

Биологические мембраны: Журнал мембранной и клеточной биологии, 2022, T. 39, № 5, стр. 321-336
Физико-химические механизмы функционирования мембраноактивных белков оболочечных вирусов
О. В. Батищев *
Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119071 Москва, Россия
* E-mail: olegbati@gmail.com
Поступила в редакцию 04.05.2022
После доработки 01.06.2022
Принята к публикации 02.06.2022
- EDN: SFNAZS
- DOI: 10.31857/S0233475522050036
Аннотация
Последние несколько лет внимание всего мира приковано к появлению новых опасных штаммов вирусов, среди которых особое место занимают коронавирусы, преодолевшие межвидовой барьер в последние 20 лет: вирусы атипичной пневмонии (SARS), ближневосточного респираторного синдрома (MERS), а также новой коронавирусной инфекции (SARS-CoV-2), вызвавшей крупнейшую пандемию со времен пандемии гриппа “испанки” в 1918 году. Коронавирусы являются представителями класса оболочечных вирусов, имеющих липопротеиновую оболочку. К подобному классу относятся также такие серьезные патогены, как вирус иммунодефицита человека (ВИЧ), гепатит, вирус Эбола, грипп и т. п. Несмотря на существенные различия в клинической картине протекания заболеваний, вызываемых оболочечными вирусами, сами они имеют ряд характерных особенностей, определяющих их общность. Независимо от способа проникновения в клетку – путем эндоцитоза или непосредственного слияния с клеточной мембраной – для оболочечных вирусов характерны следующие стадии взаимодействия с клеткой-мишенью: связывание с рецепторами на клеточной поверхности, взаимодействие поверхностных гликопротеинов вируса с мембранными структурами инфицируемой клетки, слияние липидной оболочки вириона с плазматической или эндосомальной мембраной, разрушение белкового капсида и его диссоциация от вирусного нуклеопротеина. Впоследствии внутри инфицированной клетки вновь синтезированные вирусные белки должны самоорганизоваться на ее различных мембранных структурах для формирования дочернего вириона. Таким образом, как начальные стадии вирусного инфицирования, так и сборка и выход новых вирусных частиц сопряжены с активностью белков вируса по отношению к мембране клетки и ее органелл. Данный обзор посвящен анализу физико-химических механизмов функционирования основных структурных белков ряда оболочечных вирусов с целью выявления возможных стратегий мембранной активности таких белков на различных стадиях вирусного инфицирования клетки.
ВВЕДЕНИЕ
Большинство процессов в клеточных мембранах опосредовано специальными белками. Белок-липидные взаимодействия в клетке, регулирующие топологические перестройки мембран, такие как слияние и деление клеток и их органелл, процессы эндо- и экзоцитоза, сопровождающиеся формированием мембранных везикул, их отпочковыванием и транспортом, требуют слаженной работы большого комплекса белков, что определяет сложность их изучения и экспериментального моделирования [1–4]. При этом, хотя слияние мембран в биологических системах определяется кооперативными белок-липидными взаимодействиями, механические свойства липидного матрикса, окружающего мембранные белки, определяют энергетику образования промежуточных структур, через которые реализуется слияние мембран [5]. Эксперименты, проведенные Л.В. Черномордиком и Г.Б. Меликяном на плоских липидных бислоях в лаборатории биоэлектрохимии Института электрохимии АН СССР, возглавляемой Ю.А. Чизмаджевым, показали, что процесс слияния состоит из следующих последовательных стадий: 1) установление плотного контакта мембран, 2) возникновение так называемого сталка (или перемычки) между близлежащими монослоями, 3) расширение сталка, приводящее к образованию триламинарной структуры (или диафрагмы полуслияния) и, наконец, 4) ее разрыв, знаменующий завершение слияния [6]. Было показано, что скорости упомянутых стадий существенным образом зависят от спонтанной кривизны липидных монослоев. Так, отрицательная спонтанная кривизна контактирующих монослоев (типичный пример – диолеоилфосфатидилэтаноламин) способствует образованию сталка, а разрыв диафрагмы полуслияния ускоряется при наличии положительной спонтанной кривизны (т.е. лизолипидов) в дистальных монослоях. Доказательство справедливости этой схемы слияния было получено другими сотрудниками лаборатории биоэлектрохимии В.С. Маркиным и М.М. Козловым [7]. Построенная ими теория количественно описала всю совокупность имевшихся на тот момент экспериментов. Подробное изложение вопроса и соответствующую литературу можно найти в обзоре [8].
Дальнейшее развитие работ по изучению слияния мембран закономерно требовало включение в рассмотрение белковой компоненты мембран. Первые работы в этом направлении были проведены и нацелены на исследование синаптического слияния и действия белков комплекса SNARE [9, 10]. Однако большое число белков, входящих в данный комплекс, делали затруднительным выяснение физико-химических механизмов их функционирования и построение физических моделей процессов для установления энергетики промежуточных стадий. По этой причине на первый план вышло изучение вирус-индуцированного слияния, которое в ряде систем опосредовано одним белком известной структуры, например, гемагглютинином (НА), как в случае вируса гриппа [11]. Считается, что несколько молекул HA формируют так называемую розетку слияния, а затем липидные вспучивания (димплы), на верхушках которых после их сближения образуются монослойные перемычки (сталки), приводящие к монослойному, а впоследствии к полному слиянию.
В различных модельных системах, базирующихся на использовании экспрессирующих гемагглютинин клеток [12], было показано, что влияние липидного состава мембраны-мишени на слияние хорошо описывается в рамках теории сталка. В этих же работах было показано, что роль НА сводится к образованию комплекса – “розетки” белков слияния, внутри которой происходит локальное сближение клеточных мембран на расстояние, достаточное для последующего спонтанного слияния мембран. Таким образом, была предложена гипотетическая картина слияния, в рамках которой работа молекул гемагглютинина необходима для локального сближения липидных бислоев клеточных мембран, которые далее сливаются спонтанно аналогично двум плоским бислойным липидным мембранам (БЛМ). Однако ряд экспериментальных наблюдений, полученных в клеточных системах, таких, например, как отсутствие липидного обмена через малые поры слияния, плохо описываются в рамках предложенной модели. Кроме того, последние данные по электронной томографии слияния вируса гриппа с липидными везикулами показывают формирование структур, когда изгибается только мембрана-мишень, в то время как вирусная мембрана остается практически недеформированной [13]. Анализ этих фактов приводит к выводу о том, что классическая теория сталка, хорошо описывающая слияние в случае симметричных по составу и чисто липидных мембран, может быть неприменима для белок-опосредованного слияния.
Под внешней липидной мембраной оболочечных вирусов располагается каркас из капсидных или матриксных белков. Эти белки составляют основную по количеству копий массу вирусных белков. Взаимодействуя с вирусным геномом и поверхностными гликопротеинами, они играют важную роль в сборке вирусных частиц и производстве дочерних вирионов в инфицированной клетке [14‒16]. Присущая матриксным белкам многофункциональность заключена в их двоякой роли: с одной стороны, поддержание целостности вируса, с другой – контролируемый распад образуемой данными белками оболочки для выхода генетического материала вируса в цитоплазму при попадании вируса внутрь клетки, с третьей – организация сборки вновь производимых вирионов для мультипликации инфекции. Во всех этих процессах матриксные белки взаимодействуют с липидными мембранами, будь то внешняя оболочка вируса или плазматическая мембрана клетки. И именно это взаимодействие является определяющим в жизнедеятельности вируса, хотя молекулярные механизмы таких взаимодействий по-прежнему остаются открытым вопросом. В частности, для вируса гриппа А известно, что он проникает в клетку путем эндоцитоза, попадая в результате в клеточную эндосому. Основным триггером слияния вирусной частицы и эндосомальной мембраны является изменение рН среды до 4–5, приводящее не только к конформационным переходам в белке слияния НА, но и к разрушению внутреннего белкового каркаса вириона, сформированного матриксным белком М1. При этом понижение рН внутри вирусной частицы происходит за счет работы протонных каналов М2, расположенных в оболочке вириона. В экспериментах, проводимых под руководством Ю.А. Чизмаджева было показано, что блокирование каналов М2 с помощью амантадина приводит к “замораживанию” процесса вирусного слияния на стадии малой поры слияния диаметром около 1 нм, в результате чего не происходит выход генетического материала вируса в цитоплазму клетки [17]. Таким образом, разрушение оболочки из матриксных белков является критически важным этапом вирусного инфицирования. С другой стороны, в инфицированной клетке матриксные белки должны собраться на ее плазматической мембране и самоорганизоваться в структуру нового вириона с его последующим отделением от клеточной мембраны. Процесс сборки и отпочковывания дочерней вирусной частицы подразумевает локальную деформацию клеточных мембран, важную роль в которой также должны играть матриксные или капсидные белки. Тем не менее конкретные физико-химические механизмы, определяющие протекание самосборки белкового каркаса вирусов и изменение топологии мембранных структур клетки, зачастую остаются невыясненными.
ВИРУС-ИНДУЦИРОВАННОЕ СЛИЯНИЕ МЕМБРАН
Геном оболочечных вирусов, равно как их капсидные (нуклеокапсидные) белки окружены дополнительной оболочкой – бислойной липидной мембраной, которую они приобретают от клетки-хозяина во время выхода вновь сформированного вириона из инфицированной клетки [18] (рис. 1). Оболочечные вирусы используют слияние мембран для проникновения в клетку [19]. Для осуществления данного процесса вирусы используют работу белков, называемых белками слияния. За счет конформационных перестроек данных белков, инициируемых определенным триггером, будь то связывание с рецептором на поверхности клетки или изменение рН внутри клеточной эндосомы, инициируется процесс слияние липидной оболочки вируса и мембран клетки [20]. При этом работа данных белков не требует каких-либо энергетических молекул [21].
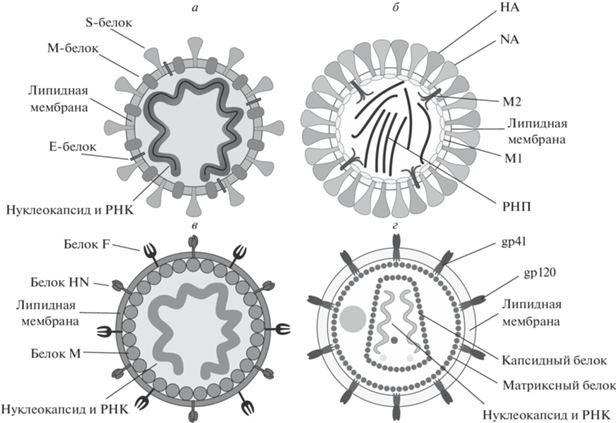
Рис. 1.
Структура оболочечных вирусов. а – Коронавирус SARS-CoV-2; б – вирус гриппа А; в – вирус болезни Ньюкасла; г – вирус иммунодефицита человека. HA – гемагглютинин, NA – нейраминидаза, РНП – рибонуклеопротеин, М1 – белок М1, М2 – протонный канал М2.
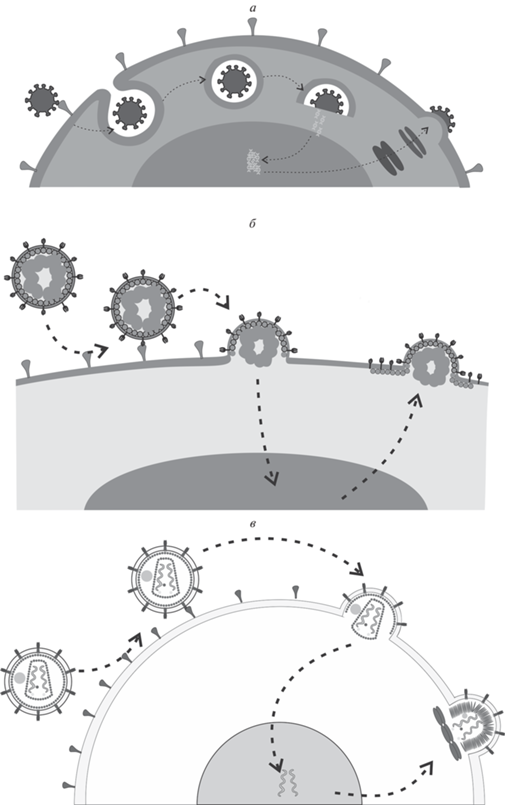
Проникновение любого вируса внутрь клетки начинается с его связывания с определенными рецепторными молекулами на поверхности клетки [22] (рис. 2). Слияние мембран является важнейшей стадией жизненного цикла оболочечных вирусов. Исходно генетический материал вируса отделен от цитоплазмы клетки двумя мембранами: вирусной и клеточной. В результате слияния этих мембран объединяются заключенные в них водные объемы, т.е. непрерывно соединяются цитоплазма клетки и внутреннее пространство вириона, содержащее вирусный генетический материал, что приводит к инфицированию клетки. Некоторые оболочечные вирусы сливаются напрямую с плазматической мембраной инфицируемой клетки, другие попадают внутрь клетки в процессе эндоцитоза.
Рис. 2.
Примеры жизненных циклов оболочечных вирусов. а – Вирус гриппа А; б – вирус болезни Ньюкасла; в – вирус иммунодефицита человека.
Слияние мембран – это один из важных элементов жизнедеятельности клеток, опосредующий многие жизненно важные процессы. Наиболее характерный пример такого процесса – это синаптическая передача, ключевым актом которой является слияние синаптических везикул, содержащих нейротрансмиттеры, с пресинаптической плазматической мембраной [23]. Кроме того, в клетке постоянно реализуется слияние органелл, которое происходит во время слияния между самими клетками, например, в процессах оплодотворения, канцерогенеза и т.п. [24–27]. Определенные белки, называемые белками слияния, осуществляют необходимую работу для реализации данных процессов [28, 29]. Например, при секреции гормонов и нейротрансмиттеров комплекс белков SNARE катализирует слияние мембран [9, 10]. Межклеточное слияние также обеспечивается специфическими белками, которые, однако, выявляются только в редких случаях и остаются неизвестными для большинства случаев [30]. Белки слияния предназначены для деформации взаимодействующих мембран с последующим их слиянием и перемешиванием изначально разделенных объемов воды. Работа, которую эти белки должны выполнять для завершения процесса слияния, в каждом случае определяет их структуру, количество, наличие вспомогательных белков и т.д.
Как правило, в вирусах слияние мембран осуществляют один или два белка; в частности, вирус гриппа A имеет только один белок слияния – гемагглютинин [31]. Слияние мембран, как и любая другая их топологическая перестройка, требует затрат энергии на работу по механической деформации липидного матрикса. Таким образом, белок-липидные взаимодействия будут определять энергетику протекания процесса вирус-индуцированного слияния мембран, а значит, регулировать данную стадию процесса инфицирования. Теоретические оценки показывают, что характерная высота энергетических барьеров слияния составляет от нескольких десятков до сотен kBT, и такие барьеры не могут быть преодолены исключительно за счет тепловых флуктуаций [32]. Механическая энергия, запасенная в белках слияния, позволяет покрывать энергетические затраты процесса.
Многочисленные экспериментальные данные показывают, что слияние мембран может происходить спонтанно, если между сливающимися мембранами добавить вещества, соединяющие две взаимодействующие мембраны (например, Ca2+) или дегидрирующие их контакт (например, полиэтиленгликоль) [33, 34]. Эти данные позволяют сделать вывод, что приведение двух мембран в тесный контакт и преодоление гидратного отталкивания являются наиболее энергоемкими фазами процесса слияния, которые не могут происходить самопроизвольно за счет энергии тепловых флуктуаций липидов. Поскольку дальнейшее слияние в таких модельных системах происходит без участия каких-либо белков, можно сделать вывод, что энергетические барьеры на остальной части траектории слияния не должны заметно влиять. Таким образом, функция белков слияния в значительной степени аналогична роли ферментов: они могут снижать основной энергетический барьер активации для слияния и, возможно, ускорять константу прямой скорости процесса.
Вирусные белки слияния I типа (например, НА вируса гриппа A и белок Env gp120/gp41 ВИЧ) являются наиболее изученными [21]. В начале цикла инфекции вирусные белки слияния связывают специфические рецепторные молекулы на поверхности клетки и сразу после этого либо при изменении pH внутри эндосомы белок слияния атакует клеточную мембрану с помощью пептида слияния, который является специфическим N‑концом, состоящий примерно из 20 аминокислот [35]. Пептид слияния выходит из гидрофобного кармана эктодомена белка слияния и встраивается в липидный бислой мембраны-мишени в качестве эффективного якоря. В последующем в ходе продолжающихся конформационных перестроек белок слияния повторно складывается, притягивая клеточную мембрану-мишень со встроенным пептидом слияния к вирусной мембране [36]. Вступая в тесный контакт, эти мембраны сливаются, что приводит к образованию поры слияния, через которую вирусный геном может высвобождаться в цитоплазму. На этой стадии белок слияния сворачивается в “пост-слиятельную” конформацию, когда его трансмембранная часть контактирует с пептидом слияния [31]. Если пептид слияния не достигает мембраны-мишени, он может встраиваться в вирусную мембрану рядом с трансмембранным доменом белка слияния, также приводя его в пост-слиятельную конформацию. Высота белка слияния в таком состоянии составляет около 10 нм [37].
Пептиды слияния вирусных белков в основном амфипатические [38, 39]. Они могут частично встраиваться в клеточную мембрану, одновременно обеспечивая и якорь, и рычаг для приложения сил и моментов, а также локально деформировать мембрану-мишень. Глубина внедрения пептида слияния регулирует его слиятельную активность [39]. Однако замена трансмембранной части белка слияния на липидный якорь препятствует расширению поры слияния [40, 41]. Следовательно, как пептид слияния, так и трансмембранный домен соответствующего белка, необходимы для эффективного слияния. Фактически, действие белков слияния приводит к образованию выпуклостей на двух контактирующих мембранах: вирусной и клеточной. Сильное гидратное отталкивание, действующее на вершины этих выпуклостей, приводит к латеральному смещению полярных липидных головок из области плотного контакта [42–44]. С некоторой вероятностью эти локальные выпуклости противоположных мембран могут проконтактировать с образованием сталка [44].
В ряде теоретических моделей было показано, что основной энергетический барьер для взаимодействия индуцированных белками слияния оболочечных вирусов выпоуклостей мембран сильно зависит от расстояния между этими мембранами [32, 45]. Это расстояние определяется толщиной слоя белков слияния в пост-слиятельной конформации, пептиды слияния которых не достигли клеточной мембраны-мишени. Для гемагглютинина эта толщина составляет около 10 нм, как показывает электронная микроскопия [37]. Таким образом, белки слияния должны локально преодолевать это расстояние, чтобы сблизить сливающиеся мембраны на небольшой площади контакта, обеспечивая относительно низкую высоту энергетического барьера для полуслияния. По разным оценкам, энергия, необходимая для такой перестройки мембран, составляет порядка нескольких десятков kBT [32, 45, 46]. Для сравнения: энергия, запасенная в одном тримере гемагглютинина, составляет около 60 kBT, т.е. 20 kBT на мономер гемагглютинина [47]. Приблизительно такая же оценка энергии верна и для тримера gp41 ВИЧ [48]. Это означает, что эффективное слияние мембран требует кооперативного действия нескольких белков слияния, формирующих так называемую розетку слияния. В случае вируса гриппа А необходимое количество тримеров НА в розетке слияния, по разным оценкам, составляет от 3 до 9 [49, 50]. Однако в случае ВИЧ известно, что одного тримера белка gp41 может быть достаточно для реализации процесса слияния [50].
Специфической функциональной особенностью белков слияния I типа является сильная деформация мембраны-мишени. Вирусная мембрана деформируется в меньшей степени из-за наличия контактирующей с липидной мембраной вируса оболочки из капсидных белков [37]. Деформация мембраны-мишени обеспечивается встраиванием в нее пептидов слияния. Подобные деформационные взаимодействия рассматривались в многочисленных теоретических работах [51–53]. В частности было показано, что при исследовании образования кооперативной розетки слияния следует учитывать зависимость упругой энергии от взаимной ориентации пептидов слияния, а не от расстояния между ними, так как положения вирусных белков слияния достаточно строго фиксированы из-за наличия в них трансмембранных доменов, взаимодействующих с вирусным капсидом. Высвобождаемые белками слияния вируса пептиды слияния, помимо локальной деформации мембраны-мишени, являются якорями, через которые белки слияния могут прилагать механические усилия – силы и моменты – к мембране-мишени [11, 54]. Таким образом, необходимые для слияния мембран выпуклости липидного матрикса могут формироваться как за счет упругих деформаций, вызванных встроенными пептидами слияния, так и за счет прямого приложения сил белками слияния, стягивающими две сливающиеся мембраны [55, 56].
Неглубокое внедрение амфипатических пептидов слияния в мембрану-мишень аналогично созданию положительной спонтанной кривизны, которая, индуцируемая, например, лизолипидами, приводит к образованию пор [57], но никак не к формированию сталка. Таким образом, физико-химический механизм функционирования белков слияния с амфипатическими пептидами слияния не может базироваться лишь на модификации контактного монослоя мембраны-мишени, и, по-видимому, неизбежно требует приложения к двум мембранам стягивающих сил. Для уменьшения высоты энергетического барьера образования сталка пептиды слияния должны эффективно индуцировать отрицательную спонтанную кривизну [57], для чего глубина их внедрения в контактный монослой мембраны-мишени должна быть увеличена. Расчеты с явным учетом глубины внедрения пептидов слияния, проведенные в работах [58, 59], полностью подтверждают этот вывод. Более того, существуют экспериментальные данные для пептида слияния белка слияния ВИЧ, согласно которым, чем глубже эти пептиды внедрены в контактный монослой мембраны-мишени, тем более вероятно слияние [39].
В случае ВИЧ слияние может катализироваться несколькими белками Env вплоть до одного тримера белка [50]. Ключевое отличие здесь заключается в том факте, что наряду со слиянием белки Env ответственны и за рецепцию вируса, то есть его связывание с клеточной плазматической мембраной [60]. Белок gp120 распознает рецептор CD4 и корецептор CXCR4 или CCR5, которые представляют собой крупные трансмембранные белки. Связывание с рецептором запускает конформационную перестройку гликопротеина Env. Такой тип триггера вирусного слияния дополнительно ограничивает количество одновременно активированных тримеров gp120/gp41 из-за относительно низкой поверхностной плотности молекул CD4 в плазматической мембране Т-лимфоцитов. Это отличается от случая вируса гриппа А, активация гемагглютинина которого вызвана понижением величины рН в поздней эндосоме, т.е. все тримеры гемагглютинина вириона активируются почти одновременно.
Традиционно предлагаются два возможных механизма действия белков слияния. Один из них основан на предположении, что встроенные пептиды слияния модифицируют упругие свойства мембраны-мишени, прежде всего, ее спонтанную кривизну [61]. Считается, что измененная спонтанная кривизна в кольцеобразной зоне розетки слияния может быть причиной образования выпуклости в мембране-мишени; сливающиеся мембраны вступают в тесный контакт в верхней части выпуклости, что существенно облегчает слияние мембран. Согласно второму механизму, вместо модификации мембраны-мишени белки слияния индуцируют изгибающие моменты, приводящие к образованию выпуклостей, и генерируют силу, непосредственно и механически приводя две плотно прилегающие мембраны к тесному контакту [31]. Анализ, проведенный в работе [62], показал, что именно этот второй механизм обеспечивает образование высокосимметричной розетки слияния, организуя кооперативность механических усилий нескольких белков слияния. Напротив, первый механизм использует симметрию розетки для объяснения образования выпуклостей на сливающихся мембранах, но не может объяснить сам факт формирования такой симметрии. Таким образом, прямая механическая активность белков слияния управляет всем процессом слияния. Эта активность может быть результатом согласованного действия различных субъединиц белка слияния, в том числе и тех участков, которые ответственны за взаимодействие с рецептором. Последнее предположение может быть ответом на вопрос о наблюдаемом различии в кооперативности действия белков слияния вируса гриппа А и ВИЧ. Таким образом, развитие упругих моделей процесса слияния мембран, позволило ответить на многие вопросы о механизмах реализации вирус-индуцированного слияния мембран, в частности, о возможных причинах кооперативного действия вирусных белков слияния.
КАПСИДНЫЕ БЕЛКИ ОБОЛОЧЕЧНЫХ ВИРУСОВ И ФОРМИРОВАНИЕ ДОЧЕРНИХ ВИРИОНОВ
Под внешней липидной мембраной оболочечных вирусов располагается каркас из капсидных или матриксных белков. Взаимодействуя с вирусным геномом и поверхностными гликопротеинами, они играют важную роль в сборке вирусных частиц и производстве дочерних вирионов в инфицированной клетке [14–16]. Матриксные белки характеризуются многофункциональностью на различных стадиях жизненного цикла вируса: с одной стороны, они должны поддерживать целостность и общую архитектуру вириона, с другой – осуществлять контролируемый распад оболочки вируса для выхода его генетического материала в цитоплазму инфицированной клетки, с третьей – организовывать сборку дочерних вирионов для мультипликации инфекции. Во всех этих процессах матриксные белки взаимодействуют с липидными мембранами, будь то внешняя оболочка вируса или плазматическая мембрана клетки. И именно это взаимодействие является определяющим в жизнедеятельности вируса, хотя молекулярные механизмы таких взаимодействий по-прежнему остаются открытым вопросом. Основная дискуссия идет о характере таких белок-липидных взаимодействий, а также их взаимосвязи с самосборкой вирусной оболочки [63–67].
Несмотря на многообразие форм оболочечных вирусов, матриксные белки в них организованы схожим способом: это либо спиральные структуры, как в случае вируса гриппа А [14, 68, 69], вируса везикулярного стоматита [15] и вируса кори [16], либо двумерные решетки, формирующие частицы околосферической формы, как в случае вируса болезни Ньюкасла [70] или ВИЧ [71]. Эволюционная и структурная близость отмечается для матриксных белков разных семейств вирусов, как например, для полипротеина Gag вируса иммунодефицита человека и белка М1 вируса гриппа А [72]. Такая близость должна свидетельствовать и об общих механизмах участия матриксных белков в формировании вирусной оболочки и отпочковывании вновь синтезированных вирионов с поверхности инфицированной клетки.
Вообще говоря, взаимосвязь между трехмерной структурой и функциональными свойствами белка является одним из важнейших базовых понятий в биохимии. Это означает, что для выполнения определенной функции белок должен иметь определенную аминокислотную последовательность, и возможно, даже более важно, что он должен быть свернут определенным образом [15]. Эта идея, однако, была пересмотрена около двух десятилетий назад, когда было найдено множество белков с частично или полностью неупорядоченной структурой. Эти белки, называемые внутренне разупорядоченными белками, существуют как динамические ансамбли конформаций, которые не имеют стабильной складчатой структуры, но тем не менее они выполняют свои (часто очень изменчивые) биологические функции [73]. Разупорядоченные участки белков могут способствовать быстрой реакции вирусов на изменяющиеся условия окружающей среды и, следовательно, их выживанию и развитию, внедряя механизмы адаптации и защиты [74]. Разупорядочение в сворачивании белка препятствует связыванию антител, что снижает иммунный ответ. Известно, что этот факт является ключом к чрезвычайной способности ВИЧ уклоняться от иммунного ответа, причем основной вклад вносят структурные особенности его матрикса [74].
Процесс вирусного отпочковывания в целом схож с клеточным экзоцитозом в том смысле, что подразумевает формирование искривленной структуры из клеточной мембраны для образования вириона. Минимальный набор белков в вирусной оболочке ограничивает вирус в выборе возможных путей воздействия на мембрану с целью ее топологической перестройки. Можно выделить следующие три возможных пути такого воздействия, реализуемые независимо друг от друга или в комбинации в различных вирусах. Первый механизм заключается в наличии определенной молекулярной геометрии у матриксных белков, позволяющей им при связывании с липидным бислоем и формировании двумерной решетки на его поверхности навязывать ему определенную кривизну, инициируя таким образом процесс формирования дочернего вириона. Рентгеноструктурный анализ показал наличие такого механизма у вируса болезни Ньюкасла [70] и вируса инфекционной анемии лососевых [75]. В обоих случаях мономеры белка в структуре каркаса объединяются под некоторым углом друг к другу, что соответствует формированию околосферических вирусных частиц. Зарождение таких частиц происходит путем формирования круглых белок-липидных доменов [76] с последующим искривлением липидной мембраны. При этом жидкое состояние липидного бислоя подразумевает, что для передачи своей кривизны липидной мембране слой матриксных белков должен быть связан с ней так, чтобы минимизировать перераспределение липидов на участках контакта мембраны с белковой оболочкой вируса. Достигаться это, по всей видимости, должно за счет гидрофобных взаимодействий, что и было показано в случае вируса болезни Ньюкасла [77]. Подобный механизм взаимодействия предполагался и в ранних работах, посвященных исследованию взаимодействия матриксного белка М1 вируса гриппа с липидным бислоем [78–81], однако не нашел подтверждения в более поздних структурных исследованиях [63], не показавших ни частичного встраивания белка М1 в липидный бислой, ни его способности навязывать мембране определенную кривизну. Эти исследования говорят, скорее, в пользу электростатической природы взаимодействия матриксного белка М1 с мембраной.
Вторым возможным механизмом является конденсация липидов под слоем белка за счет электростатических взаимодействий. Такая конденсация на внутреннем монослое плазматической мембраны клетки создает локальный дисбаланс площади мембранных монослоев, приводящий к изгибу мембраны и формированию различных выростов, призванных сбросить избыточную площадь [82]. В частности, формирование филаментозных структур, близких по диаметру к вирусным частицам, при адсорбции матриксного белка вируса гриппа С было показано в экспериментах на отрицательно заряженных гигантских однослойных везикулах [83]. Электростатическое взаимодействие с анионными липидами присуще матриксным белкам таких вирусов, как вирус гриппа [63, 84], вирус Эболы [85, 86], вирус везикулярного стоматита [87]. Для всех этих вирусов было показано, что их матриксные белки способны к формированию вирусоподобных частиц [87–90]. При этом дополнительные факторы, способствующие изменению кривизны мембраны, способствуют данному механизму вирусного отпочковывания. Так, было показано, что пальмитирование гемагглютинина вируса гриппа А, способствующее формированию правильной кривизны вирусной частицы, усиливает адсорбцию белка М1 на липидной мембране и облегчает процесс формирования вирусоподобных частиц [91].
Для ряда оболочечных вирусов было показано, что их мембрана является более устойчивой к детергентам, чем плазматическая мембрана клетки [92–96], т.е. имеет так называемую рафтовую структуру [97]. Наличие в вирусной мембране липидных рафтов дает возможность реализовать еще один механизм формирования и отпочковывания везикул: путем повышения линейного натяжения границы рафтов с их последующим выходом из плоскости мембраны [98]. И хотя механизм такого процесса пока еще не был продемонстрирован для какого-либо оболочечного вируса, его предпосылки заключаются в возможности формирования липидных доменов за счет конденсации отрицательно заряженных липидов с полностью насыщенными углеводородными цепями на внутреннем монослое плазматической мембраны клетки с последующей инициацией образования липидного домена, обогащенного сфингомиелином, на внешней части мембраны [99].
Таким образом, оболочечные вирусы из различных семейств, использующие разные пути для проникновения в клетку и отличающиеся по первичной последовательности своих структурных белков, тем не менее используют ряд общих физико-химических механизмов на различных этапах инфицирования клетки. Отмеченная выше эволюционная близость матриксных и капсидных белков различных оболочечных вирусов, а также общие элементы в их третичной структуре, также указывают на принципиальную возможность общих механизмов самосборки и дезинтеграции вирусных оболочек из данных белков. Белки слияния вирусов способны создать малую пору слияния, но, как было показано в ряде работ [13, 41], блокирование разрушения белковой оболочки вируса препятствует выходу вирусного генома в цитоплазму клетки. Вопрос о том, являются ли матриксные и капсидные белки вирусов активными участниками процесса расширения поры слияния, или они должны лишь диссоциировать от липидной оболочки вириона, по-прежнему остается открытым. Вообще говоря, многофункциональность вирусных белков говорит о том, что его матриксные белки также могут выполнять несколько ролей. Если влияние данных белков на процесс ядерного транспорта генетического материала вируса уже установлено [100–102], то их влияние на выход вирусного генома явно не было продемонстрировано. С другой стороны, эти же белки должны самоорганизоваться для формирования дочерней вирусной частицы и ее выхода из инфицированной клетки. В общем, формирование и отпочковывание вирусной частицы требует значительных топологических перестроек клеточных мембран. А значит, возникает вопрос о том, как и какие вирусные белки способны выполнять эту работу. Основной фокус здесь приходится именно на матриксные белки, так как количество их копий максимально среди белков любого оболочечного вируса, и именно они призваны создавать и поддерживать архитектуру всей вирусной частицы. Таким образом, в процессе своей самоорганизации матриксные/капсидные белки должны создавать условия для деформаций клеточных мембран и обеспечения условий для отпочковывания или выхода вирионов. Рассмотренная выше ограниченность стратегий таких модификаций структуры мембраны вирусными белками подразумевает общие функциональные механизмы, которые могут быть описаны в рамках физико-химических моделей данных процессов, однако эта область в настоящее время проработана намного слабее, чем вопросы вирус-индуцированного слияния мембран.
КОРОНАВИРУСЫ: НОВЫЙ ВЫЗОВ ДЛЯ ФИЗИКО-ХИМИЧЕСКИХ МОДЕЛЕЙ
Коронавирусы привлекли большое внимание как возбудители вспышек респираторных синдромов человека, таких как тяжелый острый респираторный синдром (SARS), ближневосточный респираторный синдром (MERS) и новая коронавирусная инфекция (COVID-19). Эти респираторные заболевания возникли в результате зоонозной передачи от животных людям, приведшей к появлению штаммов вируса, которые ранее не циркулировали в популяции людей. Среди них коронавирус SARS-CoV-2, возбудитель COVID-19, продемонстрировал быстрое глобальное распространение и привел к возникновению пандемии.
Коронавирусы, как и рассмотренные выше вирусы, относятся к классу оболочечных вирусов. Геном коронавируса содержит четыре основных структурных белка: шип (S), мембранный белок (M), оболочечный белок (E) и нуклеокапсидный белок (N), которые кодируются в 3'-конце генома [103]. Белок S опосредует прикрепление вируса к поверхностным рецепторам клетки-хозяина, что приводит к слиянию и последующему проникновению вируса. Белок М является наиболее представленным белком и определяет форму вирусной оболочки. Белок Е является наименьшим по количеству из основных структурных белков и участвует в сборке и почковании вируса. Белок N является единственным белком, который связывается с РНК-геномом, а также участвует в сборке и почковании вируса. При этом необходимо отметить, что в отличии от большинства других оболочечных вирусов, практически все структурные белки коронавирусов имеют трансмембранные фрагменты.
Несмотря на их сложность и диапазон функций [104, 105], структурные белки коронавирусов занимают только около трети кодирующей части генома. Гораздо больший участок генома, около двух третей, расположенный на 5'-конце, кодирует две длинные открытые рамки считывания 1a и 1b, которые вместе кодируют неструктурные белки вируса. Каждая последовательность сначала транслируется как предшественник полипротеина, pp1a и pp1ab. Полипротеины включают несколько вирусных протеаз, которые превращают pp1a и pp1ab в 16 неструктурных белков (nsp 1–16), необходимые на различных этапах цикла репликации вируса. Эти белки являются одними из самых консервативных белков коронавирусов [103]. Многие неструктурные белки взаимодействуют с мембранами, потому что репликация коронавируса происходит в специализированных клеточных компартментах, создаваемых вирусными белками, которые модифицируют мембраны клетки для создания областей репликации вируса, которые скрыты от клеточных факторов врожденного иммунитета [106]. Комбинация множества мембранно-взаимодействующих факторов делает коронавирусы одной из наиболее сложных моделей взаимодействия вирус-мембрана.
Репликация РНК-вирусов с положительной цепью РНК, а также ДНК-вирусов в клетках растений и животных вызывает образование субклеточной микросреды, называемой сетью репликации, вирусными фабриками или вироплазмой [107]. Хотя цель таких мембранных образований в жизненном цикле вируса до конца неизвестна, предполагается, что это облегчает, по крайней мере, стадию синтеза вирусной РНК [108, 109]. Создание таких “мини-органелл” требует деформации мембран клетки-хозяина и цитоскелета, вызывая цитопатический эффект, который использовался в качестве маркера вирусной инфекции для проверки потенциальной эффективности лекарственного средства [110]. Считается, что все положительно-цепочечные РНК-вирусы (+РНК-вирусы), которые инфицируют эукариотические клетки, образуют мембрано-связанные органеллы [111]. Одна из наиболее распространенных модификаций мембран, вызванных +РНК-вирусами, – это образование парных мембран, то есть двух близко расположенных липидных бислоев, дающих основу так называемым двухмембранным везикулам [108]. Широкое распространение таких структур при репликации +РНК-вирусов говорит о том, что это эффективная стратегия для успешного производства новых вирионов и что мембранное спаривание может каким-то образом повысить конкурентоспособность этих вирусов.
Семейство коронавирусов обладает особенно сложной репликационной мембранной сетью, происходящей из эндоплазматического ретикулума [106], аналогично вирусу Денге [112] и реовирусу [113]. Ее структура включает множество изогнутых мембран и двухмембранных везикул. Все они взаимосвязаны в непрерывную сеть друг с другом и с эндоплазматическим ретикулумом липидными нанотрубками [114]. Такая перестройка мембран клетки-хозяина рассматривается как вирусная стратегия для создания основы для репликации вируса путем локализации и концентрации необходимых факторов и обеспечения защиты от иммунного ответа [115]. Подтверждением этой гипотезы является факт того, что общий уровень вирусной РНК коррелирует с количеством двухмембранных везикул в клетке [106, 116, 117]. Однако существуют данные, которые не подтверждают взаимосвязь двухмембранных везикул в эндоплазматическом ретикулуме и синтеза вирусной РНК в инфицированной клетке [118]. Как было показано, мутации неструктурных белков вируса, препятствующие формированию ими двухмембранных везикул, не приводят к полной остановке синтеза вирусной РНК, но при этом скорость репликации вируса значительно падает.
Двухмембранные везикулы являются загадочными структурами, механизм формирования которых до сих пор неясен. Анализ физико-химических механизмов процесса образования двухмембранной везикулы из двухмембранного диска был проведен в статье [119]. В данной работе в рамках простейшей теории упругости Хельфриха [120] вычислялись и сравнивались энергии изгиба мембран двухмембранной везикулы и двухмембранного диска. Энергия изгиба сферической двухмембранной везикулы не зависит от ее радиуса, в то время как энергия изгиба двухмембранного диска линейно возрастает при росте радиуса диска. При некотором радиусе диска эти две энергии сравниваются, и двухмембранный диск может перейти в двухмембранную везикулу. В этой статье не рассматривались процессы формирования двухмембранного диска из плоской мембраны и образования двухмембранной везикулы из двухмембранного сферического сегмента; форма мембран постулировалась, а не находилась путем оптимизации упругой энергии; никак не описывались участвующие в процессе белки.
Таким образом, остается открытым вопрос о том, имеют ли вирусные репликазы еще одну критическую функцию – способны ли они вызвать искривление мембраны на необходимой органелле? Это подразумевает, что данные белки должны быть способны образовывать каркасоподобные структуры, и они должны обладать липидной специфичностью, так как отсутствует какая-либо информация о других типах специфического нацеливания этих репликаз в эндоплазматическом ретикулуме. Так, при гепатите С, который, как было показано, также образует мембранные репликационные компартменты от эндоплазматического ретикулума, белок NS5A стимулирует активность фосфатидилинозитол-4-киназы-III, следовательно, облегчает выработку фосфатидилинозитол-4-фосфата на цитоплазматической стороне эндоплазматического ретикулума и, таким образом, предположительно облегчает аккумуляцию РНК-полимеразы [121]. Было показано, что обогащение фосфатидилинозитол-4-фосфатом имеет важное значение для репликации энтеровирусной и флавивирусной РНК.
Тот факт, что неструктурные белки коронавирусов могут формировать каркасы, способствующие деформациям клеточных мембран, роднит их с матриксными белками других оболочечных вирусов и позволяет предположить наличие общих механизмов белок-липидного и белок-белкового взаимодействия. Тем более потому, что такие двухмембранные структуры не встречаются в неинфицированных клетках (за исключением аутофагосом), что свидетельствует о том, что спонтанное “сморщивание” эндоплазматического ретикулума не может быть движущей силой появления двойных мембранных структур. Понимание механизма формирования двухмембранных везикул имеет решающее значение для выяснения принципов организации компартментов вирусной репликации.
Другой предложенный механизм формирования двухмембранных везикул включает клеточную аутофагию [122, 123]. Предполагается, что белок DFCP1 (двойной белок 1, содержащий FYVE-домен) связывает фосфатидилинозитол фосфат, образуя таким образом изогнутые области эндоплазматического ретикулума, называемые омегасомами [124]. На основании данных, полученных для ряда коронавирусов, было высказано предположение, что их белки nsp 6 генерируют аутофагосомы [125].
Оболочечные белки Е коронавируса играют несколько ролей во время инфекции, включая морфогенез вируса. Они представляют собой небольшие (74–109 аминокислот) гидрофобные виропорины [126]. Белки E состоят из двух отдельных структурных доменов: более длинного, чем толщина липидного бислоя, гидрофобного домена, и заряженного цитоплазматического хвоста. Роль белков Е в сборке и выходе не до конца понятна. Потребность в них во время морфогенеза вируса варьируется в зависимости от рода вируса. Удаление белков E из трансмиссивного вируса гастроэнтерита (TGEV) приводит к репликации компетентных, но дефектных по распространению вирусов [127], как и в случае с вирусом MERS [128]. SARS-CoV продемонстрировал 200-кратное снижение выхода вируса в отсутствие белка E, которое зависело от типа клеток, используемого для инфекции [129]. Таким образом, хотя мембраноактивные белки коронавирусов, такие как nsp 3, 4, 6, а также белок E, являются высококонсервативными и критически важными для репликации вируса, механизмы их функционирования и взаимодействия с клеточными мембранами и друг с другом до сих пор остаются невыясненными.
Белок S представляет собой гликозилированный мембранный белок слияния типа I, который состоит из двух субъединиц, S1 и S2. N-концевая субъединица S1 содержит рецептор-связывающий домен (RBD), который опосредует связывание с рецептором клетки-хозяина, а именно с ангиотензин превращающим ферментом 2 (ACE2), как для SARS-CoV, так и для SARS-CoV-2 [130, 131]. Биофизические и структурные/функциональные исследования пептида слияния белка S вирусов SARS-CoV-2 [132], SARS [133] и MERS [134] дали важные сведения о функциональном значении конкретных областей пептида слияния, таких как большое количество консервативных аминокислотных остатков в двух последовательных фрагментах пептида слияния, названных FP1 и FP2 (остатки 816–835 и 835–854 соответственно в SARS-CoV-2). Эти исследования показали, что вместе фрагменты F1 и F2 образуют платформу двустороннего взаимодействия между мембранами и что аминокислотные остатки как в FP1, так и в FP2 способствуют связыванию с мембраной посредством своего взаимодействия с ионами Ca2+. Эти данные о функциональной роли Ca2+ подтверждаются результатами, полученными для пептидов слияния от других родственных вирусов, таких как MERS [134] и вирус Эбола [135]. Действительно, в пептиде слияния вируса MERS один из этих аминокислотных остатков, E891 в N-концевой (FP1) части (соответствует E819 в нумерации пептида слияния SARS-CoV-2), оказался решающим для взаимодействий с Ca2+ и обеспечения деформаций клеточной мембраны, связанных со слиянием [136]. Кроме того, было показано, что нарушение липидного бислоя за счет Ca2+-зависимых взаимодействий пептида слияния с мембраной влияет на организацию полярных групп липидов в месте взаимодействия, но не на центральную гидрофобную область мембраны [136]. Тем не менее участки связывания Ca2+ с пептидом слияния и их роль в каких-либо специфических (но неизвестных) способах взаимодействия пептида слияния с мембраной остаются неопределенными. Это затрудняет интерпретацию любых измеримых эффектов вирусных взаимодействий с мембраной и, следовательно, любых подходов к снижению инфекционности путем воздействия на эту важную область белка S.
Как было сказано выше, различие в механизме активации белков слияния и глубина встраивания пептида слияния в липидный матрикс клеточных мембран регулирует кооперативность действия белков слияния за счет деформаций липидного бислоя клетки-мишени, а не за счет прямых белок-белковых взаимодействий [62]. Построенная молекулярная модель действия пептидов слияния вируса гриппа А и вируса иммунодефицита человека позволила выяснить роль структуры рецепторов в кооперативности работы белков слияния вируса. Тот факт, что начальные стадии проникновения коронавируса SARS-CoV-2 близки к таковым у вируса иммунодефицита человека (связывание с рецептором на поверхности клетки с последующим слиянием с плазматической мембраной), а белок S SARS-CoV-2, как и белок gp41/gp120 вируса иммунодефицита человека относится к белкам слияния I типа, позволяет считать, что предпосылки этой модели верны и для описания работы пептида слияния и белка S коронавируса.
ВЫВОДЫ
Жизненный цикл всех оболочечных вирусов представляет собой сложный многостадийным процесс, начинающийся с проникновения вируса в клетку и заканчивающийся отделением вновь сформированных вирионов с ее мембраны. Он включает точно установленное взаимодействие компонентов вируса и клетки. Нарушение любого этапа жизненного цикла вириона препятствует формированию полноценных вирионов и ограничивает распространение инфекции. Ключевой особенностью всех оболочечных вирусов является наличие не только белковой оболочки, защищающей вирусный геном, но и взаимодействующей с ней липидной мембраны, в которую встроены основные структурные белки вириона. Поэтому белок-липидные взаимодействия играют одну из определяющих ролей в процессах вирусного инфицирования, репликации и выхода новых вирусных частиц. Такие взаимодействия в большинстве случаев не подразумевают формирование ковалентной химической связи, а основаны на электростатических, гидрофобных и Ван-дер-Ваальсовых взаимодействиях. Поэтому в процессах инфицирования клетки оболочечными вирусами важную роль играют различные физико-химические механизмы. Несмотря на общность описанных процессов для большинства оболочечных вирусов, по-прежнему остается много открытых вопросов о молекулярных механизмах протекания отдельных стадий, о силах, определяющих функционирование и взаимодействие различных компонентов вирусов и инфицируемой клетки. При этом очевидно, что поиск новых эффективных противовирусных препаратов должен базироваться на знании фундаментальных механизмов, регулирующих работы вирусных белков и определяющих протекание различных стадий вирусного инфицирования клетки. Модели физико-химических механизмов процессов вирусного слияния, формирования дочерних вирионов, а также новые вызовы, связанные с особенностями репликации коронавирусов, могут стать основой такого поиска. Работы по созданию подобных моделей, начатые Ю.А. Чизмаджевым и его коллегами в 1980-х годах, успешно развиваются по сей день благодаря созданной им уникальной научной школе.
Благодарности. Автор выражает благодарность М.М. Поповой за техническую помощь.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Источники финансирования. Работа выполнена при поддержке Российского научного фонда (грант № 22-13-00435).
Соответствие принципам этики. Настоящая статья не содержит описания каких-либо исследований с участием людей или животных в качестве объектов.
Список литературы
McMahon H.T., Gallop J.L. 2005. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature. 438, 590–596.
Zimmerberg J., Kozlov M.M. 2006. How proteins produce cellular membrane curvature. Nat. Rev. Mol. Cell Biol. 7, 9–19.
Schekman R., Orci L. 1996. Coat proteins and vesicle budding. Science. 271, 1526–1533.
Bremser M., Nickel W., Schweikert M., Ravazzola M., Amherdt M., Hughes C.A., Söllner T.H., Rothman J.E., Wieland F.T. 1999. Coupling of coat assembly and vesicle budding to packaging of putative cargo receptors. Cell. 96, 495–506.
Chernomordik L., Kozlov M.M., Zimmerberg J. 1995. Lipids in biological membrane fusion. J. Membr. Biol. 146, 1–14.
Меликян Г.Б., Абидор И.Г., Черномордик Л.В., Чайлахян Л.М. 1982. Электростимулированное слияние и деление бислойных липидных мембран. Докл. АН СССР. 263, 1009–1012.
Козлов М.М., Маркин В.С. 1983. Возможный механизм слияния мембран. Биофизика. 28, 242–247.
Chernomordik L.V., Kozlov M.M., Melikyan G.B., Abidor I.G., Markin V.S., Chizmadzhev Yu.A. 1985. The shape of lipid molecules and monolayer membrane fusion. Biochim. Biophys. Acta BBA – Biomembr. 812, 643–655.
Wang T., Li L., Hong W. 2017. SNARE proteins in membrane trafficking. Traffic. 18, 767–775.
Jahn R., Scheller R.H. 2006. SNAREs – engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 7, 631–643.
Skehel J.J., Wiley D.C. 2000. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 69, 531–569.
Chernomordik L.V., Frolov V.A., Leikina E., Bronk P., Zimmerberg J. 1998. The pathway of membrane fusion catalyzed by influenza hemagglutinin: Restriction of lipids, hemifusion, and lipidic fusion pore formation. J. Cell Biol. 140, 1369–1382.
Lee K.K. 2010. Architecture of a nascent viral fusion pore. EMBO J. 29, 1299–1311.
Calder L.J., Wasilewski S., Berriman J.A., Rosenthal P.B. 2010. Structural organization of a filamentous influenza A virus. Proc. Natl. Acad. Sci. USA. 107, 10685–10690.
Ge P., Tsao J., Schein S., Green T.J., Luo M., Zhou Z.H. 2010. Cryo-EM model of the bullet-shaped vesicular stomatitis virus. Science. 327, 689–693.
Liljeroos L., Huiskonen J.T., Ora A., Susi P., Butcher S.J. 2011. Electron cryotomography of measles virus reveals how matrix protein coats the ribonucleocapsid within intact virions. Proc. Natl. Acad. Sci. USA. 108, 18085–18090.
Chizmadzhev Y.A. 2004. The mechanisms of lipid-protein rearrangements during viral infection. Bioelectrochemistry. 63, 129–136.
Pettersson R.F. 1991. Protein localization and virus assembly at intracellular membranes. In: Protein traffic in eukaryotic cells. Vol. 170. Current Topics in Microbiology and Immunology. Ed. Compans R.W. Berlin, Heidelberg: Springer, p. 67–106.
White J., Kielian M., Helenius A. 1983. Membrane fusion proteins of enveloped animal viruses. Q. Rev. Biophys. 16, 151–195.
Kielian M., Rey F.A. 2006. Virus membrane-fusion proteins: more than one way to make a hairpin. Nat. Rev. Microbiol. 4, 67–76.
Colman P.M., Lawrence M.C. 2003. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 4, 309–319.
Poehlmann S., Simmons G. 2013. Viral entry into host cells. New York: Springer Science.
Südhof T.C. 2013. Neurotransmitter release: The last millisecond in the life of a synaptic vesicle. Neuron. 80, 675–690.
Hernandez L.D., Hoffman L.R., Wolfsberg T.G., White J.M. 1996. Virus-cell and cell-cell fusion. Annu. Rev. Cell Dev. Biol. 12, 627–661.
Duelli D., Lazebnik Y. 2003. Cell fusion: A hidden enemy? Cancer Cell. 3, 445–448.
Chen E.H. 2005. Unveiling the mechanisms of cell-cell fusion. Science. 308, 369–373.
Chen E.H., Grote E., Mohler W., Vignery A. 2007. Cell-cell fusion. FEBS Lett. 581, 2181–2193.
White J. 1992. Membrane fusion. Science. 258, 917–924.
Wickner W., Schekman R. 2008. Membrane fusion. Nat. Struct. Mol. Biol. 15, 658–664.
Brukman N.G., Uygur B., Podbilewicz B., Chernomordik L.V. 2019. How cells fuse. J. Cell Biol. 218, 1436–1451.
Harrison S.C. 2015. Viral membrane fusion. Virology. 479–480, 498–507.
Kuzmin P.I., Zimmerberg J., Chizmadzhev Y.A., Cohen F.S. 2001. A quantitative model for membrane fusion based on low-energy intermediates. Proc. Natl. Acad. Sci. USA. 98, 7235–7240.
Lentz B.R. 2007. PEG as a tool to gain insight into membrane fusion. Eur. Biophys. J. 36, 315–326.
Hong J., Yang H., Pang D., Wei L., Deng C. 2018. Effects of mono- and di-valent metal cations on the morphology of lipid vesicles. Chem. Phys. Lipids. 217, 19–28.
White J.M., Delos S.E., Brecher M., Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 43, 189–219.
Benhaim M.A., Lee K.K. 2020. New biophysical approaches reveal the dynamics and mechanics of type I viral fusion machinery and their interplay with membranes. Viruses. 12, 413.
Chlanda P., Mekhedov E., Waters H., Schwartz C.L., Fischer E.R., Ryham R.J., Cohen F.S., Blank P.S., Zimmerberg J. 2016. The hemifusion structure induced by influenza virus haemagglutinin is determined by physical properties of the target membranes. Nat. Microbiol. 1, 16050.
Sammalkorpi M., Lazaridis T. 2007. Configuration of influenza hemagglutinin fusion peptide monomers and oligomers in membranes. Biochim. Biophys. Acta BBA – Biomembr. 1768, 30–38.
Qiang W., Sun Y., Weliky D.P. 2009. A strong correlation between fusogenicity and membrane insertion depth of the HIV fusion peptide. Proc. Natl. Acad. Sci. USA. 106, 15314–15319.
Frolov V.A., Cho M.-S., Bronk P., Reese T.S., Zimmerberg J. 2000. Multiple local contact sites are induced by GPI-linked influenza hemagglutinin during hemifusion and flickering pore formation. Traffic. 1, 622–630.
Markosyan R.M., Cohen F.S., Melikyan G.B. 2000. The lipid-anchored ectodomain of influenza virus hemagglutinin (GPI-HA) is capable of inducing nonenlarging fusion pores. Mol. Biol. Cell. 11, 1143–1152.
Leikin S.L., Kozlov M.M., Chernomordik L.V., Markin V.S., Chizmadzhev Y.A. 1987. Membrane fusion: Overcoming of the hydration barrier and local restructuring. J. Theor. Biol. 129, 411–425.
Helm C., Israelachvili J., McGuiggan P. 1989. Molecular mechanisms and forces involved in the adhesion and fusion of amphiphilic bilayers. Science. 246, 919–922.
Kozlovsky Y., Efrat A., Siegel D.A., Kozlov M.M. 2004. Stalk phase formation: effects of dehydration and saddle splay modulus. Biophys. J. 87, 2508–2521.
Kozlovsky Y., Chernomordik L.V., Kozlov M.M. 2002. Lipid intermediates in membrane fusion: Formation, structure, and decay of hemifusion diaphragm. Biophys. J. 83, 2634–2651.
Akimov S., Polynkin M.A., Jiménez-Munguía I., Pavlov K.V., Batishchev O.V. 2018. Phosphatidylcholine membrane fusion is pH-dependent. Int. J. Mol. Sci. 19, 1358.
Kozlov M.M., Chernomordik L.V. 1998. A mechanism of protein-mediated fusion: coupling between refolding of the influenza hemagglutinin and lipid rearrangements. Biophys. J. 75, 1384–1396.
Melikyan G.B., Markosyan R.M., Hemmati H., Delmedico M.K., Lambert D.M., Cohen F.S. 2000. Evidence that the transition of HIV-1 Gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol. 151, 413–424.
Danieli T., Pelletier S.L., Henis Y.I., White J.M. 1996. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J. Cell Biol. 133, 559–569.
Yang X., Kurteva S., Ren X., Lee S., Sodroski J. 2005. Stoichiometry of envelope glycoprotein trimers in the entry of human immunodeficiency virus type 1. J. Virol. 79, 12132–12147.
Kozlovsky Y., Zimmerberg J., Kozlov M.M. 2004. Orientation and interaction of oblique cylindrical inclusions embedded in a lipid monolayer: A theoretical model for viral fusion peptides. Biophys. J. 87, 999–1012.
Pinigin K.V., Kondrashov O.V., Jiménez-Munguía I., Alexandrova V.V., Batishchev O.V., Galimzyanov T.R., Akimov S.A. 2020. Elastic deformations mediate interaction of the raft boundary with membrane inclusions leading to their effective lateral sorting. Sci. Rep. 10, 4087.
Kondrashov O.V., Galimzyanov T.R., Jiménez-Munguía I., Batishchev O.V., Akimov S.A. 2019. Membrane-mediated interaction of amphipathic peptides can be described by a one-dimensional approach. Phys. Rev. E. 99, 022401.
Harrison J.S., Higgins C.D., O’Meara M.J., Koellhoffer J.F., Kuhlman B.A., Lai J.R. 2013. Role of electrostatic repulsion in controlling pH-dependent conformational changes of viral fusion proteins. Structure. 21, 1085–1096.
McMahon H.T., Kozlov M.M., Martens S. 2010. Membrane curvature in synaptic vesicle fusion and beyond. Cell. 140, 601–605.
Kozlov M.M., McMahon H.T., Chernomordik L.V. 2010. Protein-driven membrane stresses in fusion and fission. Trends Biochem. Sci. 35, 699–706.
Chernomordik L. 1996. Non-bilayer lipids and biological fusion intermediates. Chem. Phys. Lipids. 81, 203–213.
Molotkovsky R., Galimzyanov T., Jiménez-Munguía I., Pavlov K., Batishchev O., Akimov S. 2017. Switching between successful and dead-end intermediates in membrane fusion. Int. J. Mol. Sci. 18, 2598.
Акимов С.А., Молотковский Р.Ю., Галимзянов Т.Р., Радаев А.В., Шилова Л.А., Кузьмин П.И., Батищев О.В., Воронина Г.Ф., Чизмаджев Ю.А. 2014. Модель слияния мембран: непрерывный переход в пору слияния с учетом гидрофобных и гидратационных взаимодействий. Биол. мембраны. 31, 14–24.
Merk A., Subramaniam S. 2013. HIV-1 envelope glycoprotein structure. Curr. Opin. Struct. Biol. 23, 268–276.
Martens S., Kozlov M.M., McMahon H.T. 2007. How synaptotagmin promotes membrane fusion. Science. 316, 1205–1208.
Akimov S.A., Kondrashov O.V., Zimmerberg J., Batishchev O.V. 2020. Ectodomain pulling combines with fusion peptide inserting to provide cooperative fusion for influenza virus and HIV. Int. J. Mol. Sci. 21, 5411.
Baudin F., Petit I., Weissenhorn W., Ruigrok R.W.H. 2001. In vitro dissection of the membrane and RNP binding activities of influenza virus M1 protein. Virology. 281, 102–108.
Leser G.P., Lamb R.A. 2017. Lateral organization of influenza virus proteins in the budozone region of the plasma membrane. J. Virol. 91, e02104-16.
Ruigrok R.W.H., Barge A., Durrer P., Brunner J., Ma K., Whittaker G.R. 2000. Membrane interaction of influenza virus M1 protein. Virology. 267, 289–298.
Ruigrok R.W.H., Schoehn G., Dessen A., Forest E., Volchkov V., Dolnik O., Klenk H.-D., Weissenhorn W. 2000. Structural characterization and membrane binding properties of the matrix protein VP40 of Ebola virus. J. Mol. Biol. 300, 103–112.
Wijesinghe K.J., Urata S., Bhattarai N., Kooijman E.E., Gerstman B.S., Chapagain P.P., Li S., Stahelin R.V. 2017. Detection of lipid-induced structural changes of the Marburg virus matrix protein VP40 using hydrogen/deuterium exchange-mass spectrometry. J. Biol. Chem. 292, 6108–6122.
Shtykova E.V., Dadinova L.A., Fedorova N.V., Golanikov A.E., Bogacheva E.N., Ksenofontov A.L., Baratova L.A., Shilova L.A., Tashkin V.Yu., Galimzyanov T.R., Jeffries C.M., Svergun D.I., Batishchev O.V. 2017. Influenza virus matrix protein M1 preserves its conformation with pH, changing multimerization state at the priming stage due to electrostatics. Sci. Rep. 8, e82431.
Peukes J., Xiong X., Erlendsson S., Qu K., Wan W., Calder L.J., Schraidt O., Kummer S., Freund S.M.V., Kräusslich H.G., Briggs J.A.G. 2020. The native structure of the assembled matrix protein 1 of influenza A virus. Nature. 587, 495–498.
Battisti A.J., Meng G., Winkler D.C., McGinnes L.W., Plevka P., Steven A.C., Morrison T.G., Rossmann M.G. 2012. Structure and assembly of a paramyxovirus matrix protein. Proc. Natl. Acad. Sci. USA. 109, 13996–4000.
Briggs J.A.G., Wilk, T., Welker, R., Krausslich, H-G., Fuller S.D. 2003. Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 22, 1707–1715.
Harris A., Sha B., Luo M. 1999. Structural similarities between influenza virus matrix protein M1 and human immunodeficiency virus matrix and capsid proteins: an evolutionary link between negative-stranded RNA viruses and retroviruses. J. Gen. Virol. 80, 863–869.
Dunker A.K., Babu M.M., Barbar E., Blackledge M., Bondos S.E., Dosztányi Z., Dyson H.J., Forman-Kay J., Fuxreiter M., Gsponer J., Han K.H., Jones D.T., Longhi S., Metallo S.J., Nishikawa K., Nussinov R., Obradovic Z., Pappu R.V., Rost B., Selenko P., Subramaniam V., Sussman J.L., Tompa P., Uversky V.N. 2013. What’s in a name? Why these proteins are intrinsically disordered: Why these proteins are intrinsically disordered. Intrinsically Disord. Proteins. 1, e24157.
Goh G., Dunker A.K., Uversky V.N. 2008. A comparative analysis of viral matrix proteins using disorder predictors. Virol. J. 5, 126.
Zhang W., Zheng W., Toh Y., Betancourt-Solis M.A., Tu J., Fan Y., Vakharia V.N., Liu J., McNew J.A., Jin M., Tao Y.J. 2017. Crystal structure of an orthomyxovirus matrix protein reveals mechanisms for self-polymerization and membrane association. Proc. Natl. Acad. Sci. USA. 114, 8550–8555.
Shnyrova A.V., Ayllon J., Mikhalyov I.I., Villar E., Zimmerberg J., Frolov V.A. 2007. Vesicle formation by self-assembly of membrane-bound matrix proteins into a fluidlike budding domain. J. Cell Biol. 179, 627–633.
Faaberg K.S., Peeples M.E. 1988. Association of soluble matrix protein of newcastle disease virus with liposomes is independent of ionic conditions. Virology. 166, 123–132.
Sha B., Luo M. 1997. Structure of a bifunctional membrane-RNA binding protein, influenza virus matrix protein M1. Nat. Struct. Biol. 4, 239–244.
Gregoriades A. 1980. Interaction of influenza M protein with viral lipid and phosphatidylcholine vesicles. J. Virol. 36, 470–479.
Gregoriades A., Frangione B. 1981. Insertion of influenza M protein into the viral lipid bilayer and localization of site of insertion. J. Virol. 40, 323–328.
Ye Z.P., Pal R., Fox J.W., Wagner R.R. 1987. Functional and antigenic domains of the matrix (M1) protein of influenza A virus. J. Virol. 61, 239–246.
Shi Z., Baumgart T. 2015. Membrane tension and peripheral protein density mediate membrane shape transitions. Nat. Commun. 6, 5974.
Saletti D., Radzimanowski J., Effantin G., Midtvedt D., Mangenot S., Weissenhorn W., Bassereau P., Bally M. 2017. The Matrix protein M1 from influenza C virus induces tubular membrane invaginations in an in vitro cell membrane model. Sci. Rep. 7, 40801.
Hilsch M., Goldenbogen B., Sieben C., Höfer C.T., Rabe J.P., Klipp E., Herrmann A., Chiantia S. 2014. Influenza A matrix protein M1 multimerizes upon binding to lipid membranes. Biophys. J. 107, 912–923.
Adu-Gyamfi E., Johnson K.A., Fraser M.E., Scott J.L., Soni S.P., Jones K.R., Stahelin R.V. 2015. Host cell plasma membrane phosphatidylserine regulates the assembly and budding of Ebola virus. J. Virol. 89, 9440–9453.
Gc J.B., Gerstman B.S., Stahelin R.V., Chapagain P.P. 2016. The Ebola virus protein VP40 hexamer enhances the clustering of PI(4,5)P2 lipids in the plasma membrane. Phys. Chem. Chem. Phys. 18, 28409–28417.
Justice P.A., Sun W., Li Y., Ye Z., Grigera P.R., Wagner R.R. 1995. Membrane vesiculation function and exocytosis of wild-type and mutant matrix proteins of vesicular stomatitis virus. J. Virol. 69, 3156–3160.
Gómez-Puertas P., Albo C., Pérez-Pastrana E., Vivo A., Portela A. 2000. Influenza virus matrix protein is the major driving force in virus budding. J. Virol. 74, 11538–11547.
Latham T., Galarza J.M. 2001. Formation of wild-type and chimeric influenza virus-like particles following simultaneous expression of only four structural proteins. J. Virol. 75, 6154–6165.
Jasenosky L.D., Neumann G., Lukashevich I., Kawaoka Y. 2001. Ebola virus VP40-induced particle formation and association with the lipid bilayer. J. Virol. 75, 5205–5214.
Chlanda P., Mekhedov E., Waters H., Sodt A., Schwartz C., Nair V., Blank P.S., Zimmerberg J. 2017. Palmitoylation contributes to membrane curvature in influenza A virus assembly and hemagglutinin-mediated membrane fusion. J. Virol. 91, e00947-17.
Scheiffele P., Rietveld A., Wilk T., Simons K. 1999. Influenza Viruses select ordered lipid domains during budding from the plasma membrane. J. Biol. Chem. 274, 2038–2044.
Veit M., Thaa B. 2011. Association of influenza virus proteins with membrane rafts. Adv. Virol. 2011, 370606.
Gerl M.J., Sampaio J.L., Urban S., Kalvodova L., Verbavatz J.M., Binnington B., Simons K. 2012. Quantitative analysis of the lipidomes of the influenza virus envelope and MDCK cell apical membrane. J. Cell Biol. 196, 213–221.
Nayak D.P., Balogun R.A., Yamada H., Zhou Z.H., Barman S. 2009. Influenza virus morphogenesis and budding. Virus Res. 143, 147–161.
Pohl C., Duprex W.P., Krohne G., Rima B.K., Schneider-Schaulies S. 2007. Measles virus M and F proteins associate with detergent-resistant membrane fractions and promote formation of virus-like particles. J. Gen. Virol. 88, 1243–1250.
Simons K., Ikonen E. 1997. Functional rafts in cell membranes. Nature. 387, 569–572.
Baumgart T., Hess S.T., Webb W.W. 2003. Imaging coexisting fluid domains in biomembrane models coupling curvature and line tension. Nature. 425, 821–824.
Bobone S., Hilsch M., Storm J., Dunsing V., Herrmann A., Chiantia S. 2017. Phosphatidylserine lateral organization influences the interaction of influenza virus matrix protein 1 with lipid membranes. J. Virol. 91, e00267-17.
Peeples M.E., Wang C., Gupta K.C., Coleman N. 1992. Nuclear entry and nucleolar localization of the Newcastle disease virus (NDV) matrix protein occur early in infection and do not require other NDV proteins. J. Virol. 66, 3263–3269.
Wang Y.E., Pernet O., Lee B. 2012. Regulation of the nucleocytoplasmic trafficking of viral and cellular proteins by ubiquitin and small ubiquitin-related modifiers. Biol. Cell 104, 121–138.
Bui M., Wills E.G., Helenius A., Whittaker G.R. 2000. Role of the influenza virus M1 protein in nuclear export of viral ribonucleoproteins. J. Virol. 74, 1781–1786.
Masters P.S. 2006. The molecular biology of coronaviruses. Adv. Virus Res. 66, 193–292.
Perlman S., Netland J. 2009. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 7, 439–450.
Reguera J., Mudgal G., Santiago C., Casasnovas J.M. 2014. A structural view of coronavirus-receptor interactions. Virus Res. 194, 3–15.
Ulasli M., Verheije M.H., de Haan C.A.M., Reggiori F. 2010. Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell. Microbiol. 12, 844–861.
Miller S., Krijnse-Locker J. 2008. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 6, 363–374.
Netherton C.L., Wileman T. 2011. Virus factories, double membrane vesicles and viroplasm generated in animal cells. Curr. Opin. Virol. 1, 381–387.
Hsu N.Y., Ilnytska O., Belov G., Santiana M., Chen Y.H., Takvorian P.M., Altan-Bonnet N. 2010. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell. 141, 799–811.
de Wilde A.H., Raj V.S., Oudshoorn D., Bestebroer T.M., van Nieuwkoop S., Limpens R.W., van den Hoogen B.G. 2013. MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-α treatment. J. Gen. Virol. 94, 1749–1760.
Neuman B.W., Angelini M.M., Buchmeier M.J. 2014. Does form meet function in the coronavirus replicative organelle? Trends Microbiol. 22, 642–647.
Welsch S., Miller S., Romero-Brey I., Merz A., Bleck C.K., Walther P., Bartenschlager R. 2009. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe. 5, 365–375.
Tenorio R., Fernández de Castro I., Knowlton J.J., Zamora P.F., Lee C.H., Mainou B.A., Dermody T.S., Risco C. 2018. Reovirus σNS and μNS proteins remodel the endoplasmic reticulum to build replication neo-organelles. mBio. 9, e01253-18.
Knoops K., Kikkert M., Worm S.H.E. van den, Zevenhoven-Dobbe J.C., van der Meer Y., Koster A.J., Mommaas A.M., Snijder E.J. 2008. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 6, e226.
Gosert R., Kanjanahaluethai A., Egger D., Bienz K., Baker S.C. 2002. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 76, 3697–3708.
Verheije M.H., Raaben M., Mari M., Te Lintelo E.G., Reggiori F., van Kuppeveld F.J.M., Rottier P.J.M., de Haan C.A.M. 2008. Mouse hepatitis coronavirus RNA replication depends on GBF1-mediated ARF1 activation. PLoS Pathog. 4, e1000088.
Stokes H.L., Baliji S., Hui C.G., Sawicki S.G., Baker S.C., Siddell S.G. 2010. A new cistron in the murine hepatitis virus replicase gene. J. Virol. 84, 10148–10158.
Sola I., Almazán F., Zúñiga S., Enjuanes L. 2015. Continuous and discontinuous RNA synthesis in coronaviruses. Annu. Rev. Virol. 2, 265–288.
Knorr R.L., Dimova R., Lipowsky R. 2012. Curvature of double-membrane organelles generated by changes in membrane size and composition. PloS One. 7, e32753.
Helfrich W. 1973. Elastic properties of lipid bilayers: Theory and possible experiments. Z. Für Naturforschung C. 28, 693–703.
Reiss S., Rebhan I., Backes P., Romero-Brey I., Erfle H., Matula P., Bartenschlager R. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe. 9, 32–45.
Reggiori F., Monastyrska I., Verheije M.H., Calì T., Ulasli M., Bianchi S., Bernasconi R., de Haan C.A.M., Molinari M. 2010. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe. 7, 500–508.
Cottam E.M., Maier H.J., Manifava M., Vaux L.C., Chandra-Schoenfelder P., Gerner W., Britton P., Ktistakis N.T., Wileman T. 2011. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy. 7, 1335–1347.
Axe E.L., Walker S.A., Manifava M., Chandra P., Roderick H.L., Habermann A., Griffiths G., Ktistakis N.T. 2008. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182, 685–701.
Prentice E., Jerome W.G., Yoshimori T., Mizushima N., Denison M.R. 2004. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 279, 10136–10141.
Ruch T.R., Machamer C.E. 2012. The coronavirus E protein: Assembly and beyond. Viruses. 4, 363–382.
Curtis K.M., Yount B., Baric R.S. 2002. Heterologous gene expression from transmissible gastroenteritis virus replicon particles. J. Virol. 76, 1422–1434.
Almazán F., DeDiego M.L., Sola I., Zuñiga S., Nieto-Torres J.L., Marquez-Jurado S., Andrés G., Enjuanes L. 2013. Engineering a replication-competent, propagation-defective Middle East respiratory syndrome coronavirus as a vaccine candidate. mBio. 4, e00650-00613.
DeDiego M.L., Alvarez E., Almazán F., Rejas M.T., Lamirande E., Roberts A., Enjuanes L. 2007. A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in vitro and in vivo. J. Virol. 81, 1701–1713.
Bosch B.J., van der Zee R., de Haan C.A.M., Rottier P.J.M. 2003. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 77, 8801–8811.
Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Pöhlmann S. 2020. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 181, 271–280.
Lai A.L., Freed J.H. 2021. SARS-CoV-2 Fusion peptide has a greater membrane perturbating effect than SARS-CoV with highly specific dependence on Ca2. J. Mol. Biol. 433, 166946.
Lai A.L., Millet J.K., Daniel S., Freed J.H., Whittaker G.R. 2017. The SARS-CoV fusion peptide forms an extended bipartite fusion platform that perturbs membrane order in a calcium-dependent manner. J. Mol. Biol. 429, 3875–3892.
Straus M.R., Tang T., Lai A.L., Flegel A., Bidon M., Freed J.H., Daniel S., Whittaker G.R. 2020. Ca2+ ions promote fusion of middle east respiratory syndrome coronavirus with host cells and increase infectivity. J. Virol. 94, e00426-20.
Nathan L., Lai A.L., Millet J.K., Straus M.R., Freed J.H., Whittaker G.R., Daniel S. 2020. Calcium ions directly interact with the Ebola virus fusion peptide to promote structure-function changes that enhance infection. ACS Infect. Dis. 6, 250–260.
Madu I.G., Belouzard S., Whittaker G.R. 2009. SARS-coronavirus spike S2 domain flanked by cysteine residues C822 and C833 is important for activation of membrane fusion. Virology. 393, 265–271.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии