

Биологические мембраны: Журнал мембранной и клеточной биологии, 2022, T. 39, № 4, стр. 307-318
Изменения pH в матриксе митохондрий и цитозоле при индуцированной глутаматом дисрегуляции Са2+-гомеостаза в культивируемых нейронах гиппокампа крысы
А. М. Сурин a, b, *, Л. Р. Горбачева c, d, И. Г. Савинкова c, Р. Р. Шарипов a, В. Г. Пинелис b
a НИИ общей патологии и патофизиологии
125315 Москва, Россия
b “НМИЦ здоровья детей” Минздрава России
119991 Москва, Россия
c Российский национальный исследовательский медицинский университет им. Н.И. Пирогова
117513 Москва, Россия
d Московский государственный университет им. М.В. Ломоносова, биологический факультет
119234 Москва, Россия
* E-mail: surin_am@mail.ru
Поступила в редакцию 02.03.2022
После доработки 21.03.2022
Принята к публикации 27.03.2022
- EDN: FHVFZG
- DOI: 10.31857/S0233475522040089
Аннотация
Воздействие высоких концентраций глутамата (Glu) на первичные культуры нейронов из мозга крысы приводит к сильной деполяризации митохондрий, развивающейся синхронно со вторичным подъемом внутриклеточной концентрации свободного Са2+ (отсроченной кальциевой дисрегуляцией, ОКД). В данной работе одновременно с измерениями внутриклеточной концентрации свободного Ca2+ ([Ca2+]i) были измерены pH в матриксе митохондрий (рНm) и цитозоле (рНс) нейронов, при действии токсической дозы Glu (100 мкМ). Для этого в первичных культурах из гиппокампа новорожденных крыс была достигнута экспрессия pH-чувствительного зеленого белка mtYFP в митохондриях и рН-чувствительного красного белка mKate в цитозоле. Полученную нейрональную культуру нагружали Ca2+-индикатором Fura-FF и в тех нейронах, которые экспрессировали совместно mtYFP и mKate, одновременно измеряли [Ca2+]i, рНm и рНс. Обнаружено, что во время первой фазы Ca2+-ответа на Glu, когда наблюдается частичная деполяризация митохондрий, происходит увеличение градиента pH между матриксом митохондрий и цитозолем (ΔpH), которое компенсирует снижение электрического компонента митохондриального потенциала (∆Ψm), поддерживая тем самым постоянство электрохимического потенциала митохондрий. Развитие ОКД приводит к резкому снижению ∆Ψm и ΔpH в соме нейронов, однако полного коллапса ΔpH не наблюдается. Это может означать, что ОКД не обусловлена неспецифической мегапорой во внутренней мембране митохондрий (мРТР), как это принято считать. Либо часть митохондрий в соме нейронов сохраняет барьерные свойства внутренней мембраны и не формирует мРТР даже при развитии ОКД и достижении высокого Ca2+-плато.
ВВЕДЕНИЕ
Глутамат (Glu) – основной возбуждающий нейротрансмиттер в центральной нервной системе, контролирует различные клеточные и синаптические функции [1]. Glu принимает участие в развитии таких заболеваний, как эпилепсия и нейродегенеративные расстройства [2–5]. В области, окружающей зону поражения при черепно-мозговой травме и инсульте, Glu превращается в сильнейший нейротоксин [6–8].
Детальное изучение изменений внутриклеточной концентрации свободных ионов кальция ([Ca2+]i) при действии Glu привело к открытию явления, названного отсроченной кальциевой дисрегуляцией (ОКД) [9, 10]. Динамика изменений [Ca2+]i имеет сложный трехфазный характер и тесно связана с изменениями электрического компонента трансмембранного потенциала внутренней мембраны митохондрий (∆Ψm) [11, 12]. В настоящее время считается, что митохондрии, как основной производитель ATP и самое емкое внутриклеточное Са2+-депо, играют ведущую роль в развитии ОКД [11–15].
Применение флуоресцентного белкового ATP-сенсора позволило обнаружить ряд ранее неизвестных особенностей воздействия Glu на нейроны в культуре [16]. Сопоставление кинетики изменений концентрации ATP в цитозоле ([ATP]c) и [Ca2+]i показало, что падение [ATP]c в каждом нейроне, экспрессировавшем ATP-сенсор, всегда опережало развитие ОКД, которое начиналось при достижении [ATP]c ~16% от уровня в покоящихся нейронах. В фазе высокого [Ca2+]i-плато [ATP]с опускалась до ~10% от уровня покоя. Эти величины в 4–6 раза ниже значений [ATP]с, полученных с помощью физико-химических или биохимических методов анализа в среднем по клеточной популяции [17–20]. Причина расхождения, по крайней мере отчасти, связана с ATP, поступающим из глиальных клеток, 15–35% которых всегда присутствуют в нейрональных культурах [21]. Полностью подавить рост глиальных клеток не удается, даже применяя цитостатик Ara-C при концентрациях, не убивающих нейроны (см., например, рис. 2A в [21] и Материалы и методы, первый абзац).
Стимуляция ионотропных глутаматных рецепторов при патологических процессах вызывает не только мощный вход Са2+, но и сильное закисление цитоплазмы и митохондрий, обусловленное работой Са2+-ATP-аз и Na+/H+-обменников плазмалеммы [22] и митохондриальным Са2+/H+-обменом [23, 24]. В этой связи pH-зависимость сигналов флуоресцентных белковых сенсоров является не только недостатком, осложняющим интерпретацию сигналов [16], но может быть и достоинством, позволяющим использовать их в качестве pH-индикаторов в интересующем компартменте клетки, например, только в матриксе митохондрий или только в цитозоле [24–26].
Главные функциональные характеристики митохондрий, а именно, способность синтезировать ATP, захватывать из цитоплазмы Ca2+ и другие катионы, транспортировать белки в матрикс, удерживать факторы апоптоза, обусловлены трансмембранным электрохимическим потенциалом их внутренней мембраны (ΔµH) [27, 28]. Этот важнейший интегральный показатель функционального состояния митохондрий определяется суммой электрического трансмембранного потенциала (ΔΨm) и трансмембранного градиента рН между матриксом митохондрий и цитозолем (ΔpH). Изменения ∆Ψm в нейронах во время первой фазы Ca2+-ответа на нейротоксическое действие Glu и при развитии ОКД почти “зеркально” отражают изменения [Ca2+]i [10, 11], тогда как относительно изменений ΔpH в индивидуальных нейронах, насколько нам известно, нет экспериментальных данных. Поэтому в данной работе одновременно с измерениями [Ca2+]i были измерены pH в матриксе митохондрий (рНm) и цитозоле (рНс) нейронов, подвергнутых токсическому воздействию Glu. Для этого в первичных культурах из гиппокампа новорожденных крыс экспрессировали pH-чувствительный зеленый белок mtYFP в митохондриях и рН-чувствительный красный белок mKate в цитозоле. Обнаружено, что во время первой фазы Ca2+-ответов нейронов на глутамат происходит увеличение ΔpH, которое компенсирует, по крайней мере отчасти, снижение ΔΨm. При развитии ОКД и установлении высокого [Ca2+]i-плато ΔpH резко падает, однако полного коллапса ΔpH не происходит. Вероятно, развитие ОКД не обусловлено образованием неспецифической поры высокой проводимости, либо такая пора возникает не во всех митохондриях в соме нейронов.
МАТЕРИАЛЫ И МЕТОДЫ
Первичные нейрональные культуры из гиппокампа новорожденных крыс получали, как описано в [29]. Кратко, животных анестезировали, декапитировали, извлекали мозг и затем гиппокампы. Суспензию клеток (106 клеток/мл) получали, обрабатывая гиппокампы папаином (10 ед/мл), диссоциируя 15-кратным пипетированием, и отмывали от разрушенных клеток двукратным осаждением в центрифуге (1000 об/мин). Суспензию (200 мкл) переносили на покровные стекла, прикрепленные к лункам 35 мм пластиковых чашек Петри (MatTek, США). Стекла предварительно покрывали полиэтиленимином (10 мг/мл). Через час добавляли 1.5 мл нейробазальной среды, содержащей 2% Supplement B-27 и 0.5 мМ L-глутамина. Клетки содержали при 37°C в атмосфере 5% СО2/95% воздуха при 100% влажности. На 3–4 день добавляли арабинозид С (Ara-C, 2 мкМ) для подавления роста глиальных клеток. Культуры использовали на 8–10 день после посева (3–4 день после трансфекции).
Эксперименты на животных выполняли в соответствии с этическими принципами и нормативными документами, рекомендованными Европейской конвенцией о защите позвоночных животных, используемых в экспериментах (Guide for the Care and Use of Laboratory Animals: Eighth Edition, 2010), а также в соответствии с “Правилами надлежащей лабораторной практики”, утвержденными приказом Министерства здравоохранения РФ № 199н от 01.04.2016 г.
Микрофлуориметрические измерения выполнены при температуре 27–29°C в буфере, содержащем (мM): 135 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 20 HEPES, 5 D-глюкозы; pH 7.4 устанавливали, добавляя 20 мМ HEPES и дотитровывая 1 М HCl. Номинально бескальциевые растворы вместо CaCl2 содержали 0.1мМ EGTA и 2 мМ MgCl2. Смену растворов осуществляли 2 × 2 мл заменой содержимого чашки с клетками за время не более 30 с.
Для измерения [Ca2+]i клетки нагружали Fura-FF в форме ацетоксиметилового (АМ) эфира (концентрация маточного раствора 2 мМ в ДМСО). Fura-FF/АМ предварительно смешивали с неионным детергентом Pluronic F-127 (Molecular Probes, США) для предотвращения образования в буфере суспензий АМ-эфиров индикаторов. Нагрузку проводили при температуре 25–26°C в указанном выше буфере в течение 40–50 мин. Концентрации Fura-FF/АМ и Pluronic F-127 составляли соответственно 3–5 мкМ и 0.02%.
Для измерения pH митохондрий и цитозоля плазмиды, несущие гены соответствующих белков, доставляли в клетки, используя Lipofectamine-2000 (LF-2000). Плазмиду (2–3 мкг ДНК/100 мкл OptiMem) смешивали с LF-2000 (4–6 мкл/100 мкл OptiMem) и через 20 мин, когда завершалось образование комплекса ДНК с LF-2000, вносили смесь в клеточную культуру. Перед этим кондиционированную клеточную среду временно заменяли на Transfect Medium (200 мкл в лунке с клетками) и в каждую лунку вносили 50 мкл комплекса ДНК с LF-2000, после чего помещали клетки в СО2-инкубатор. Через 2 ч удаляли Transfect Medium, ополаскивали клетки нейробазальной средой и возвращали клеткам ту кондиционную среду, в которой они находились перед трансфекцией. Калибровку pH-зависимости сигналов флуоресцентных белковых сенсоров в нейронах проводили как описано в [24] с небольшими изменениями в способе расчета, используя растворы с ионофорами, обеспечивающими выравнивание pH между буфером и внутриклеточной средой. Калибровочные растворы имели состав (мМ): 0.005 нигерицина, 0.001 FCCP, 134 глюконата калия, 1 MgCl2, 20 HEPES. Для доведения pH использовали 1 М растворы HCl или КОН. Для предотвращения работы митохондриальной ATP-азы в прямом или реверсивном режиме, что могло потенциально повлиять на воспроизводимость калибровочных кривых, в калибровочные растворы добавляли олигомицин (2.5 мкг/мл).
В тех нейронах, которые экспрессировали одновременно mtYFP и mKate, выполняли измерения [Ca2+]i, рНm и рНс. Флуоресцентно-микроскопические измерения проводили на установках, включающих инвертированный микроскоп Olympus IX-71, систему освещения Sutter Labmda 10-2 с ксеноновой лампой 175 Вт (Sutter Instruments) и CCD-камеру CoolSNAP HQ2 (Photometrics), управляемых через компьютерную программу MetaFluor (Molecular Devices, США). Возбуждающий свет от лампы проходил поочередно через светофильтры 340 ± 8 и 380 ± 8 нм для Fura-FF и через 485 ± 10 и 565 ± 15 нм соответственно для mtYFP и mKate и отражался на образец трехполосным зеркалом (максимумы отражения 300–400 нм, 485 ± 15 и 560 ± 20 нм; полосы пропускания 460 ± 20, 525 ± 20 и выше 580 нм). Для регистрации сигналов Fura-FF и mtYFP использовали один и тот же эмиссионный светофильтр 525 ± 15 нм. Для регистрации эмиссии mKate использовали пороговый светофильтр, имеющий пропускание 590–700 нм. Светофильтры и дихроичные зеркала производства Omega (США).
Все реагенты фирмы Sigma (США). Флуоресцентный Ca2+-индикатор Fura-FF приобретен у Invitrogen (США). Плазмиды, кодирующие флуоресцентные белковые сенсоры, были любезно предоставлены Dr. R. Rizzuto (University of Ferrara, Италия) и проф. Д. Чудаковым (ИБХ им. М.М. Шемякина и Ю.А. Овчинникова РАН).
Для обсчета флуоресцентных сигналов и построения графиков, а также для статистической обработки данных использовали программы MetaFluor Analyst, Excel, GraphPad Prizm 6.1 и Origin 7.0.
РЕЗУЛЬТАТЫ
Применение смеси двух плазмид, содержащих гены флуоресцентных белковых рН-сенсоров (имеющих разные спектры возбуждения и эмиссии), приводит к трансфекции как нейронов, так и астроцитов. Следует отметить, что трансфекция терминально дифференцированных клеток с применением Lipofectamine-2000 не очень высокая. В наших экспериментах доля трансфицированных нейронов в культурах гиппокампа новорожденных крыс обычно не превышала 0.1%. Эффективность трансфекции астроцитов была в 3–5 раз выше. Соотношение флуоресцентных сигналов может значительно различаться как между индивидуальными клетками, так и в каждой трансфицированной клетке, если она экспрессирует более одного флуоресцентного белка. На рис. 1 и 2 представлены изображения нейронов, в митохондриях которых экспрессируется зеленый флуоресцентный белок mtYFP [24] (рис. 1а и 2а), а в цитозоле красный флуоресцентный белок mKate [30] (рис. 1б и 2б). При использовании объектива с относительно высоким увеличением и разрешением хорошо видно, что mtYFP заполняет часть перинуклеарного пространства в соме и в виде пунктиров расположен в дендритах и аксоне (рис. 1а). Аналогичный характер флуоресценции наблюдается при окрашивании нейронов митотрекерами [31] или митохондриальными потенциал-чувствительными зондами [21, 32].
Рис. 1.
Экспрессия одновременно двух флуоресцентных белков не изменяет характер Ca2+-ответов на глутамат. Флуоресцентные изображения первичной культуры клеток гиппокампа крысы, экспрессирующих в одном из нейронов зеленый pH-чувствительный белок mtYFP в митохондриях (а) и красный pH-чувствительный белок mKate в цитозоле (б). На панели в приведено изображение всех клеток в поле наблюдения, нагруженных низкоаффинным флуоресцентным Ca2+-индикатором Fura-FF. Графики изменений [Ca2+]i, индуцированные глутаматом (Glu, 100мкМ в безмагниевом буфере в присутствии 10 мкМ глицина), представлены на панели г; пунктиром выделен график изменений [Ca2+]i в нейроне, экспрессировавшем одновременно mtYFP и mKate. Цифрами 1, 2 и 3 отмечены клетки, в которых за время действия Glu развилась ОКД. Изображение в соответствует 1770 с на панели г или 1170 с с момента добавления Glu. Длины волн возбуждения и испускания флуоресценции см. Материалы и методы; объектив (40×/NA = 1.35 oil). Масштабный отрезок на панелях а, б, в соответствует 20 мкм.
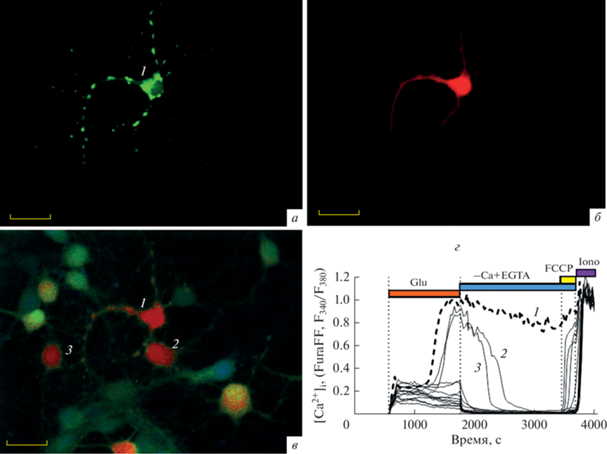
Рис. 2.
Экспрессия одновременно двух флуоресцентных белков и окрашивание нейронов Ca2+-индикатором позволяет сопоставить изменения pH в митохондриях и цитозоле (pHm и pHc) с изменениями [Ca2+]i. Изображения гиппокампальных нейронов, (а) экспрессирующих зеленый pH-чувствительный белок mtYFP в митохондриях, (б) красный pH-чувствительный белок mKate в цитозоле и (в) нагруженных низкоаффинным Са2+-индикатором Fura-FF (возб. 340, 380, испуск. 525 нм). г – Графики изменений pHm и pHc одного из нейронов, показанных на изображениях (ROI 9; обведен кружком) при действии токсических доз глутамата (Glu), а также ингибитора дыхания цианида (CN, 3 мМ), ингибитора митохондриальной ATP-азы олигомицина (Oligo, 2.5 мкг/мл) и протонофора FCCP (1 мкМ). д – Графики изменения [Ca2+]i и градиента pH между матриксом митохондрий и цитозолем (ΔpH) того же нейрона (ROI 9). Объектив 20×/NA = 0.70. Концентрация Glu и условия регистрации флуоресцентных сигналов как на рис. 1. Масштабный отрезок на панелях а, б, в соответствует 40 мкм.
В отличие от mtYFP, красный флуоресцентный белок mKate, не имеющий адресной пептидной последовательности, заполняет цитозоль и ядро (рис. 1б). В ядерной оболочке имеются поры, размер которых позволяет диффундировать из цитозоля в ядро и обратно молекулам размером до 20–40 кДа [33, 34], что согласуется с размером мономерных флуоресцентных белков, в частности mKate [30].
На рис. 1г, 3а, 3б показаны графики изменений [Ca2+]i для группы нейронов, окрашенных Fura-FF (рис. 1в). Видно, что процедура трансфекции (см. Материалы и методы) не отменила индивидуальности ответов нейронов на Glu. Часть нейронов смогла противостоять токсическому действию Glu и в них не возникла ОКД (11 из 14 на рис. 1г). В трех нейронах развилась ОКД (обозначены цифрами 1, 2 и 3 на рис. 1в; их графики отмечены 1, 2 и 3 на рис. 1г), в том числе и в том, который экспрессировал mtYFP и mKate (рис. 1). Индивидуальность развития ОКД в нейронах в ответ на Glu хорошо документирована [11]. Если процедура трансфекции подобрана корректно, то индивидуальность Ca2+-ответов сохраняется даже в тех случаях, когда используют субъединичные флуоресцентные белковые сенсоры, молекулярная масса которых в 3 раза превышает массу mtYFP и mKate [16, 35].
Чтобы выяснить, как соотносятся изменения рНm и рНс с изменениями [Ca2+]i в индивидуальных нейронах, клеточную культуру нагружали Fura-FF и выбирали участок, в котором находилось как можно больше нейронов, экспрессировавших одновременно mtYFP и mKate. Пример участка нейрональной культуры, в котором содержится группа таких нейронов, представлена на рис. 2а, 2б, 2в. Изменения рНm, рНс, их разности (ΔpH) и [Ca2+]i в этих нейронах, а также в части клеток, не экспрессировавших белковые сенсоры, показаны на рис. 3.
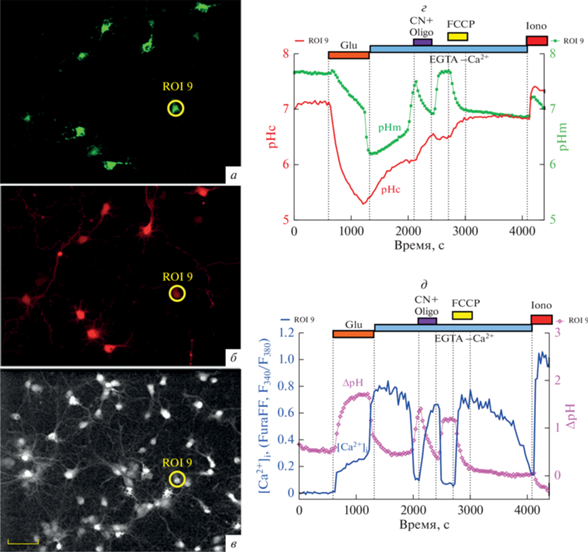
Рис. 3.
Изменения [Ca2+]i и pH в цитозоле (pHc) и в матриксе митохондрий (pHm) клеток гиппокампа крысы, изображения которых представлены на рис. 2. а – Изменения [Ca2+]i в клетках, экспрессировавших одновременно флуоресцентные белковые pH-сенсоры в цитозоле (mKate) и митохондриях (mtYFP). б – Изменения [Ca2+]i в клетках, не экспрессировавших mtYFP и mKate. в – Изменения сигналов mtYFP, (г) mKate и (д) ∆pH. е – Максимальные значения ∆pH во время действия Glu и через ~200 с после отмывки. Для сравнения сигналы одного из нейронов, приведенных на рис. 2 (ROI 9), выделены цветными линиями; сигналы остальных нейронов представлены серыми линиями. На панели е точки, соответствующие нейрону (ROI 9), отмечены цветными точками; столбики “∆pH макс с ОКД” и “∆pH макс без ОКД” соответствуют максимальным значениям ∆pH в каждом индивидуальном нейроне, не имевшем ОКД или во время первой фазы подъема [Ca2+]i, предшествующей ОКД в тех нейронах, где ОКД развилась; столбики “∆pH после ОКД (1510 с)” соответствуют значениям ∆pH при максимальном подъеме [Ca2+]i в результате развития ОКД (примерно 1510 с на панелях а–д); “∆pH без ОКД (1510с)” – значения ∆pH в тех нейронах, в которых ОКД не произошла.
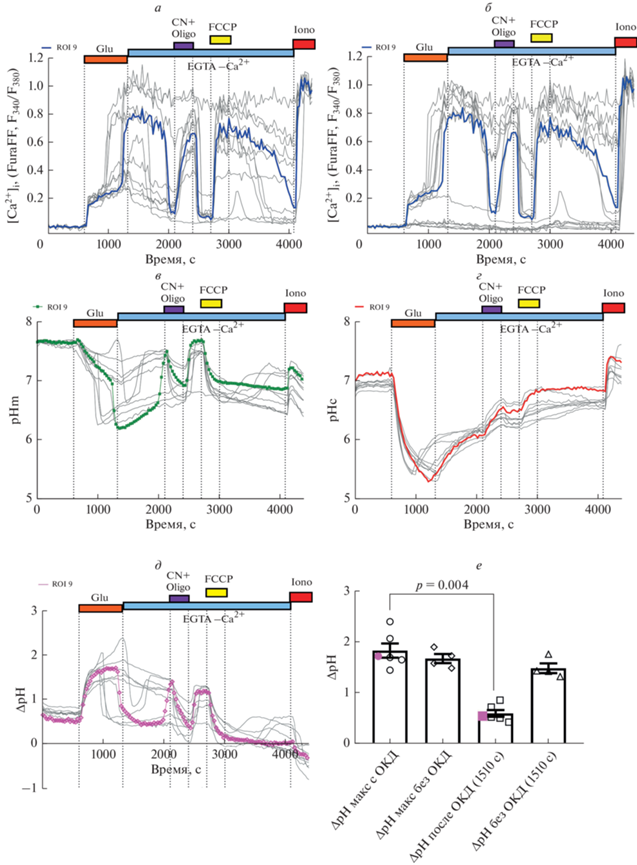
Сразу после добавления Glu одновременно со скачком [Ca2+]i (рис. 2д, 3а, 3б) происходило снижение рНm и рНс (рис. 2г, 3в, 3г). Динамика изменений рНm и рНс заметно различалась, что приводило к росту ΔpH. По мере стабилизации изменений [Ca2+]i во время лаг-периода, предшествующего началу ОКД, стабилизировались также изменения ΔpH (рис. 2д, 3д).
Выполненные ранее нами [11, 36] и другими [37] исследования показали, что изменения [Ca2+]i в ответ на токсическую концентрацию Glu имеют “зеркальный” характер относительно изменений ΔΨm. В момент добавления Glu происходит сравнительно небольшой скачок [Ca2+]i от базального ~100 нМ [38] до нескольких микромолей/литр (первая фаза подъема [Ca2+]i) [11, 38, 39]. Этот небольшой подъем [Ca2+]i стабилизируется на период от единиц до десятков минут и сопровождается снижением ΔΨm на ~60 мВ [10, 32]. Это умеренное снижение ΔΨm во время первой фазы Ca2+-ответа на Glu компенсируется ростом ΔpH (рис. 2д, 3д, 3е).
При продолжительном действии Glu наступает вторая фаза изменений [Ca2+]i, названная ОКД, при которой [Ca2+]i поднимается до десятков микромолей/литр [39]. Этот подъем осуществляется с лаг-периодом, индивидуальным для каждого нейрона (например, рис. 1г и 3а, 3б) и происходит всегда синхронно с сильным падением ΔΨm [11, 29]. До конца не ясно, от чего в большей степени зависит вторичный подъем [Ca2+]i – происходит ли выброс Ca2+ из митохондрий одновременно с деполяризацией или же больше сказывается поступление Ca2+ извне. В развитие ОКД и стабилизацию [Ca2+]i на уровне высокого плато вовлечены, по-видимому, различающиеся процессы, поскольку удаление Ca2+ из буфера в самом начале ОКД быстро снижает [Ca2+]i, тогда как смена буфера на бескальциевый в фазе высокого [Ca2+]i-плато влияет на [Ca2+]i гораздо слабее [11, 36].
При возникновении ОКД происходило дополнительное снижение pHm, тогда как падение pHc замедлялось или даже сменялось его ростом (рис. 2г, 3в, 3г), в результате чего ΔpH быстро падала (рис. 2д). Обращает внимание то, что ΔpH уменьшался до ~0.5 единиц рН (рис. 2д и 3д, 3е), т.е. до уровня, наблюдаемого в покоящихся нейронах.
Отмывание Glu бескальциевым буфером приводило к снижению [Ca2+]i в тех клетках, в которых не наступила ОКД (рис. 3а, 3б). Если в клетках развилась ОКД, то восстановление низкой [Ca2+]i происходило с задержкой, а в некоторых не восстанавливалась за время измерений. Во время Ca2+-плато происходило постепенное защелачивание матрикса, которое ускорялось в постглутаматный период при восстановлении низкой [Ca2+]i (рис. 2г и 3в).
В цитозоле падение рН сменялось ростом при наступлении ОКД, а также при прекращении действия Glu (рис. 2г и 3г). Другими словами, в постглутаматный период происходило постепенное выравнивание градиента рН между матриксом митохондрий и цитозолем, однако ни в одной из клеток за время действия Glu не наступило коллапса ∆pH (рис. 3д, 3е).
Добавление в постглутаматный период ингибитора комплекса IV дыхательной цепи цианида (NaCN, 3 мМ) в сочетании с ингибированием F1F0ATP-азы олигомицином (Oligo, 2.5 мкг/мл), вызывающее коллапс ΔΨm, быстро увеличивал [Ca2+]i в результате мобилизации Ca2+ из митохондрий в цитозоль (рис. 2д и 3а, 3б). К подобному подъему [Ca2+]i приводила замена цианида на ингибиторы комплексов I или III дыхательной цепи (соответственно, ротенон или антимицин A; данные не показаны). Причиной роста [Ca2+]i являлась, очевидно, ликвидация электрофоретической силы, удерживавшей Ca2+ в матриксе митохондрий [11, 13].
Цианид резко закислял матрикс и ускорял защелачивание цитозоля, сближая величины pHm и pHc (рис. 2г и 3в, 3г). Это указывает на то, что при остановке дыхательной цепи утечка протонов сквозь внутреннюю мембрану митохондрий способна ликвидировать ∆pH за несколько минут (рис. 2д и 3д).
Отмывание NaCN быстро понижало [Ca2+]i в большинстве клеток (в 16 из 26), в которых Glu индуцировал подъем [Ca2+]i (рис. 3а, 3б). Очевидно, в результате разблокирования комплекса IV дыхательной цепи происходило восстановление ∆Ψm и возобновление электрофоретического захвата Са2+ митохондриями.
Отмывание цианида также быстро восстанавливало pHm до уровня, предшествовавшего действию митохондриального яда (рис. 3в). Это наблюдение согласуется с тем, что дыхательная цепь возобновила выкачивание протонов из матрикса в цитозоль, повышая тем самым pHm.
Протонофор FCCP (1мкМ), добавленный через 5 мин после отмывания цианида, резко увеличивал [Ca2+]i и уменьшал разницу между pHm и pHc (рис. 3). Напомним, что добавление FCCP часто используют как методический прием, чтобы вызвать коллапс ΔΨm и проверить наличие Са2+ в митохондриях [11, 13]. Скачок [Ca2+]i, индуцированный FCCP, согласуется с тем, что понижение [Ca2+]i после удаления NaCN обусловлено именно поглощением Ca2+ митохондриями, а не откачиванием наружу.
Примечательно, что при отмывании FCCP часть клеток смогла начать понижение [Ca2+]i (рис. 3а, 3б). В этих же нейронах происходило повышение ∆pH (рис. 3д). Это показывает, что даже после таких разнообразных воздействий, как токсические дозы глутамата и митохондриальные яды, клетки сохранили способность поддерживать функциональную активность митохондрий. Другими словами, процедура трансфекции и экспрессия чужеродных белков в цитозоле и в митохондриях не нарушила работоспособность нейронов, по крайней мере в течение ~2 ч в минимальном солевом буфере при температуре, близкой к комнатной.
Необходимо отметить, что часть клеток не реагировала ростом [Ca2+]i на Glu и, соответственно, на митохондриальные яды в постглутаматный период (рис. 3б). Эти клетки реагировали только на добавление Са2+-ионофора иономицина (Iono) на завершающей стадии эксперимента. Вероятно, эти нейроны не содержали ионотропных глутаматных рецепторов, либо были клетками глии. Отметим, что добавление иономицина в конце эксперимента для определения максимального сигнала Ca2+-индикатора (Fura-FF), неизменно вызывало защелачивание митохондрий и цитозоля (рис. 3). Этот ионофор является Са2+/H+-обменником, поэтому перенос Са2+ в цитозоль и затем в митохондрии сопровождается удалением протонов в буфер, т.е. повышением как pHm, так и pHc.
ОБСУЖДЕНИЕ
Одновременные измерения [Ca2+]i, pHm и pHc в одном и том же нейроне позволили обнаружить рост ΔpH, компенсирующий снижение ΔΨm во время первой фазы Ca2+-ответа на глутамат. За счет процессов дыхания и Ca2+/H+-обмена между матриксом митохондрий и цитозолем умеренное снижение ΔΨm на ~60 мВ во время первой фазы Ca2+-ответа на Glu [10, 32] компенсируется ростом ΔpH на ~1 (рис. 2д, 3в, 3г). Согласно уравнению Нернста [13, 27, 28] такой рост ΔpH соответствует росту электрохимического потенциала митохондрий на ~60 мВ и происходит за счет Ca2+/H+-обмена, осуществляемого совокупным действием Ca2+-унипортера, протонных насосов дыхательной цепи (комплексов I, III и IV) и системой Са2+/3Na+- и Na+/H+-обменников [13, 27, 28].
Недавно появились данные о том, что полость внутри крист внутренней мембраны митохондрий не является резервуаром для протонов, поступающих из межмембранного пространства и служащих “субстратом” для F1F0-ATP-азы ([40] и ссылки там). Предполагается, что плотная упаковка ферментов окислительного фосфорилирования в мембранах крист обеспечивает локальную кинетическую связь экструзии протонов дыхательной цепью с F1F0-ATP-азой, потребляющей эти протоны. В этом случае значения ΔpH, полученные в настоящей работе, завышены. На наш взгляд, истина где-то посередине и обмен протонами между цитозолем, межмембранным пространством митохондрий и внутренним пространством крист, соединенным с межмембранным пространством, существует, по крайней мере, при действии глутамата на нейроны. Иначе поступление Са2+ в митохондрии не влияло бы на рН цитозоля ([16, 23, 24] и данное исследование).
Такая компенсация сохраняет (а возможно, даже увеличивает) способность митохондрий поддерживать необходимую скорость синтеза ATP в условиях его повышенного потребления ионными насосами плазматической мембраны. При развитии ОКД этот компенсаторный эффект, если не исчезает, то значительно ослабевает, однако полного коллапса ΔpH не происходит, по крайней мере в начальный период ОКД. Это свидетельствует о том, что, если повышение протонной проводимости внутренней мембраны митохондрий вносит вклад в развитие ОКД и сильной митохондриальной деполяризации, то повышение этой проводимости едва ли является классической мегапорой (mitochondrial permeability transition pore, мРТР). В противном случае ΔpH стал бы равен нулю [14, 27, 28, 41]. Либо мРТР формируется не во всех митохондриях, локализованных в соме нейронов. В пользу отсутствия “классической” циклоспорин-А чувствительной поры, по крайней мере на начальном этапе ОКД, свидетельствует также то, что замена Ca2+ на Sr2+, который препятствует мРТР [41, 42], не отменяла возникновения отсроченной “стронциевой дисрегуляции” [24, 43]. Более того, в присутствии Sr2+ и циклоспорина-А наблюдалось кратковременное увеличение градиента pH между матриксом и цитозолем, совпадающее с началом ОКД [24], чего не должно быть, если бы развитие ОКД было полностью обусловлено мРТР. Более детальный анализ формирования поры и ее структуры дан, например, в обзорах [44, 45], в том числе при нейродегенеративных заболеваниях [46].
Стоит также отметить, что меньшее снижение pHm, чем pHc во время первой фазы Ca2+-ответа на Glu, препятствует сильному увеличению растворимости Ca2+-фосфатных комплексов в матриксе митохондрий, предотвращая более значительное падение их Ca2+-буферной емкости в условиях снижения ΔΨm [46].
Деполяризация митохондрий в бескальциевом буфере в постглутаматный период, когда они накопили много Са2+, вызывает высвобождение Са2+ в цитозоль, сопряженное с захватом митохондриями протонов. Возникающее закисление матрикса митохондрий вызывает, по-видимому, растворение Са2+-фосфатных комплексов [47] и дополнительное высвобождение Са2+ из митохондрий, обеспечивая дополнительное поступление протонов в матрикс. Формально, подобный процесс работает по принципу положительной обратной связи, или “порочного круга” (vicious cycle). Эта гипотеза механизма развития ОКД, при которой одновременно происходит рост [Ca2+]i, деполяризация митохондрий и закисление их матрикса, была предложена Борисом Израилевичем Ходоровым за несколько лет до описанных в данной работе результатов [11].
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Источники финансирования. Работа выполнена по планам Государственных заданий Министерства здравоохранения Российской Федерации № AAAA-A19-119012590191-3 и Министерства науки и высшего образования Российской Федерации № FGFU-2022-0012.
Соответствие принципам этики. Эксперименты с животными выполняли в соответствии с этическими принципами и нормативными документами, рекомендованными Европейским научным фондом (ESF) и декларацией о гуманном отношении к животным и в соответствии с Приказом Минздравсоцразвития России № 708н от 23.08.2010 г. “Об утверждении правил лабораторной практики”.
Список литературы
Wang Y., Qin Z. 2010. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis. 15 (11), 1382–1402. https://doi.org/10.1007/s10495-010-0481-0
Zhou Y., Danbolt N.C. 2014. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 121 (8), 799–817.https://doi.org/10.1007/s00702-014-1180-8
Gudiño-Cabrera G., Ureña-Guerrero M.E., Rivera-Cervantes M.C., Feria-Velasco A.I., Beas-Zárate C. 2014. Excitotoxicity triggered by neonatal monosodium glutamate treatment and blood–brain barrier function. Arch. Med. Res. 45 (8), 653–659. https://doi.org/10.1016/j.arcmed.2014.11.014
Jewett B.E., Thapa B. 2021. Physiology, NMDA receptor. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing.
Zöllner J.P., Schmitt F.C., Rosenow F., Kohlhase K., Seiler A., Strzelczyk A., Stefan H. 2021. Seizures and epilepsy in patients with ischaemic stroke. Neurol. Res. Pract. 3 (1), 63. https://doi.org/10.1186/S42466-021-00161-W
Verkhratsky A., Kirchhoff F. 2007. NMDA receptors in glia. Neuroscientist. 13 (1), 28–37. https://doi.org/10.1177/1073858406294270
Gerkau N.J., Rakers C., Petzold G.C., Rose C.R. 2017. Differential effects of energy deprivation on intracellular sodium homeostasis in neurons and astrocytes. J. Neurosci. Res. 95 (11), 2275–2285. https://doi.org/10.1002/jnr.23995
Huo Y., Feng X., Niu M., Wang L., Xie Y., Wang L., Ha J., Cheng X., Gao Z., Sun Y. 2021. Therapeutic time windows of compounds against NMDA receptors signaling pathways for ischemic stroke. J. Neurosci. Res. 99 (12), 3204–3221. https://doi.org/10.1002/JNR.24937
Tymianski M., Charlton M.P., Carlen P.L., Tator C.H. 1993. Secondary Ca2+ overload indicates early neuronal injury which precedes staining with viability indicators. Brain Res. 607 (1–2), 319–323. https://doi.org/10.1016/0006-8993(93)91523-U
Nicholls D.G., Ward M.W. 2000. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: Mortality and millivolts. Trends Neurosci. 23 (4), 166–174. https://doi.org/10.1016/S0166-2236(99)01534-9
Khodorov B. 2004. Glutamate-induced deregulation of calcium homeostasis and mitochondrial dysfunction in mammalian central neurones. Progr. Biophys. Mol. Biol. 86 (2), 279–351. https://doi.org/10.1016/j.pbiomolbio.2003.10.002
Abramov A.Y., Duchen M.R. 2010. Impaired mitochondrial bioenergetics determines glutamate-induced delayed calcium deregulation in neurons. Biochim. Biophys. Acta. 1800 (3), 297–304. https://doi.org/10.1016/j.bbagen.2009.08.002
Nicholls D.G., Budd S.L. 2000. Mitochondria and neuronal survival. Physiol. Rev. 80 (1), 315–360.
Duchen M.R. 2012. Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflügers Arch. 464 (1), 111–121. https://doi.org/10.1007/s00424-012-1112-0
Plotegher N., Filadi R., Pizzo P., Duchen M.R. 2021. Excitotoxicity revisited: Mitochondria on the verge of a nervous breakdown. Trends Neurosci. 44 (5), 342–351. https://doi.org/10.1016/J.TINS.2021.01.001
Surin A.M., Gorbacheva L.R., Savinkova I.G., Sharipov R.R., Khodorov B.I., Pinelis V.G. 2014. Study on ATP concentration changes in cytosol of individual cultured neurons during glutamate-induced deregulation of calcium homeostasis. Biochemistry (Moscow). 79 (2), 146–157. https://doi.org/10.1134/S0006297914020084
Budd S.L., Nicholls D.G. 1996. A reevaluation of the role of mitochondria in neuronal Ca2+ homeostasis. J. Neurochem. 66 (1), 403–411. https://doi.org/10.1046/J.1471-4159.1996.66010403.X
Pinelis V.G., Bykova L.P., Bogachev A.P., Isaev N.K., Viktorov I.V, Khodorov B.I. 1997. Toxic effect of glutamate on cultured cerebellar granular cells reduces the intracellular level of ATP. The role of Ca2+ ions. Bull. Exp. Biol. Med. 123 (2), 162–164.
Ioudina M., Uemura E., Greenlee H.W. 2004. Glucose insufficiency alters neuronal viability and increases susceptibility to glutamate toxicity. Brain Res. 1004 (1–2), 188–192. https://doi.org/10.1016/J.BRAINRES.2003.12.046
Sorokina E.G., Reutov V.P., Senilova Y.E., Khodorov B.I., Pinelis V.G. 2007. Changes in ATP content in cerebellar granule cells during hyperstimulation of glutamate receptors: possible role of NO and nitrite ions. Bull. Exp. Biol. Med. 143 (4), 442–445. https://doi.org/10.1007/S10517-007-0151-6
Surin A.M., Khiroug S.S., Gorbacheva L.R., Khodorov B.I., Pinelis V.G., Khiroug L. 2013. Comparative analysis of cytosolic and mitochondrial ATP synthesis in embryonic and postnatal hippocampal neuronal cultures. Front. Mol. Neurosci. 5, 102. https://doi.org/10.3389/fnmol.2012.00102
Hwang S.-M., Koo N.-Y., Jin M., Davies A.J., Chun G.-S., Choi S.-Y., Kim J.-S., Park K. 2011. Intracellular acidification is associated with changes in free cytosolic calcium and inhibition of action potentials in rat trigeminal ganglion. J. Biol. Chem. 286 (3), 1719–1729.
Wang G.J., Randall R.D., Thayer S.A. 1994. Glutamate-induced intracellular acidification of cultured hippocampal neurons demonstrates altered energy metabolism resulting from Ca2+ loads. J. Neurophysiol. 72 (6), 2563–2569. https://doi.org/10.1152/JN.1994.72.6.2563
Bolshakov A.P., Mikhailova M.M., Szabadkai G., Pinelis V.G., Brustovetsky N., Rizzuto R., Khodorov B.I. 2008. Measurements of mitochondrial pH in cultured cortical neurons clarify contribution of mitochondrial pore to the mechanism of glutamate-induced delayed Ca2+ deregulation. Cell Calcium. 43 (6), 602–614. https://doi.org/10.1016/j.ceca.2007.10.005
Chudakov D.M., Matz M.V., Lukyanov S., Lukya-nov K.A. 2010. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol. Rev. 90 (3), 1103–1163. https://doi.org/10.1152/PHYSREV.00038.2009
Okumoto S. 2010. Imaging approach for monitoring cellular metabolites and ions using genetically encoded biosensors. Curr. Opin. Biotechnol. 21 (1), 45–54. https://doi.org/10.1016/J.COPBIO.2010.01.009
Nicholls D.G., Ferguson S.J. 2013. Bioenergetics. 4th ed. Middletown, USA: Acad. Press. 434 p. https://doi.org/10.1017/CBO9781107415324.004
Скулачев В.П., Богачев А.В., Каспаринский Ф.О. 2011. Мембранная биоэнергетика. М.: Изд-во МГУ. 368 стр.
Сурин А.М., Красильникова И.А., Пинелис В.Г., Ходоров Б.И. 2014. Исследование взаимосвязи между индуцированной глутаматом отсроченной Са2+ дизрегуляцией, митохондриальной деполяризацией и последующей гибелью нейронов. Патогенез. 12 (4), 40–46.
Shcherbo D., Merzlyak E.M., Chepurnykh T.V., Fradkov A.F., Ermakova G.V., Solovieva E.A., Lukyanov K.A., Bogdanova E.A., Zaraisky A.G., Lukyanov S., Chudakov D.M. 2007. Bright far-red fluorescent protein for whole-body imaging. Nat. Methods. 4 (9), 741–746. https://doi.org/10.1038/NMETH1083
Buckman J.F., Hernandez H., Kress G.J., Votyakova T.V., Pal S., Reynolds I.J. 2001. MitoTracker labeling in primary neuronal and astrocytic cultures: Influence of mitochondrial membrane potential and oxidants. J. Neurosci. Methods. 104 (2), 165–176. https://doi.org/10.1016/S0165-0270(00)00340-X
Duchen M.R., Surin A., Jacobson J. 2003. Imaging mitochondrial function in intact cells. Methods Enzymol. 361, 353–389. https://doi.org/10.1016/S0076-6879(03)61019-0
Keminer O., Peters R. 1999. Permeability of single nuclear pores. Biophys. J. 77 (1), 217–228. https://doi.org/10.1016/S0006-3495(99)76883-9
Mattaj I.W., Englmeier L. 1998. Nucleocytoplasmic transport: The soluble phase. Annu. Rev. Biochem. 67, 265–306. https://doi.org/10.1146/ANNUREV.BIOCHEM.67.1.265
Brustovetsky N., Dubinsky J.M. 2000. Dual responses of CNS mitochondria to elevated calcium. J. Neurosci. 20 (1), 103–113.
Шарипов Р.Р., Красильникова И.А., Пинелис В.Г., Горбачева Л.Р., Сурин А.М. 2018. Исследование механизма сенситизации нейронов к повторному действию глутамата. Биол. мембраны. 35 (5), 384–397.
Ходоров Б.И., Сторожевых Т.П., Сурин А.М., Сорокина Е.Г., Юравичус А.И., Бородин А.В., Винская Н.П., Каспеков Л.Г., Пинелис В.Г. 2001. Митохондриальная деполяризация играет доминирующую роль в механизме нарушения нейронального кальциевого гомеостаза вызванного глутаматом. Биол. мембраны. 18 (6), 421–432.
Abramov A.Y., Duchen M.R. 2008. Mechanisms underlying the loss of mitochondrial membrane potential in glutamate excitotoxicity. Biochim. Biophys. Acta. 1777 (7–8), 953–964. https://doi.org/10.1016/j.bbabio.2008.04.017
Kiedrowski L. 1999. N-methyl-D-aspartate excitotoxicity: Relationships among plasma membrane potential, Na+/Ca2+ exchange, mitochondrial Ca2+ overload, and cytoplasmic concentrations of Ca2+, H+, and K+. Mol. Pharmacol. 56 (3), 619–632.
Toth A., Meyrat A., Stoldt S., Santiago R., Wenzel D., Jakobs S., von Ballmoos C., Ott M. 2020. Kinetic coupling of the respiratory chain with ATP synthase, but not proton gradients, drives ATP production in cristae membranes. Proc. Natl. Acad. Sci. USA. 117(5), 2412–2421. https://doi.org/10.1073/pnas.1917968117
Bernardi P. 1999. Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol. Rev. 79 (4), 1127–1155. https://doi.org/10.1038/370621a0
Dubinin M.V., Adakeeva S.I., Samartsev V.N. 2013. Long-chain α,ω-dioic acids as inducers of cyclosporin A-insensitive nonspecific permeability of the inner membrane of liver mitochondria loaded with calcium or strontium ions. Biochemistry (Moscow). 78 (4), 412–417. https://doi.org/10.1134/S000629791304010X
Вабниц А.В., Сторожевых Т., Пинелис В.Г., Ходоров Б.И. 2005. Открывание митохондриальной поры не является необходимым условием для деполяризации митохондрий нарушения кальциевой регуляции, вызываемых глутаматом в нейронах мозга. Биол. мембраны. 2 (4), 378–382.
Bauer T.M., Murphy E. 2020. Role of mitochondrial calcium and the permeability transition pore in regulating cell death. Circ. Res. 126 (2), 280–293. https://doi.org/10.1161/CIRCRESAHA.119.316306
Carinci M., Vezzani B., Patergnani S., Ludewig P., Lessmann K., Magnus T., Casetta I., Pugliatti M., Pinton P., Giorgi C. 2021. Different roles of mitochondria in cell death and inflammation: Focusing on mitochondrial quality control in ischemic stroke and reperfusion. Biomedicines. 9 (2), 1–28. https://doi.org/10.3390/BIOMEDICINES9020169
Abramov A.Y., Berezhnov A. V., Fedotova E.I., Zinchenko V.P., Dolgacheva L.P. 2017. Interaction of misfolded proteins and mitochondria in neurodegenerative disorders. Biochem. Soc. Trans. 45 (4), 1025–1033. https://doi.org/10.1042/BST20170024
Chalmers S., Nicholls D.G. 2003. The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J. Biol. Chem. 278 (21), 19 062–19 070. https://doi.org/10.1074/jbc.M212661200
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии