупность всех этих молекул как единый скаффолд “фибрин–внеклеточный матрикс”.
Минимальный список задач для этого скаффолда приведен ниже.
1. Фибрин сорбирует тромбин, который при этом сохраняет часть своей активности даже в иммобилизированном виде. Возможные роли в норме и патологии включают в себя ограничение пространственного распространения тромбина путем защиты от диффузии, а также возможность позднее отсоединиться и проявить свою активность.
2. Фибрин связывает фактор Виллебранда, к которому привязан фактор VIII. Кроме того, фибрин сам формирует эффективные сайты связывания для фактора VIII, которые могут быть важнее для связывания с тромбоцитами, чем фосфатидилсерин [21]. Недавние работы показывают, что фибрин может быть важен для связывания не только этого фактора [28]. В “шапках” прокоагулянтных тромбоцитов этот скаффолд переплетается с фосфатидилсерином и GPIb-ассоциированным фактором Виллебранда, а также тромбоспондином. В богатых тромбоцитами тромбах фактор Виллебранда оказывается критически важным элементом архитектуры, берущим на себя механическую функцию и препятствующим фибринолизу [20, 26].
3. Фибрин контролирует анти- и профибринолитические процессы. Он радикально ускоряет активацию фактора XIII тромбином, активацию плазминогена его тканевым активатором, а также защищает плазмин от быстрой инактивации в плазме (по интересной аналогии с защитой для мембранно-связанных ферментов свертывания).
Таким образом, фибрин можно рассматривать не только как механическую основу тромба и мишень для фибринолиза, но и как одну из “нерастворимых” фаз, контролирующих скорость работы, пространственное распространение и инактивацию ферментов свертывания крови и фибринолиза. Физический и физиологический смысл этого матрикса требует уточнений. Сейчас считается, что пространственное распределение фибрина в тромбах далеко не гомогенное, что открывает возможности для гибкой регуляции [29, 30]. Отметим, что фибрин может также служить механическим барьером как для проникновения в кровь патогенов при ранениях, так и для хемотаксиса иммунных клеток из крови в ткань, что позволяет регулировать локальное воспаление [48].
Коллаген
Среди скаффолдов внеклеточного матрикса, не ассоциированных напрямую с фибрином, стоит отдельно выделить фибриллярный коллаген – важнейший участник инициации гемостатического ответа при повреждении сосудистой стенки. После обнажения субэндотелиального матрикса происходит адгезия мультимеров фактора фон Виллебранда к коллагену стенок сосуда, а затем обратимое взаимодействие этих мультимеров с тромбоцитами через рецептор гликопротеин GPIb, необходимое для остановки тромбоцитов [49]. После этого происходит взаимодействие тромбоцитарного рецептора GPVI с коллагеном, активация тромбоцитов и их прочное закрепление на субэндотелии благодаря интегринам (a2β1 и a2bβ3) [50]. Известно, что локальные биомеханические (механическая жесткость субстрата) [51], гемодинамические (скорость сдвига и ее градиент) и биохимические (молекулярный состав области повреждения) [52, 53] параметры определяют не только то, в каких условиях протекают эти этапы, но и доминирующие молекулярные механизмы соответствующих процессов, что, в свою очередь, играет важную роль в дальнейшем росте сгустка [53–55]. Следует отметить, что фибриллярный коллаген также может участвовать в контактном пути свертывания за счет прямой активации XII фактора [56].
Как фибрин, так и фибриллярный коллаген обладают сложной пространственной организацией, и ключевые параметры этих скаффолдов (вязкоэластичные свойства и пространственная ориентация фибрилл) могут играть существенную роль в молекулярных процессах, которые разворачиваются на этих структурах. Следует отметить, что объединение коллагена и фибрина в единый тип фибриллярных скаффолдов в данном разделе не подразумевает их функциональную и пространственную ассоциацию в ходе гемостатического ответа, а выделяет фибриллярные скаффолды в отдельный тип внеклеточных матриц, которые играют важнейшую роль в разворачивании и регуляции процессов гемостаза.
Полифосфаты и ДНК в тромбах
Третий важнейший скаффолд, значимость которого была в полной мере раскрыта в последние десять лет, – это разнообразные полимеры фосфатов.
1. Неорганические полифосфаты плотных гранул тромбоцитов представляют собой плохо растворимые комплексы с кальцием, имеют среднюю длину в 60–100 фосфатных единиц и обладают набором биохимических свойств: они ускоряют активацию фактора V, блокируют работу ингибитора пути тканевого фактора, модулируют структуру сгустка, а также радикально ускоряют активацию фактора XI тромбином [23]. Они также являются кандидатами на роль активатора фактора XII на поверхности тромбоцитов [24], хотя есть данные против этого механизма [57] и предположения, что на самом деле это делают компоненты альфа-гранул [17]. Полифосфаты также замедляют фибринолиз через действие на тромбин-активируемый ингибитор фибринолиза [25].
2. Внеклеточные ловушки нейтрофилов представляют собой комплексы ДНК и гистонов, выбрасываемые нейтрофилами при активации. В частности, в процессе тромбообразования нейтрофилы активируются тромбоцитами через Р-селектин. Далее эти ловушки способны активировать свертывание по контактному пути, рекрутировать тромбоциты, внеклеточные везикулы и фактор фон Виллебранда, выступая как универсальные скаффолды [58]. Они играют важную роль в венозном и артериальном тромбозе; их роль в нормальном гемостазе не ясна.
3. Наконец, анализ тромбов при ишемическом инсульте выявил значительное количество ДНК предположительно лейкоцитарного происхождения; эти ДНК стабилизируют тромбы и препятствуют их лизису [26, 27]. Эти ДНК частично могут быть внеклеточными ловушками нейтрофилов [22].
ВЫВОДЫ
Свертывание крови и фибринолиз происходят в нескольких фазах, помимо растворимой. Можно выделить как минимум три пространственно разнесенных матрикса или скаффолда, которые служат основой для связывания белков и реализации определенных процессов в этих системах. Расширение наших знаний об этих процессах и формирование четких теорий, объясняющих разнесение и группирование процессов фибринолиза и свертывания между жидкой фазой плазмы крови и тремя “нерастворимыми” фазами необходимо для принципиального прогресса в нашем понимании гемостаза, тромбоза и регенерации ранений.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Источники финансирования. Работа была поддержана грантом Российского научного фонда 20-45-01014.
Соответствие принципам этики. Настоящая статья не содержит описания каких-либо исследований с участием людей или животных в качестве объектов.
Список литературы
Mann K.G., Orfeo T., Butenas S., Undas A., Brummel-Ziedins K. 2009. Blood coagulation dynamics in haemostasis. Hämostaseologie. 29 (1), 7–16.
Shibeko A.M., Panteleev M.A. 2016. Untangling the complexity of blood coagulation network: Use of computational modelling in pharmacology and diagnostics. Brief Bioinform. 17 (3), 429–439.
Panteleev M.A., Andreeva A.A., Lobanov A.I. 2020. Differential drug target selection in blood coagulation: What can we get from computational systems biology models? Curr. Pharm. Des. 26 (18), 2109–2115.
Panteleev M.A., Balandina A.N., Lipets E.N., Ovanesov M.V., Ataullakhanov F.I. 2010. Task–oriented modular decomposition of biological networks: Trigger mechanism in blood coagulation. Biophys. J. 98 (9), 1751–1761.
Shibeko A.M., Chopard B., Hoekstra A.G., Panteleev M.A. 2020. Redistribution of TPA fluxes in the presence of PAI-1 regulates spatial thrombolysis. Biophys. J. 119 (3), 638–651.
Panteleev M.A., Dashkevich N.M., Ataullakhanov F.I. 2015. Hemostasis and thrombosis beyond biochemistry: Roles of geometry, flow and diffusion. Thromb. Res. 136 (4), 699–711.
Dashkevich N.M., Ovanesov M.V., Balandina A.N., Karamzin S.S., Shestakov P.I., Soshitova N.P., Tokarev A.A., Panteleev M.A., Ataullakhanov F.I. 2012. Thrombin activity propagates in space during blood coagulation as an excitation wave. Biophys. J. 103 (10), 2233–2240.
Balandina A.N., Shibeko A.M., Kireev D.A., Novikova A.A., Shmirev I.I., Panteleev M.A., Ataullakhanov F.I. 2011. Positive feedback loops for factor V and factor VII activation supply sensitivity to local surface tissue factor density during blood coagulation. Biophys. J. 101 (8), 1816–1824.
Shibeko A.M., Lobanova E.S., Panteleev M.A., Ataullakhanov F.I. 2010. Blood flow controls coagulation onset via the positive feedback of factor VII activation by factor Xa. BMC Syst. Biol. 4, 5.
Mann K.G., Nesheim M.E., Church W.R., Haley P., Krishnaswamy S. 1990. Surface-dependent reactions of the vitamin K-dependent enzyme complexes. Blood. 76 (1), 1–16.
Podoplelova N.A., Nechipurenko D.Y., Ignatova A.A., Sveshnikova A.N., Panteleev M.A. 2021. Procoagulant platelets: Mechanisms of generation and action. Hämostaseologie. 41 (2), 146–153.
Kovalenko T.A., Panteleev M.A., Sveshnikova A.N. 2017. Substrate delivery mechanism and the role of membrane curvature in factor X activation by extrinsic tenase. J. Theor. Biol. 435, 125–133.
Panteleev M.A., Ananyeva N.M., Greco N.J., Ataullakhanov F.I., Saenko E.L. 2006. Factor VIIIa regulates substrate delivery to the intrinsic factor X-activating complex. FEBS J. 273 (2), 374–387.
Panteleev M.A., Saenko E.L., Ananyeva N.M., Ataullakhanov F.I. 2004. Kinetics of Factor X activation by the membrane-bound complex of Factor IXa and Factor VIIIa. Biochem. J. 381 (Pt 3), 779–794.
Podoplelova N.A., Sveshnikova A.N., Kotova Y.N., Eckly A., Receveur N., Nechipurenko D.Y., Obydennyi S.I., Kireev I.I., Gachet C., Ataullakhanov F.I., Mangin P.H., Panteleev M.A. 2016. Coagulation factors bound to procoagulant platelets concentrate in cap structures to promote clotting. Blood. 128 (13), 1745–1755.
Podoplelova N.A., Sveshnikova A.N., Kurasawa J.H., Sarafanov A.G., Chambost H., Vasil’ev S.A., Demina I.A., Ataullakhanov F.I., Alessi M.C., Panteleev M.A. 2016. Hysteresis-like binding of coagulation factors X/Xa to procoagulant activated platelets and phospholipids results from multistep association and membrane-dependent multimerization. Biochim. Biophys. Acta. 1858 (6), 1216–1227.
Zakharova N.V., Artemenko E.O., Podoplelova N.A., Sveshnikova A.N., Demina I.A., Ataullakhanov F.I., Panteleev M.A. 2015. Platelet surface-associated activation and secretion-mediated inhibition of coagulation factor XII. PLoS One. 10 (2), e0116665.
Terentyeva V.A., Sveshnikova A.N., Panteleev M.A. 2015. Kinetics and mechanisms of surface-dependent coagulation factor XII activation. J. Theor. Biol. 382, 235–243.
Lipets E., Vlasova O., Urnova E., Margolin O., Soloveva A., Ostapushchenko O., Andersen J., Ataullakhanov F., Panteleev M. 2014. Circulating contact-pathway-activating microparticles together with factors IXa and XIa induce spontaneous clotting in plasma of hematology and cardiologic patients. PLoS One. 9 (1), e87692.
Denorme F., Langhauser F., Desender L., Vandenbulcke A., Rottensteiner H., Plaimauer B., Francois O., Andersson T., Deckmyn H., Scheiflinger F., Kleinschnitz C., Vanhoorelbeke K., De Meyer S.F. 2016. ADAMTS13-mediated thrombolysis of t-PA-resistant occlusions in ischemic stroke in mice. Blood. 127 (19), 2337–2345.
Gilbert G.E., Novakovic V.A., Shi J., Rasmussen J., Pipe S.W. 2015. Platelet binding sites for factor VIII in relation to fibrin and phosphatidylserine. Blood. 126 (10), 1237–1244.
Laridan E., Denorme F., Desender L., Francois O., Andersson T., Deckmyn H., Vanhoorelbeke K., De Meyer S.F. 2017. Neutrophil extracellular traps in ischemic stroke thrombi. Ann. Neurol. 82 (2), 223–232.
Morrissey J.H., Choi S.H., Smith S.A. 2012. Polyphosphate: An ancient molecule that links platelets, coagulation, and inflammation. Blood. 119 (25), 5972–5979.
Muller F., Mutch N.J., Schenk W.A., Smith S.A., Esterl L., Spronk H.M., Schmidbauer S., Gahl W.A., Morrissey J.H., Renne T. 2009. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 139 (6), 1143–1156.
Smith S.A., Mutch N.J., Baskar D., Rohloff P., Docampo R., Morrissey J.H. 2006. Polyphosphate modulates blood coagulation and fibrinolysis. Proc. Natl. Acad. Sci. USA. 103 (4), 903–908.
Staessens S., Denorme F., Francois O., Desender L., Dewaele T., Vanacker P., Deckmyn H., Vanhoorelbeke K., Andersson T., De Meyer S.F. 2020. Structural analysis of ischemic stroke thrombi: Histological indications for therapy resistance. Haematologica. 105 (2), 498–507.
Staessens S., Francois O., Desender L., Vanacker P., Dewaele T., Sciot R., Vanhoorelbeke K., Andersson T., De Meyer S.F. 2021. Detailed histological analysis of a thrombectomy-resistant ischemic stroke thrombus: A case report. Thromb. J. 19 (1), 11.
Dohrmann M., Makhoul S., Gross K., Krause M., Pillitteri D., von Auer C., Walter U., Lutz J., Volf I., Kehrel B.E., Jurk K. 2020. CD36-fibrin interaction propagates FXI-dependent thrombin generation of human platelets. FASEB J. 34 (7), 9337–9357.
Nechipurenko D.Y., Receveur N., Yakimenko A.O., Shepelyuk T.O., Yakusheva A.A., Kerimov R.R., Obydennyy S.I., Eckly A., Leon C., Gachet C., Grishchuk E.L., Ataullakhanov F.I., Mangin P.H., Panteleev M.A. 2019. Clot contraction drives the translocation of procoagulant platelets to thrombus surface. Arterioscler. Thromb. Vasc. Biol. 39 (1), 37–47.
Kovalenko T.A., Giraud M.N., Eckly A., Ribba A.S., Proamer F., Fraboulet S., Podoplelova N.A., Valentin J., Panteleev M.A., Gonelle-Gispert C., Cook S., Lafanechere L., Sveshnikova A.N., Sadoul K. 2021. Asymmetrical forces dictate the distribution and morphology of platelets in blood clots. Cells. 10 (3), 584.
Zhou G.Q., Zhong W.Z. 1982. Diffusion-controlled reactions of enzymes. A comparison between Chou’s model and Alberty-Hammes-Eigen’s model. Eur. J. Biochem. 128 (2–3), 383–387.
Nesheim M.E., Tracy R.P., Mann K.G. 1984. “Clotspeed,” a mathematical simulation of the functional properties of prothrombinase. J. Biol. Chem. 259 (3), 1447–1453.
Obydennyy S.I., Sveshnikova A.N., Ataullakhanov F.I., Panteleev M.A. 2016. Dynamics of calcium spiking, mitochondrial collapse and phosphatidylserine exposure in platelet subpopulations during activation. J. Thromb. Haemost. 14 (9), 1867–1881.
Panteleev M.A., Ananyeva N.M., Greco N.J., Ataullakhanov F.I., Saenko E.L. 2005. Two subpopulations of thrombin-activated platelets differ in their binding of the components of the intrinsic factor X-activating complex. J. Thromb. Haemost. 3 (11), 2545–2553.
Sveshnikova A.N., Ataullakhanov F.I, Panteleev M.A. 2015. Compartmentalized calcium signaling triggers subpopulation formation upon platelet activation through PAR1. Mol. Biosyst. 11 (4), 1052–1060.
Sveshnikova A.N., Balatskiy A.V., Demianova A.S., Shepelyuk T.O., Shakhidzhanov S.S., Balatskaya M.N., Pichugin A.V., Ataullakhanov F.I., Panteleev M.A. 2016. Systems biology insights into the meaning of the platelet’s dual-receptor thrombin signaling. J. Thromb. Haemost. 14 (10), 2045–2057.
Topalov N.N., Kotova Y.N., Vasil’ev S.A., Panteleev M.A. 2012. Identification of signal transduction pathways involved in the formation of platelet subpopulations upon activation. Br. J. Haematol. 157 (1), 105–115.
Topalov N.N., Yakimenko A.O., Canault M., Artemenko E.O., Zakharova N.V., Abaeva A.A., Loosveld M., Ataullakhanov F.I., Nurden A.T., Alessi M.C., Panteleev M.A. 2012. Two types of procoagulant platelets are formed upon physiological activation and are controlled by integrin alpha(IIb)beta(3). Arterioscler. Thromb. Vasc. Biol. 32 (10), 2475–2483.
Yakimenko A.O., Verholomova F.Y., Kotova Y.N., Ataullakhanov F.I., Panteleev M.A. 2012. Identification of different proaggregatory abilities of activated platelet subpopulations. Biophys. J. 102 (10), 2261–2269.
Majumder R., Weinreb G., Lentz B.R. 2005. Efficient thrombin generation requires molecular phosphatidylserine, not a membrane surface. Biochemistry. 44 (51), 16998–17006.
Hoffman M., Monroe D.M., 3rd. 2001. A cell-based model of hemostasis. Thromb. Haemost. 85 (6), 958–965.
Panteleev M.A., Ovanesov M.V., Kireev D.A., Shibeko A.M., Sinauridze E.I., Ananyeva N.M., Butylin A.A., Saenko E.L., Ataullakhanov F.I. 2006. Spatial propagation and localization of blood coagulation are regulated by intrinsic and protein C pathways, respectively. Biophys. J. 90 (5), 1489–1500.
Reitsma S.E., Pang J., Raghunathan V., Shatzel J.J., Lorentz C.U., Tucker E.I., Gruber A., Gailani D., McCarty O.J.T., Puy C. 2021. Role of platelets in regulating activated coagulation factor XI activity. Am. J. Physiol. Cell Physiol. 320 (3), C365–C374.
Hathcock J.J., Nemerson Y. 2004. Platelet deposition inhibits tissue factor activity: In vitro clots are impermeable to factor Xa. Blood. 104 (1), 123–127.
Koklic T., Majumder R., Weinreb G.E., Lentz B.R. 2009. Factor XA binding to phosphatidylserine-containing membranes produces an inactive membrane-bound dimer. Biophys. J. 97 (8), 2232–2241.
Yuan Y., Alwis I., Wu M.C., Kaplan Z., Ashworth K., Bark D., Pham A., Mcfadyen J., Schoenwaelder S.M., Josefsson E.C., Kile B.T. 2017. Neutrophil macroaggregates promote widespread pulmonary thrombosis after gut ischemia. Sci. Trans. Med. 9 (409), eaam5861.
Abaeva A.A., Canault M., Kotova Y.N., Obydennyy S.I., Yakimenko A.O., Podoplelova N.A., Kolyadko V.N., Chambost H., Mazurov A.V., Ataullakhanov F.I., Nurden A.T., Alessi M.C., Panteleev M.A. 2013. Procoagulant platelets form an alpha-granule protein-covered “cap” on their surface that promotes their attachment to aggregates. J. Biol. Chem. 288 (41), 29 621–29 632.
Kaplan Z.S., Zarpellon A., Alwis I., Yuan Y., McFadyen J., Ghasemzadeh M., Schoenwaelder S.M., Ruggeri Z.M., Jackson S.P. 2015. Thrombin-dependent intravascular leukocyte trafficking regulated by fibrin and the platelet receptors GPIb and PAR4. Nat. Commun. 6, 7835.
Ruggeri Z.M. 2007. The role of von Willebrand factor in thrombus formation. Thromb. Res. 120 Suppl 1, S5–9.
Nieswandt B., Watson S.P. 2003. Platelet-collagen interaction: Is GPVI the central receptor? Blood. 102 (2), 449–461.
Qiu Y., Brown A.C., Myers D.R., Sakurai Y., Mannino R.G., Tran R., Ahn B., Hardy E.T., Kee M.F., Kumar S., Bao G., Barker T.H., Lam W.A. 2014. Platelet mechanosensing of substrate stiffness during clot formation mediates adhesion, spreading, and activation. Proc. Natl. Acad. Sci. USA. 111 (40), 14 430–14 435.
Barnes M.J., MacIntyre D.E. 1979. Platelet-reactivity of isolated constituents of the blood vessel wall. Haemostasis. 8 (3–5), 158–170.
Gutierrez E., Petrich B.G., Shattil S.J., Ginsberg M.H., Groisman A., Kasirer-Friede A. 2008. Microfluidic devices for studies of shear-dependent platelet adhesion. Lab. Chip. 8 (9), 1486–1495.
van Geffen J.P., Brouns S.L.N., Batista J., McKinney H., Kempster C., Nagy M., Sivapalaratnam S., Baaten C., Bourry N., Frontini M., Jurk K., Krause M., Pillitteri D., Swieringa F., Verdoold R., Cavill R., Kuijpers M.J.E., Ouwehand W.H., Downes K., Heemskerk J.W.M. 2019. High-throughput elucidation of thrombus formation reveals sources of platelet function variability. Haematologica. 104 (6), 1256–1267.
Welsh J.D., Poventud-Fuentes I., Sampietro S., Diamond S.L., Stalker T.J., Brass L.F. 2017. Hierarchical organization of the hemostatic response to penetrating injuries in the mouse macrovasculature. J. Thromb. Haemost. 15 (3), 526–537.
van der Meijden P.E., Munnix I.C., Auger J.M., Govers-Riemslag J.W., Cosemans J.M., Kuijpers M.J., Spronk H.M., Watson S.P., Renne T., Heemskerk J.W. 2009. Dual role of collagen in factor XII-dependent thrombus formation. Blood. 114 (4), 881–890.
Faxalv L., Boknas N., Strom J.O., Tengvall P., Theodorsson E., Ramstrom S., Lindahl T.L. 2013. Putting polyphosphates to the test: Evidence against platelet-induced activation of factor XII. Blood. 122 (23), 3818–3824.
Thalin C., Hisada Y., Lundstrom S., Mackman N., Wallen H. 2019. Neutrophil extracellular traps: Villains and targets in arterial, venous, and cancer-associated thrombosis. Arterioscler. Thromb. Vasc. Biol. 39 (9), 1724–1738.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии