

Биологические мембраны: Журнал мембранной и клеточной биологии, 2023, T. 40, № 1, стр. 3-18
Роль липидных доменов и физических свойств мембран в развитии возрастных нейродегенеративных заболеваний
В. Д. Краснобаев a, b, О. В. Батищев a, *
a Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119071 Москва, Россия
b Московский физико-технический институт (НИУ)
141701 г. Долгопрудный, Россия
* E-mail: olegbati@gmail.com
Поступила в редакцию 03.08.2022
После доработки 31.08.2022
Принята к публикации 01.09.2022
- EDN: NTTUMF
- DOI: 10.31857/S023347552301005X
Аннотация
Растущее количество исследований указывает на взаимосвязь развития нейродегенеративных заболеваний со структурой и липидным составом мембран нейронов. Одними из элементов структуры клеточных мембран, которым в этой связи уделяется особое внимание, являются жидко-упорядоченные липидные домены, или рафты. Изучение рафтов и возрастных изменений липидного состава нейрональных клеток становится все более актуальным и постоянно пополняется новыми исследованиями. В данном обзоре мы постарались осветить возможную роль липидной компоненты клеточных мембран, их структуры и физико-химических характеристик в развитии заболеваний, связанных со старением. Рассматриваются свидетельства, подтверждающие возможную роль рафтов при заболеваниях, приводящих к долговременным нарушениям функционирования нейронов. Есть основания предполагать, что терапевтические эффекты различных молекул, таких как лизолипиды и ганглиозиды, обусловлены их физико-химическими свойствами и реализуются опосредованно, через влияние на организацию липидных доменов в мембранах. По мере более полного определения роли липидных доменов и вообще механизмов взаимодействия и взаимного влияния липидного состава и развития заболеваний, эти знания можно будет использовать для разработки новых терапевтических или профилактических методов борьбы с заболеваниями, связанными со старением.
ВВЕДЕНИЕ
Значительное увеличение средней продолжительности жизни в мире, произошедшее за последние 100 лет, вывело на передний план проблему возрастных нейродегенеративных заболеваний. По данным Всемирной организации здравоохранения, в настоящее время около 8% населения Земли старше 60 лет страдает теми или иными формами деменции, и этот показатель может удвоиться к 2030 году. При этом наше понимание причин развития нейродегенеративных заболеваний все еще недостаточно. Так, например, болезнь Альцгеймера в настоящее время является самым распространенным нейродегенеративным заболеванием в мире (каждые три секунды обнаруживается новый пациент с этим диагнозом), что приводит к колоссальному ущербу для экономики. На сегодняшний день не существует ни способа лечения этой болезни, ни возможности ее диагностирования на ранних стадиях, а используемые для борьбы с ней терапевтические стратегии могут лишь незначительно замедлить скорость ее развития. Последние исследования ставят под сомнение распространенные в течение последних 16 лет представления о ключевой роли олигомерной формы бета-амилоидного пептида в механизмах возникновения болезни Альцгеймера. В июле 2022 года в журнале Science была опубликована статья, согласно которой наличие особых форм амилоидных олигомеров, являющееся подтверждением основной гипотезы развития болезни Альцгеймера, с большой вероятностью является результатом недобросовестности авторов [1]. Поэтому становится ясна причина неэффективности клинических испытаний, пытавшихся через таргетное воздействие на бета-амилоид (монотерапию) вылечить заболевание [2].
Одновременно с этим растущее количество свидетельств указывает на связь развития различных нейродегенеративных заболеваний со структурой и липидным составом мембран нейрональных клеток. В связи с этим особое внимание уделяется жидко-упорядоченным липидным доменам, или рафтам [3, 4]. Поэтому изучение рафтов и возрастных изменений липидного состава нейрональных клеток становится все более актуальным и постоянно пополняется новыми исследованиями различного характера, результаты которых часто противоречат друг другу [5–9]. Тем не менее эти работы действительно показывают взаимосвязь развития возрастных нейродегенеративных заболеваний с процессами, влияющими и на липидный состав мембраны, и на ее физико-химические свойства. В данном обзоре мы постарались осветить возможную роль липидной компоненты клеточных мембран, их структуры и физико-химических характеристик в развитии заболеваний, связанных со старением.
ЖИДКО-УПОРЯДОЧЕННЫЕ ЛИПИДНЫЕ ДОМЕНЫ (РАФТЫ)
Гетерогенность липидного состава клеточных мембран приводит к тому, что даже при физиологических значениях температуры в них могут формироваться более упорядоченные липидные домены (рафты), отличающиеся по структуре и свойствам от остальной части мембраны [10–12]. Эти домены влияют на функционирование различных клеточных белков [13–16], участвуют в передаче сигналов в клетке [17, 18], процессах эндоцитоза [19] и экзоцитоза при синаптической передаче [20–22]. Было показано, что ассоциация ряда нейрональных мембранных белков с липидными рафтами необходима для активации сигнальных каскадов [23–25]. По этой причине рафты могут играть важную роль в передаче сигнала в нервных клетках и значительно влиять на нейрональные функции [26].
Впервые липид-белковые домены в мембранах клеток были обнаружены в 1973 году в печени кролика с помощью метода спиновых меток [27]. В 1982 году Клаузнер и Карновский выпустили статью, в которой проанализировали данные экспериментов, свидетельствующих о наличии в мембране липидных доменов с использованием различных методов: рентгеновской дифракции, электронной микроскопии, диффузионных и калориметрических измерений [28]. Обнаруженные домены различались по составу; один из типов доменов характеризовался повышенным (по сравнению с окружающей мембраной) содержанием холестерина и сфинголипидов. Такие домены формируются из-за того, что данные липиды собираются в отдельную фазу, что было продемонстрировано с помощью калориметрических измерений на модельных мембранах [29]. Следующим шагом было обнаружение таких доменов с помощью рентгеновской дифракции и нескольких методов флуоресцентной микроскопии [30].
Позже Кай Симмонс и Геррит ван Меер исследовали домены клеточных мембран, обогащенные холестерином, гликолипидами и сфинголипидами [11]. Они заинтересовались такими доменами, чтобы объяснить механизм переноса холестерина в эпителиальной клетке из комплекса Гольджи внутри клетки в наружную мембрану клетки. Впоследствии они назвали эти домены липидными “рафтами” (от английского raft – плот). Идея была разработана в 1997 году Симонсом и Элиной Иконен [13]. В своей статье, ставшей одной из фундаментальных для дальнейшего изучения рафтов, они подробно обсудили специфическую растворимость рафтов, а также основания для следующих предполагаемых функций рафтов в клетке:
1. Мембранный транспорт
○ биосинтетический – перенос различных липидных и белковых молекул внутри клетки (главным образом изнутри на клеточную мембрану);
○ эндоцитозный – захват наружной мембраной клетки различных специфических веществ.
2. Транспортная селективность. В процессе биосинтетического транспорта из комплекса Гольджи на мембрану идут два принципиально разных пути с разными промежуточными этапами, которые, по-видимому, определяются благодаря различию между рафтовыми и нерафтовыми везикулами.
3. Передача сигнала – накопление различных сигнальных молекул в силу особенностей состава и структуры рафта.
На симпозиуме 2006 года по липидным рафтам и их клеточным функциям сами рафты были формально определены как малые (10–200 нм), гетерогенные, высокодинамичные, обогащенные стеринами и сфинголипидами домены, которые опосредуют клеточные процессы. Небольшие рафты иногда взаимодействуют с образованием более крупных платформ посредством белок-белковых и белок-липидных взаимодействий [15].
В последние годы исследования липидных рафтов, в частности размер и время жизни рафтов, остаются предметом дискуссий [5]. Экспериментальные наблюдения макроскопических доменов в модельных мембранах с составом, подобным липидному составу внешнего монослоя плазматических мембран, свидетельствуют в пользу идеи липидных доменов [31–33]. Однако в модельных системах липидные домены находились в жидком, но более упорядоченном состоянии, чем окружающая мембрана. Это состояние часто называется в литературе жидко-упорядоченной фазой (Lo, liquid-ordered). Попытки оптически визуализировать липидные домены в клетке не привели к успеху [34]. Авторы отмечают, что при оптическом наблюдении разрешение определяется пределом дифракции света, и неоднородности размером менее сотен нанометров обнаружить невозможно. Другим важным фактором для существования рафтов и фазового разделения является температура. Отмечено, что характерная критическая температура мембраны составляет около 20°C [33], т.е. при более высоких температурах, например температуре человеческого тела, фазовое разделение невозможно. Однако, даже если физиологическая температура слишком высока для полного фазового разделения липидной подсистемы, домены упорядоченной фазы все еще могут образовываться локально, например, вокруг мембранных белков благодаря эффекту “смачивания” [35, 36]. Другой моделью формирования рафтов является флуктуационная модель, предполагающая, что рафты представляют собой локальные флуктуации состава мембран, в результате которых на короткие промежутки времени собираются упорядоченные липидные домены [33].
Несмотря на вышеупомянутые трудности, исследования показывают, что липидные домены играют важную роль во многих клеточных процессах, таких как эндо- и экзоцитоз, апоптоз и т.д., а также при вирусном инфицировании клетки [37–42]. Роль липидных доменов отмечается в функционировании множества рецепторных систем [43]. Например, в работе рецептора иммуноглобулина E [44], белка инсулиноподобного фактора роста (IGF – insulin-like growth factor), роль липидных доменов заключается не только в расположении самого рецептора IGF-IR внутри упорядоченной фазы, но и в накоплении его сигнальных молекул [45]. Также известно о связывании с рафтами сигнальных молекул и самого комплекса T-клеточного рецептора [46, 47] и B-клеточного рецептора [48]. О роли рафтов в различных процессах, непосредственно связанных с развитием нейродегенеративных заболеваний, речь пойдет в следующих разделах обзора.
Отметим, что жидко-упорядоченные липидные домены, или рафты, – не единственный фактор неоднородности биомембран. Например, плазматическая мембрана эукариот соединена с цитоскелетом посредством множества опорных белков, создающих таким образом более плотные участки мембраны [49]. Одним из наиболее изученных подобных мембранных каркасов является плазматическая мембрана эритроцитов, белковый каркас которой организован в виде правильной гексагональной решетки [50]. Известно также об относительно стабильных доменах, сформированных белками, связанными с гликозилфосфатидилинозитолом [51], и множестве других структур и моделей функциональных неоднородностей мембран [52]. В данном обзоре рассматриваются физико-химические свойства жидко-упорядоченных липидных доменов, обогащенных сфинголипидами и холестерином, и их взаимодействия с различными молекулами. Мы будем использовать термины “рафты” и “липидные домены” как синонимы.
РОЛЬ СТРУКТУРЫ И СОСТАВА ЛИПИДНОГО МАТРИКСА КЛЕТОЧНЫХ МЕМБРАН В РАЗВИТИИ ВОЗРАСТНЫХ ЗАБОЛЕВАНИЙ
Болезнь Альцгеймера (БА) является самым распространенным нейродегенеративным заболеванием в мире. Хотя прошло более 100 лет с тех пор, как впервые были описаны симптомы этой болезни [53], все еще мало известно о последовательности явлений, которые приводят к ее развитию. В настоящее время установлено, что амилоидный пептид (бета-амилоид, или Аβ), который агрегирует с образованием амилоидных бляшек в головном мозге, является продуктом последовательного расщепления мембранного гликопротеина, белка-предшественника бета-амилоида (PBA или APP, amyloid-precursor protein) β- и γ-секретазами в плазматической мембране нейронов [54]. Трансмембранный гликопротеин APP экспрессируется почти во всех изученных в настоящее время клетках животных. Хотя существуют тысячи исследований, посвященных изучению APP, его клеточные функции еще не выяснены до конца. Недавно было сделано несколько предположений, а именно, что APP является рецептором холестерина в рафтах мембран нейронов [55, 56], регулирует метаболизм железа в нейронах [57, 58], а также в некотором смысле является одним из компонентов врожденной иммунной системы в нервной системе человека [59, 60].
При БА изменяется соотношение Aβ-пептидов разной длины, и их количество сильно увеличивается, что впоследствии приводит к формированию амилоидных бляшек, образование которых основано на структурном изменении бета-амилоида [61]. Именно с образованием бляшек обычно связывают изменения в тканях при БА. Однако в настоящее время также имеются экспериментальные данные о том, что некоторые формы Aβ оказывают слабое токсическое действие еще до образования фибрилл и бляшек [62, 63]. Согласно ряду исследований, нейротоксическое действие зависит от степени агрегирования молекул Aβ [64]. При этом во множестве работ не было найдено свидетельств того, что сами по себе амилоидные бляшки являются патогенными: во-первых, они могут присутствовать и в тканях людей, не страдающих БА, во-вторых, клинические исследования показали, что воздействия, направленные на устранение амилоидных бляшек, не приносят удовлетворительных терапевтических результатов [65–67]. В связи с этим в настоящее время существует две точки зрения: болезнь Альцгеймера либо никак не связана с Аβ, либо связана, но не на этапе формирования бляшек.
Несмотря на определенные успехи в структурных и динамических исследованиях Αβ пептидов, подробный молекулярный механизм инициирования их образования из APP еще не предложен, а результаты и выводы различных исследовательских групп не всегда согласуются друг с другом. Вопрос о структуре, динамике и характере димеризации фрагментов APP в норме и патологии остается открытым. Нет определенности и в понимании конформационных перестроек мембранных фрагментов APP после их расщепления в мембране даже в мономерном состоянии: например, разворачивается ли C-концевая часть спирали, или сегмент с околомембранной областью в виде амфифильной спирали последовательно погружается в мембрану. APP имеет, по всей видимости, два типа процессинга: неамилоидогенный (участвует в регуляции нейронных функций: регулирует возбудимость, синаптическую пластичность, рост и выживание клеток) и собственно амилоидогенный, в котором образуются свободные пептиды Аβ, состоящие, как правило, из 38–43 аминокислот. Примечательным в данном случае является то, что процессинг APP зависит от его локализации в рафтовой или нерафтовой части мембраны. Трансмембранный участок APP может быть расщеплен двумя основными путями: ɑ-секретазой или ансамблем β- и γ-секретазы. При этом ɑ-секретаза расщепляет APP, находящийся в нерафтовой фазе мембраны, а β-секретаза BACE-1 и γ-секретаза – в рафтовой (рис. 1) [68]. Отщепленные участки Аβ относятся к белкам с природной (внутренней) неупорядоченностью (intrinsically disordered proteins, IDP [69]), и во внеклеточной водной среде они формируют структуры с большим количеством бета-листов, которые составляют большую часть тех самых амилоидных бляшек [70]. Примечательно, что более половины мутаций семейства APP, которые связывают с предрасположенностью к болезни Альцгеймера, происходят именно в его трансмембранном домене, приблизительно соответствующем остаткам 700–723 белка APP (например, мутации A713V, T714I/A, V717F/I/L/G), а также часть на мембранной области 688–694, которая располагается в виде амфифильной альфа-спирали сразу после металл-связывающего домена (например, мутации A692G, E693Q/K/G, D694N) [71–73]. Таким образом, выяснение пространственной структуры трансмембранных фрагментов APP, мутации в которых коррелируют с развитием БА, все еще является актуальной темой исследований, необходимых для выявления молекулярных механизмов начальных стадий патогенеза данной болезни.
Рис. 1.
Взаимодействие APP с ферментами в зависимости от положения в мембране: а – APP в рафте, б – APP вне рафта.
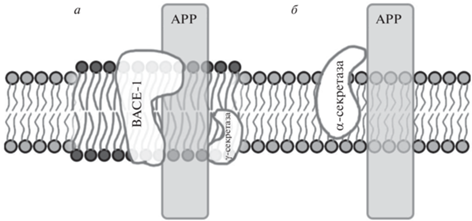
Помимо образования амилоидных бляшек, другие аспекты нейротоксичности продуктов последовательного протеолиза белка APP остаются нераскрытыми, в частности неизвестен молекулярный механизм образования ион-проводящих пор бета-амилоидом [74, 75], хотя существуют молекулярные модели, основанные на экспериментальных структурных данных [76]. Формирование сквозных пор в мембранах нейронов считается одним из возможных механизмов нейротоксичности бета-амилоидов. По результатам различных исследований фибриллам приписываются как нейротоксичные свойства (участие в гибели нейронов по не вполне понятному механизму), так и протекторные: они рассматриваются в качестве резервуара, связывающего свободные бета-амилоиды, либо как защитная реакция организма в попытке закрыть образующиеся под действием бета-амилоидов поры в нейрональных мембранах [74, 75].
Известно, что цепочки Аβ имеют свойство связываться с гликосфинголипидами [77]. Жидко-упорядоченные липидные домены являются теми мембранными структурами, в которых содержание таких липидов выше по сравнению с остальной (жидко-неупорядоченной) мембраной. Таким образом, находящимся в межклеточном пространстве Аβ-пептидам должно быть выгодно связываться с липидными доменами в мембранах нейронов. В этом случае из-за локального повышения концентрации Аβ-пептидов по сравнению с внеклеточной средой данные пептиды, по-видимому, могут приобретать вторичную структуру амилоидной природы [78]. Кроме того, есть основания полагать, что “нормальный” процессинг APP происходит тогда, когда его трансмембранная часть располагается в жидко-неупорядоченной области мембраны, а амилоидогенный – если трансмембранная часть APP оказывается в жидко-упорядоченном домене мембраны [79, 80]. В некоторых исследованиях это объясняют относительно высоким содержанием в рафтах холестерина, уменьшение доли которого в мембране может уменьшать и количество получаемого амилоида [79]. Примечательно, что в качестве генетического фактора риска для БА была предложена дисфункция аполипопротеина E4 (apoE4) [81], роль которого заключается в перераспределении липидов между клетками и регуляции уровня холестерина в мембране.
Биофизическим исследованиям болезни Альцгеймера и, в частности, амилоидной агрегации и взаимодействию бета-амилоидов с мембранами посвящено большое количество научных работ [82–87]. Основные результаты получены в модельных системах различной степени сложности: от мицелл детергента до клеточных культур. На мицеллах, сформированных их различных детергентов, было показано, что бета-амилоиды Аβ-40 и Аβ-42, хотя и отличаются лишь двумя гидрофобными аминокислотными остатками, ведут себя совершенно различно [88]. При одних и тех же соотношениях концентраций пептида и детергента Аβ-40 формирует протяженные агрегаты – фибриллы, хорошо видимые на электронных микрофотографиях, в то время как Аβ-42 образует относительно небольшие олигомеры. При встраивании мицелл с такими олигомерами в плоскую липидную мембрану образуются сквозные поры. В большинстве случаев мембрана с реконструированными мицеллами имеет дискретные уровни проводимости, что соответствует фиксированной стехиометрии пор и олигомеров. Характерно, что при аналогичном встраивании Аβ-40 поры никогда не образовывались [88]. В этой же работе было установлено, что формирующий пору олигомер имеет структуру бета-листа. Однако при наличии в мембране холестерина адсорбированный пептид, по-видимому, может принимать альфа-спиральную конформацию [89, 90]. Поры имеют диаметр приблизительно 1.5 нм [91] и обладают селективной проницаемостью для ионов кальция [92].
Рассмотрим возможные нарушения в работе сигнальных систем, связанных с липидными рафтами и развитием БА, но не связанных напрямую с процессами расщепления APP.
Примером липидов, которым посвящено множество работ в связи с нейродегенеративными и онкологическими заболеваниями, являются церамиды, также участвующие в формировании и регуляции работы липидных доменов [93]. Исследования указывают на повышенные уровни церамидов на самой ранней клинической стадии БА [94], некоторых случаях бокового амиотрофического склероза [95] и в общем случае с возрастом [96]. Подчеркнем, что церамид и церамид-1-фосфат, накапливающиеся в головном мозге при БА, являются производными сфинголипидов. Их накопление может активировать цитозольную фосфолипазу А2 (cPLA2), что приводит к изменениям текучести и проницаемости мембран, а продукты работы cPLA2 могут стимулировать воспалительные процессы [97]. Воспалительные процессы такого рода являются характерными для БА [98], и таким образом подтверждается, что компоненты липидных рафтов, в данном случае сфинголипиды, влияют на БА в том числе и не зависимым от APP образом.
Другим примером изменений в функционировании рафтов, связанных с развитием БА, является холинергическая система (холинергический путь передачи сигнала). Холинергическая система играет важную роль в нейроиммунной коммуникации: участвует в регуляции иммунного ответа. Она включает в себя нейромедиаторы, ацетилхолин, его рецепторы (AChR) и различные ферменты [99]. Известно, что холестерин, являясь одним из основных компонентов липидных рафтов, влияет на структурные и функциональные свойства AChR [100]. Более того, липидные рафты участвуют в кластеризации AChR, необходимой для передачи сигнала, и изменения размеров и характера взаимодействия рафтов уменьшают кластеризацию и функционирование рецепторов [101]. Наконец, согласно многим исследованиям, именно гипофункция холинергической системы является одним из главных признаков БА [102–104].
Болезнь Хантингтона (БХ), характеризующаяся нейродегенерацией полосатого тела и коры головного мозга [105], обусловлена экспрессией тринуклеотида CAG в экзоне 1 гена БХ, что приводит к включению удлиненной последовательности остатков глутамина в N-концевую часть белка хантингтина (htt) [106]. Исследования постсинаптических мембран, выделенных из мышей с моделью БХ, показали, что данный белок до появления симптомов накапливался в мембранах клеток и, как было показано впоследствии, связывался с липидными доменами [107]. Те же исследователи затем обнаружили, что мутантный htt от мышей с бессимптомной БХ более явно связывался с рафтами, чем htt дикого типа.
Болезнь Паркинсона (БП) характеризуется прогрессирующей потерей дофаминергических нейронов. Примерно у трети больных на завершающей стадии развивается деменция [108]. В то время как большинство случаев перечислены как спорадические, 10–15% имеют определенную генетическую причину, согласно исследованию В. Бонифати [109]. Всего им было определено шесть генов, мутации в которых вызывают БП. Согласно предположению автора, взаимодействие мутантного белка с липидными рафтами влияет на передачу сигнала и со временем способствует разрушению нейронов в черной субстанции (substantia nigra). Эта идея поддерживается наблюдениями, что по крайней мере четыре белка, мутации в которых связаны с БП, ассоциированы с рафтами. В частности, было установлено, что причиной аутосомно-доминантной БП, одной из наиболее распространенных форм наследуемой БП, являются мутации в гене LRRK2 [110]. Преобладающая мутантная форма LRRK2 (G2019S) обладает повышенной киназной активностью, вызывающей нейротоксичность [111]. Белок (мутантный и дикого типа) преимущественно локализуется в рафтах, где, согласно предположению, мутант нарушает нормальную передачу сигнала, что приводит к дегенерации нейронов substantia nigra [110]. Причиной редкого аутосомно-доминантного типа БП могут также являться мутации в α-синуклеине [112], а также сверхэкспрессия α-синуклеина дикого типа [113]. В то время как α-синуклеин связывается с молекулами липидов в рафтах, в частности фосфатидилсерином, имеющим остаток олеиновой кислоты и остаток полиненасыщенной жирной кислоты, мутант A30P α-синуклеин нарушает эту связь [114], что приводит к потере функции. Исследования 2007 года показали, что когда α-синуклеин связывается с ганглиозидом GM1, он способствует сворачиванию белка в α-спираль и уменьшает образование α-фибрилл синуклеина [115]. Согласно этому исследованию, GM1 оказывает подобное действие на мутант A53T α-синуклеина, однако его влияние на мутант A30P минимально. Можно предположить, что изменения в связи взаимодействий GM1–рафт могут вызывать изменения в α-синуклеине, которые способствуют симптомам БП. Это также может объяснить положительное действие GM1 на некоторых больных БП в клинических испытаниях [116].
Боковой амиотрофический склероз (БАС) – нейродегенеративное заболевание, характеризующееся прогрессирующей потерей функции двигательных нейронов в головном и спинном мозге, что приводит к параличу мышц, ответственных за произвольные движения. Известно, что только около 10% случаев обусловлены генетическими нарушениями [117]. На возможную роль липидных рафтов в БАС указывают наблюдения, что активация TrkB и экспрессия нейротрофина в мышцах крыс дифференцированно регулируется сигналами, получаемыми от моторных нейронов [118]. После разрушения рафтов с использованием метил-бета-циклодекстрина (mβCD) культура исследуемых двигательных нейронов стала нечувствительной к эксайтотоксичности BDNF. Эти наблюдения в сочетании с рядом ранних исследований, указывающих на то, что люди с двигательными невропатиями могут экспрессировать антитела к ганглиозидам [119–121], поддерживают идею о том, что нормальное функционирование липидных доменов необходимо для деятельности двигательных нейронов. Чрезмерная экспрессия BDNF, малое количество GM1 или другие изменения могут способствовать медленному снижению функциональности нейронов, что в конечном итоге приводит к гибели клеток.
ВОЗМОЖНЫЕ МЕХАНИЗМЫ ЛИПИДНОЙ РЕГУЛЯЦИИ ВОЗРАСТНЫХ НЕЙРОДЕГЕНЕРАТИВНЫХ ЗАБОЛЕВАНИЙ
Различные реакции, происходящие снаружи и внутри клетки, могут изменять липидный состав ее мембраны, например, в случае перекисного окисления липидов при воспалении и окислительном стрессе, нарушении функции белков и нарушении липидного обмена. Установлено, что эти изменения для нескольких видов липидов коррелируют с уже упомянутыми заболеваниями. Например, показано, что фосфолипиды модифицируются в мозге, пораженном БА [122]. В частности, изменения состава липидов коры головного мозга происходят на ранней стадии спорадической БА [123]. Липидный состав также изменяется в лобной и зрительной коре при БП [124–127], а в спинном мозге – при БАС [128, 129]. Существует несколько исследований, в которых сообщается об использовании изменения уровня фосфолипидов, циркулирующих в плазме крови, в качестве возможного биомаркера БА [130–132]. В частности, большое внимание привлекают лизолипиды [133].
Изменение уровня лизолипидов в клетках и в плазме крови может быть результатом окислительного стресса и воспалений, связанных с развитием нейродегенеративных заболеваний [134, 135]. Более того, хорошо известно, что высокий уровень лизолипидов способствует образованию пор в липидном матриксе клеточных мембран [136]. Однако до сих пор неясно, каким может быть действие небольшого количества лизолипидов в клетках, которые с помощью разных механизмов поддерживают целостность своих мембран за счет динамического изменения липидного состава, например, за счет изменения содержания холестерина [137]. Мембраны нейронов содержат относительно большое количество разнообразных гликолипидов, и в первую очередь, ганглиозидов [138]. В структуре Аβ-42 имеется участок 5–16, обеспечивающий связывание пептида с ганглиозидом GM1 [139]. Сам по себе короткий пептид Аβ-5-16 поры не формирует. Однако он эффективно связывает ганглиозид GM1, и при последующей добавке полноразмерного пептида Аβ-42 к клеточной культуре эффективность образования пор оказывается значительно снижена, по-видимому, за счет слабой адсорбции Аβ-42 на мембрану, не имеющей свободного ганглиозида. Схожий эффект достигается при подавлении синтеза ганглиозидов в клетках [140].
Состав ганглиозидов в тканях головного мозга также подвержен возрастным изменениям [141–143]. В целом, содержание ганглиозидов постепенно снижается с возрастом, с некоторой дифференциацией по разделам головного мозга. Интересно, что в масштабном исследовании испытуемых в возрастном диапазоне от 20 до 100 лет общее содержание ганглиозидов оказалось примерно постоянным в период от 20 до 70 лет, однако менялось соотношение различных молекул ганглиозида, а именно – наблюдалось уменьшение доли GM1 и GD1a [138]. GM1 является важным регуляторным липидом, и изменение его уровня связано с нейродегенеративными расстройствами, особенно с БА [145–147], и индуцированным опухолью апоптозом Т-клеток [148]. Кроме того, GM1 может быть фактором, способствующим агрегации Aβ на плазматической мембране [149–151].
В работах [91, 152] было показано, что в структуре Аβ-42 имеется участок 22–35, ответственный за связывание с холестерином за счет образования водородной связи с его OH-группой. Более эффективное связывание Аβ-42 с мембраной, содержащей холестерин, было также продемонстрировано методами молекулярной динамики [153]. Было показано, что короткий пептид Аβ-22–35 способен самостоятельно формировать поры в мембранах клеток SH-SY5Y [90]. Если из мембран предварительно частично удалить холестерин, то поры под действием как Аβ-22-35, так и Аβ-42 образуются значительно реже [140]. В работе [91] показано, что структурно схожий с холестерином бексаротен (bexarotene) может конкурентно связываться с участком 22–35. При инкубации клеток с бексаротеном в концентрации 220 нМ эффективность порации мембран коротким пептидом Аβ-22–35 снижалась приблизительно в 10 раз, а эффективность порации полноразмерным пептидом Аβ-42 снижалась в 2 раза.
На основании данных о структуре Аβ-42 в работе [140] была предложена так называемая двойная терапевтическая стратегия, направленная на подавление связывания Аβ-42 как с ганглиозидом GM1, так и с холестерином. Для этого предлагалось применять химерный пептид Аβ-5–16, связывающий практически весь свободный ганглиозид на мембране, а также бексаротен, конкурирующий с холестерином за связывание с Аβ-42. Было показано, что инкубация клеток SH-SY5Y с химерным пептидом Аβ-5–16 снижает эффективность образования пор полноразмерным Аβ-42 приблизительно в 3 раза, инкубация с бексаротеном – в 2 раза [90], а “двойная инкубация” как с химерным пептидом, так и с бексаротеном – в 10 раз. Однако, хотя предложенная стратегия оказалась эффективна в культуре клеток SH-SY5Y, ее применение для профилактики и лечения БА, по-видимому, практически не осуществимо. Ганглиозид GM1 является линейно-активным веществом: даже в малых концентрациях (доли мольных процентов) он способен существенно (на порядок) изменять граничную энергию мембранных доменов, участвующих в передаче клеточных сигналов [154, 155], что, в свою очередь, приводит к значительному изменению распределения доменов по размерам. Кроме того, установлено, что вызываемые ганглиозидами изменения физико-химических свойств мембранных доменов могут приводить к запуску апоптоза клеток [138, 156, 157]. Связывание ганглиозидов химерными пептидами в рамках применения терапевтической стратегии, вероятнее всего, приведет к хаотической потере нейронами их функций или гибели вследствие глобальной перестройки сложного равновесия мембранных доменов. Поскольку бексаротен в относительно высокой концентрации (220 нМ) снижает эффективность образования пор Аβ-42 всего в 2 раза, для эффективной защиты нейронов необходимо использование более высоких концентраций бексаротена. Согласно результатам работ [158, 159], замена холестерина его ближайшим аналогом и метаболическим предшественником 7-дегидрохолестерином, отличающимся от холестерина лишь одной двойной связью, приводит к значительным изменениям динамики мембранных доменов. Если домены, образованные в мембране, содержащей холестерин, сливаются практически при каждом столкновении, то после замены холестерина на 7-дегидрохолестерин домены при столкновениях не сливаются и не расходятся, образуя протяженные цепные агрегаты круглых доменов. Накопление 7-дегидрохолестерина у человека происходит при синдроме Смита–Лемли–Опица, проявление которого варьирует от мягких нарушений поведения и проблем с обучением до летального исхода. Таким образом, эффективность интенсивной терапии нейронов высокими концентрациями бексаротена, структурно схожего с холестерином, сомнительна.
Липидные рафты представляют собой динамические структуры, обогащенные липидами с насыщенными углеводородными цепями (в основном, сфингомиелином) в комплексе с холестерином [13, 31, 160, 161]. Они толще остальной части мембраны [160, 162, 163], и потому на их границе возникают деформации мембраны, призванные минимизировать контакт гидрофобной части липидного бислоя с водой. Кроме того, различия в плотности упаковки и составе рафта и окружающей мембраны приводят к различию химических потенциалов липидов внутри домена и вне его. Сумма этих вкладов в свободную энергию системы, отнесенная к длине границы липидного домена, называется линейным натяжением [31, 32]. Линейное натяжение регулирует равновесные размеры и форму рафтов и зависит от химической структуры липидов и состава мембраны [159, 164]. По сути, этот энергетический параметр представляет собой двумерный аналог поверхностного натяжения, и существуют липидные компоненты, аккумулирующиеся на границе рафтов и действующие как двумерные поверхностно-активные вещества. Такие молекулы были названы линейно-активными компонентами [165, 166], и они, в зависимости от концентрации, могут значительно изменять размеры рафтов в мембране. Ранее было показано, что вблизи границы липидного домена существует переходная зона размером несколько нанометров, в пределах которой монослои рафта смещены друг относительно друга [167]. Амфифильным молекулам с определенной геометрией, а именно большей полярной частью по сравнению с гидрофобной, энергетически выгодно накапливаться в этих зонах, за счет чего они способны значительно изменять линейное натяжение и размеры рафтов, присутствуя в мембране в достаточно малых количествах [155, 168]. Характерным примером таких молекул является моносиалганглиозид GM1, широко встречающийся в нервных клетках [169], и, как было показано, способный вызывать агрегацию бета-амилоидов [170]. Этот гликолипид может значительно изменять характерные размеры рафтов, причем в концентрациях порядка десятых долей мольного процента [154, 155]. Этот процесс зависит от соотношения между количеством основных рафтовых липидов, сфингомиелина и холестерина. При низком относительном содержании холестерина линейное натяжение границы рафтов и, соответственно, их размер снижаются с ростом концентрации ганглиозида [154]. При более высоком относительном содержании холестерина линейное натяжение снижается при увеличении концентрации GM1 до 0.5 мол. %, а затем повышается, превосходя значения для системы без ганглиозида [155]. Такое поведение системы может объяснить наблюдаемые противоречия в функциях ганглиозида при развитии БА: с одной стороны, кластеры GM1 могут вызывать формирование амилоидных фибрилл [170], с другой – ганглиозид GM1 в малых концентрациях может выполнять и нейропротекторную функцию [171, 172]. Если в мембране наблюдается избыток сфингомиелина, равно как и других липидов с насыщенными углеводородными хвостами, по отношению к холестерину, то присутствие ганглиозида, имеющего высокое сродство к липидным доменам, приведет к практически полному исчезновению рафтов [154] и, как следствие, формированию кластеров ганглиозида, вызывающих амилоидную агрегацию, даже при физиологических концентрациях [151, 173]. С другой стороны, при равных долях сфингомиелина и холестерина в мембране рафты в присутствии GM1 должны укрупняться [155], что будет препятствовать образованию кластеров ганглиозида и мешать формированию амилоидных фибрилл. Таким образом, возрастные и патологические изменения содержания холестерина, сфингомиелина и насыщенных липидов в мембранах нейрональных клеток [26] могут изменять роль GM1 с нейропротекторной на амилоидогенную по рафтозависимому механизму.
Как уже было сказано выше, влияние GM1 на морфологию рафтов определяется в основном его молекулярной геометрией, а именно соотношением размеров полярной и гидрофобной частей молекулы, за счет которых данный гликолипид, располагаясь в переходной зоне вблизи границы рафта, может заметно влиять на его размеры. Аналогичным образом любые амфипатические пептиды должны собираться вблизи границы липидных доменов в мембране, причем такое сродство будет регулироваться соотношением числа гидрофобных и полярных аминокислот в структуре пептида. Если на границе рафтов происходит аккумуляция бета-амилоидов, то этот процесс будет способствовать росту амилоидных фибрилл в этих зонах, причем, как и в случае с ганглиозидами, этот процесс будет зависеть от липидного состава мембраны.
Лизолипиды имеют только один углеводородный хвост в каждой молекуле вместо двух, как у обычных липидов. Следовательно, аналогично ганглиозидам, монослои лизолипидов будут характеризоваться более высокой положительной спонтанной кривизной, чем таковые из обычных липидов и, согласно предложенному механизму, также должны влиять на линейное натяжение границы липидных доменов. Таким образом, с физической точки зрения их влияние на распределение размеров липидных доменов должно быть сходно с влиянием GM1. Эта гипотеза согласуется с имеющимися экспериментальными свидетельствами влияния лизофосфолипидов непосредственно на образование жидко-упорядоченных доменов в липидных мембранах [174, 175].
ЗАКЛЮЧЕНИЕ
Рассмотренные в настоящем обзоре исследования указывают на возможную роль рафтов и состава липидного матрикса клеточных мембран при заболеваниях, которые приводят к нарушению функционирования нейронов. Из этих работ следует, что изменения структуры липидных рафтов могут привести к нарушениям в передаче сигнала и, как следствие, к гибели клеток. В этом контексте мы находимся в начале пути к пониманию возможной связи некоторых мутаций и болезней, обусловленных ими. Можно видеть, что изменения содержания холестерина и гликолипидов в мембранах могут существенно влиять на функционирование клеток. Так, изменения концентрации холестерина в клетках коррелируют с повреждением нейронов [176, 177]; ганглиозиды, присутствующие в высокой концентрации в сером веществе мозга, участвуют в развитии нейродегенеративных заболеваний [178–180]. Хотя причинно-следственная связь возрастных изменений липидного состава мембран нейрональных клеток и развития нейродегенеративных заболеваний пока не установлена, есть основания предполагать, что различные молекулы, такие как обсужденные выше лизолипиды и ганглиозиды могут оказывать терапевтическое действие, обусловленное их физическими свойствами (в данном случае, например, выраженной положительной спонтанной кривизной монослоя их молекул), и их влияние на развитие заболеваний опосредовано организацией липидных доменов в мембранах клеток.
По мере более полного определения роли липидных доменов и вообще механизмов взаимодействия и взаимного влияния липидного состава и развития заболеваний, эти знания можно будет использовать для разработки новых терапевтических или профилактических методов борьбы с заболеваниями, связанными со старением.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Источники финансирования. Работа выполнена при поддержке Министерства науки и высшего образования Российской Федерации.
Соответствие принципам этики. Настоящая статья не содержит описания каких-либо исследований с участием людей или животных в качестве объектов.
Список литературы
Piller C. 2022. Blots on a field? Science. 377, 358–363.
Lobello K., Ryan J.M., Liu E., Rippon G., Black R. 2012. Targeting beta amyloid: A clinical review of immunotherapeutic approaches in Alzheimer’s disease. Int. J. Alzheimerx2019s Dis. 2012, e628070.
Schengrund C.-L. 2010. Lipid rafts: Keys to neurodegeneration. Brain Res. Bull. 82, 7–17.
Mollinedo F., Gajate C. 2015. Lipid rafts as major platforms for signaling regulation in cancer. Adv. Biol. Regul. 57, 130–146.
Munro S. 2003. Lipid rafts: Elusive or illusive? Cell. 115, 377–388.
Levental I., Levental K.R., Heberle F.A. 2020. Lipid rafts: Controversies resolved, mysteries remain. Trends Cell Biol. 30, 341–353.
Ferrara A., Barrett-Connor E., Shan J. 1997. Total, LDL, and HDL cholesterol decrease with age in older men and women. The Rancho Bernardo Study 1984–1994. Circulation. 96, 37–43.
Berns M.A., de Vries J.H., Katan M.B. 1988. Determinants of the increase of serum cholesterol with age: A longitudinal study. Int. J. Epidemiol. 17, 789–796.
Shiomi M., Ito T., Fujioka T., Tsujita Y. 2000. Age-associated decrease in plasma cholesterol and changes in cholesterol metabolism in homozygous Watanabe heritable hyperlipidemic rabbits. Metabolism. 49, 552–556.
van Meer G., Voelker D.R., Feigenson G.W. 2008. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124.
Simons K., van Meer G. 1988. Lipid sorting in epithelial cells. Biochemistry. 27, 6197–6202.
Lagerholm B.C., Weinreb G.E., Jacobson K., Thompson N.L. 2005. Detecting microdomains in intact cell membranes. Annu. Rev. Phys. Chem. 56, 309–336.
Simons K., Ikonen E. 1997. Functional rafts in cell membranes. Nature. 387, 569–572.
Anderson R.G.W., Jacobson K. 2002. A role for lipid shells in targeting proteins to caveolae, rafts, and other lipid domains. Science. 296, 1821–1825.
Pike L.J. 2006. Rafts defined: A report on the Keystone symposium on lipid rafts and cell function. J. Lipid Res. 47, 1597–1598.
Epand R.M. 2008. Proteins and cholesterol-rich domains. Biochim. Biophys. Acta. 1778, 1576–1582.
Mañes S., Mira E., Gómez-Moutón C., Lacalle R.A., Keller P., Labrador J.P., Martínez-A C. 1999. Membrane raft microdomains mediate front-rear polarity in migrating cells. EMBO J. 18, 6211–6220.
Aman M.J., Ravichandran K.S. 2000. A requirement for lipid rafts in B cell receptor induced Ca2+ flux. Curr. Biol. 10, 393–396. https://doi.org/10.1016/s0960-9822(00)00415-2
Lamaze C., Dujeancourt A., Baba T., Lo C.G., Benmerah A., Dautry-Varsat A. 2001. Interleukin 2 receptors and detergent-resistant membrane domains define a clathrin-independent endocytic pathway. Mol. Cell. 7, 661–671.
Jahn R., Scheller R.H. 2006. SNAREs–engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 7, 631–643.
Salaün C., Gould G.W., Chamberlain L.H. 2005. Lipid raft association of SNARE proteins regulates exocytosis in PC12 cells. J. Biol. Chem. 280, 19449–19453.
Suzuki T., Zhang J., Miyazawa S., Liu Q., Farzan M.R., Yao W.-D. 2011. Association of membrane rafts and postsynaptic density: Proteomics, biochemical, and ultrastructural analyses. J. Neurochem. 119, 64–77.
Suzuki S., Numakawa T., Shimazu K., Koshimizu H., Hara T., Hatanaka H., Mei L., Lu B., Kojima M. 2004. BDNF-induced recruitment of TrkB receptor into neuronal lipid rafts: Roles in synaptic modulation. J. Cell Biol. 167, 1205–1215.
Pereira D.B., Chao M.V. 2007. The tyrosine kinase Fyn determines the localization of TrkB receptors in lipid rafts. J. Neurosci. Off. J. Soc. Neurosci. 27, 4859–4869.
Pryor S., McCaffrey G., Young L.R., Grimes M.L. 2012. NGF causes TrkA to specifically attract microtubules to lipid rafts. PloS One. 7, e35163.
Colin J., Gregory-Pauron L., Lanhers M.-C., Claudepierre T., Corbier C., Yen F.T., Malaplate-Armand C., Oster T. 2016. Membrane raft domains and remodeling in aging brain. Biochimie. 130, 178–187.
Stier A., Sackmann E. 1973. Spin labels as enzyme substrates heterogeneous lipid distribution in liver microsomal membranes. Biochim. Biophys. Acta BBA – Biomembr. 311, 400–408.
Karnovsky M.J., Kleinfeld A.M., Hoover R.L., Dawidowicz E.A., McIntyre D.E., Salzman E.A., Klausner R.D. 1982. Lipid domains in membranes. Ann. N. Y. Acad. Sci. 401, 61–74. https://doi.org/10.1083/jcb.94.1.1
Estep T.N., Mountcastle D.B., Barenholz Y., Biltonen R.L., Thompson T.E. 1979. Thermal behavior of synthetic sphingomyelin-cholesterol dispersions. Biochemistry. 18, 2112–2117.
Goodsaid-Zalduondo F., Rintoul D., Carlson J., Hansel W. 1982. Luteolysis-induced changes in phase composition and fluidity of bovine luteal cell membranes. Proc. Natl. Acad. Sci. USA.79 (14), 4332–4336. https://doi.org/10.1073/pnas.79.14.4332
Samsonov A.V., Mihalyov I., Cohen F.S. 2001. Characterization of cholesterol-sphingomyelin domains and their dynamics in bilayer membranes. Biophys. J. 81, 1486–1500.
Baumgart T., Hess S.T., Webb W.W. 2003. Imaging coexisting fluid domains in biomembrane models coupling curvature and line tension. Nature. 425, 821–824.
Veatch S.L., Cicuta P., Sengupta P., Honerkamp-Smith A., Holowka D., Baird B. 2008. Critical fluctuations in plasma membrane vesicles. ACS Chem. Biol. 3, 287–293.
Veatch S.L., Keller S.L. 2005. Seeing spots: Complex phase behavior in simple membranes. Biochim. Biophys. Acta. 1746, 172–185.
Gil T., Sabra M.C., Ipsen J.H., Mouritsen O.G. 1997. Wetting and capillary condensation as means of protein organization in membranes. Biophys. J. 73, 1728–1741.
Akimov S.A., Frolov V.A.J., Kuzmin P.I., Zimmerberg J., Chizmadzhev Y.A., Cohen F.S. 2008. Domain formation in membranes caused by lipid wetting of protein. Phys. Rev. E. 77, 051901.
Nichols B. 2003. Caveosomes and endocytosis of lipid rafts. J. Cell Sci. 116, 4707–4714.
Allen J.A., Halverson-Tamboli R.A., Rasenick M.M. 2007. Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 8, 128–140.
Scheiffele P., Rietveld A., Wilk T., Simons K. 1999. Influenza viruses select ordered lipid domains during budding from the plasma membrane. J. Biol. Chem. 274, 2038–2044.
Gniadecki R., Poumay Y. 2009. Lipid rafts and keratinocyte apoptosis: Regulation death receptors and Akt. Open Dermatol. J. 3, 163–165.
Campbell S.M., Crowe S.M., Mak J. 2001. Lipid rafts and HIV-1: From viral entry to assembly of progeny virions. J. Clin. Virol. 22, 217–227.
Suomalainen M. 2002. Lipid rafts and assembly of enveloped viruses. Traffic. 3, 705–709.
Simons K., Toomre D. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1, 31–39.
Baird B., Sheets E.D., Holowka D. 1999. How does the plasma membrane participate in cellular signaling by receptors for immunoglobulin E? Biophys. Chem. 82, 109–119.
Hong S., Huo H., Xu J., Liao K. 2004. Insulin-like growth factor-1 receptor signaling in 3T3-L1 adipocyte differentiation requires lipid rafts but not caveolae. Cell Death Differ. 11, 714–723.
Janes P.W., Ley S.C., Magee A.I., Kabouridis P.S. 2000. The role of lipid rafts in T cell antigen receptor (TCR) signalling. Semin. Immunol. 12, 23–34.
Langlet C., Bernard A.M., Drevot P., He H.T. 2000. Membrane rafts and signaling by the multichain immune recognition receptors. Curr. Opin. Immunol. 12, 250–255.
Cheng P.C., Dykstra M.L., Mitchell R.N., Pierce S.K. 1999. A role for lipid rafts in B cell antigen receptor signaling and antigen targeting. J. Exp. Med. 190, 1549–1560.
Kapus A., Janmey P. 2013. Plasma membrane–cortical cytoskeleton interactions: A cell biology approach with biophysical considerations. Compr. Physiol. 3 (3), 1231–1281. https://doi.org/10.1002/cphy.c120015
Fowler V.M. 2013. The human erythrocyte plasma membrane: A Rosetta Stone for decoding membrane–cytoskeleton structure. Curr Top Membr. 72, 39–88. https://doi.org/10.1016/B978-0-12-417027-8.00002-7
Varma R., Mayor S. 1998. GPI-anchored proteins are organized in submicron domains at the cell surface. Nature. 394, 798–801.
Sezgin E., Levental I., Mayor S., Eggeling C. 2017. The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 18, 361–374.
Berchtold N.C., Cotman C.W. 1998. Evolution in the conceptualization of dementia and Alzheimer’s disease: Greco-Roman period to the 1960s. Neurobiol. Aging. 19, 173–189.
Tiraboschi P., Hansen L.A., Thal L.J., Corey-Bloom J. 2004. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology. 62, 1984–1989.
Liu Q., Zerbinatti C.V., Zhang J., Hoe H.-S., Wang B., Cole S.L., Herz J., Muglia L., Bu G. 2007. Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron. 56, 66–78.
Beel A.J., Mobley C.K., Kim H.J., Tian F., Hadziselimovic A., Jap B., Prestegard J.H., Sanders C.R. 2008. Structural studies of the transmembrane C-terminal domain of the amyloid precursor protein (APP): Does APP function as a cholesterol sensor? Biochemistry. 47, 9428–9446.
Masaldan S., Bush A.I., Devos D., Rolland A.S., Moreau C. 2019. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic. Biol. Med. 133, 221–233.
Duce J.A., Tsatsanis A., Cater M.A., James S.A., Robb E., Wikhe K., Leong S.L., Perez K., Johanssen T., Greenough M.A., Cho H., Galatis D., Moir R.D., Masters C.L., McLean C., Tanzi R.E., Cappai R., Barnham K.J., Ciccotosto G.D., Rogers J.T., Bush A.I. 2010. Iron-export ferroxidase activity of β-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell. 142, 857–867.
Webers A., Heneka M.T., Gleeson P.A. 2020. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 98, 28–41.
Soscia S.J., Kirby J.E., Washicosky K.J., Tucker S.M., Ingelsson M., Hyman B., Burton M.A., Goldstein L.E., Duong S., Tanzi R.E., Moir R.D. 2010. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PloS One. 5, e9505.
Gouras G.K., Olsson T.T., Hansson O. 2015. β-Amyloid peptides and amyloid plaques in Alzheimer’s disease. Neurotherapeutics.12 (1), 3–11. https://doi.org/10.1007/s13311-014-0313-y
Koh J.Y., Yang L.L., Cotman C.W. 1990. Beta-amyloid protein increases the vulnerability of cultured cortical neurons to excitotoxic damage. Brain Res. 533, 315–320.
Miranda S., Opazo C., Larrondo L.F., Muñoz F.J., Ruiz F., Leighton F., Inestrosa N.C. 2000. The role of oxidative stress in the toxicity induced by amyloid beta-peptide in Alzheimer’s disease. Prog. Neurobiol. 62, 633–648.
Pike C.J., Burdick D., Walencewicz A.J., Glabe C.G., Cotman C.W. 1993. Neurodegeneration induced by beta-amyloid peptides in vitro: The role of peptide assembly state. J. Neurosci. 13 (4), 1676–1687. https://doi.org/10.1523/JNEUROSCI.13-04-01676.1993
Morris G.P., Clark I.A., Vissel B. 2014. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2, 135.
Jang H., Connelly L., Arce F.T., Ramachandran S., Lal R., Kagan B.L., Nussinov R. 2013. Alzheimer’s disease: Which type of amyloid-preventing drug agents to employ? Phys. Chem. Chem. Phys. 15 (23), 8868–8877. https://doi.org/10.1039/c3cp00017f
Rosenblum W.I. 2014. Why Alzheimer trials fail: Removing soluble oligomeric beta amyloid is essential, inconsistent, and difficult. Neurobiol. Aging. 35, 969–974.
Cole S.L., Vassar R. 2007. The Alzheimer’s disease beta-secretase enzyme, BACE1. Mol. Neurodegener. 2, 22. https://doi.org/10.1186/1750-1326-2-22
Uversky V.N. 2009. Intrinsic disorder in proteins associated with neurodegenerative diseases. Front. Biosci. (Landmark Ed). 14 (14), 5188–5238. https://doi.org/10.2741/3594
Lührs T., Ritter C., Adrian M., Riek-Loher D., Bohrmann B., Döbeli H., Schubert D., Riek R. 2005. 3D structure of Alzheimer’s amyloid-beta(1-42) fibrils. Proc. Natl. Acad. Sci. USA. 102, 17342–17347.
Kumar-Singh S., Dewachter I., Moechars D., Lübke U., Jonghe C.D., Ceuterick C., Checler F., Naidu A., Cordell B., Cras P., Broeckhoven C.V., Leuven F.V. 2000. Behavioral disturbances without amyloid deposits in mice overexpressing human amyloid precursor protein with Flemish (A692G) or Dutch (E693Q) mutation. Neurobiol. Dis. 7, 9–22.
Murrell J.R., Hake A.M., Quaid K.A., Farlow M.R., Ghetti B. 2000. Early-onset Alzheimer disease caused by a new Mutation (V717L) in the amyloid precursor protein gene. Arch. Neurol. 57, 885.
Dimitrov M., Alattia J.-R., Lemmin T., Lehal R., Fligier A., Houacine J., Hussain I., Radtke F., Dal Peraro M., Beher D., Fraering P.C. 2013. Alzheimer’s disease mutations in APP but not γ-secretase modulators affect epsilon-cleavage-dependent AICD production. Nat. Commun. 4, 2246.
Peters C., Bascuñán D., Opazo C., Aguayo L.G. 2016. Differential membrane toxicity of amyloid-β fragments by pore forming mechanisms. J. Alzheimers Dis. JAD. 51, 689–699.
Wärmländer S.K.T.S., Österlund N., Wallin C., Wu J., Luo J., Tiiman A., Jarvet J., Gräslund A. 2019. Metal binding to the amyloid-β peptides in the presence of biomembranes: Potential mechanisms of cell toxicity. J. Biol. Inorg. Chem. 24 (8), 1189–1196. https://doi.org/10.1007/s00775-019-01723-9
Ngo S.T., Derreumaux P., Vu V.V. 2019. Probable transmembrane amyloid α-helix bundles capable of conducting Ca2+ ions. J. Phys. Chem. B. 123, 2645–2653.
Ariga T., Kobayashi K., Hasegawa A., Kiso M., Ishida H., Miyatake T. 2001. Characterization of high-affinity binding between gangliosides and amyloid beta-protein. Arch. Biochem. Biophys. 388, 225–230.
Fantini J., Yahi N. 2010. Molecular insights into amyloid regulation by membrane cholesterol and sphingolipids: Common mechanisms in neurodegenerative diseases. Expert Rev. Mol. Med. 12, e27.
Ehehalt R., Keller P., Haass C., Thiele C., Simons K. 2003. Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 160, 113–123.
Rushworth J.V., Hooper N.M. 2010. Lipid rafts: Linking Alzheimer’s amyloid-β production, aggregation, and toxicity at neuronal membranes. Int. J. Alzheimers. Dis. 2011, 603052. https://doi.org/10.4061/2011/603052
Strittmatter W.J., Saunders A.M., Schmechel D., Pericak-Vance M., Enghild J., Salvesen G.S., Roses A.D. 1993. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA. 90, 1977–1981.
Qiang W., Yau W.-M., Lu J.-X., Collinge J., Tycko R. 2017. Structural variation in amyloid-β fibrils from Alzheimer’s disease clinical subtypes. Nature. 541, 217–221.
Takahashi R.H., Nagao T., Gouras G.K. 2017. Plaque formation and the intraneuronal accumulation of β-amyloid in Alzheimer’s disease. Pathol. Int. 67, 185–193.
Srivastava A.K., Pittman J.M., Zerweck J., Venkata B.S., Moore P.C., Sachleben J.R., Meredith S.C. 2019. β-Amyloid aggregation and heterogeneous nucleation. Protein Sci. Publ. Protein Soc. 28, 1567–1581.
Ivanova M.I., Lin Y., Lee Y.-H., Zheng J., Ramamoorthy A. 2021. Biophysical processes underlying cross-seeding in amyloid aggregation and implications in amyloid pathology. Biophys. Chem. 269, 106507.
Reiss A.B., Arain H.A., Stecker M.M., Siegart N.M., Kasselman L.J. 2018. Amyloid toxicity in Alzheimer’s disease. Rev. Neurosci. 29, 613–627.
Tiwari S., Atluri V., Kaushik A., Yndart A., Nair M. 2019. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomedicine. 14, 5541–5554.
Serra-Batiste M., Ninot-Pedrosa M., Bayoumi M., Gairí M., Maglia G., Carulla N. 2016. Aβ42 assembles into specific β-barrel pore-forming oligomers in membrane-mimicking environments. Proc. Natl. Acad. Sci. USA. 113, 10866–10871.
Ji S.-R., Wu Y., Sui S. 2002. Cholesterol is an important factor affecting the membrane insertion of β-amyloid peptide (Aβ1–40), which may potentially inhibit the fibril formation. J. Biol. Chem. 277, 6273–6279. https://doi.org/10.1074/jbc.M104146200
Di Scala C., Chahinian H., Yahi N., Garmy N., Fantini J. 2014. Interaction of Alzheimer’s β-amyloid peptides with cholesterol: Mechanistic insights into amyloid pore formation. Biochemistry. 53, 4489–4502.
Fantini J., Di Scala C., Yahi N., Troadec J.-D., Sadelli K., Chahinian H., Garmy N. 2014. Bexarotene blocks calcium-permeable ion channels formed by neurotoxic Alzheimer’s β-amyloid peptides. ACS Chem. Neurosci. 5, 216–224.
Shafrir Y., Durell S., Arispe N., Guy H.R. 2010. Models of membrane-bound Alzheimer’s Abeta peptide assemblies. Proteins. 78, 3473–3487.
van Blitterswijk W.J., van der Luit A.H., Veldman R.J., Verheij M., Borst J. 2003. Ceramide: Second messenger or modulator of membrane structure and dynamics? Biochem. J. 369, 199–211.
Han X., M Holtzman D., McKeel D.W., Kelley J., Morris J.C. 2002. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: Potential role in disease pathogenesis. J. Neurochem. 82, 809–818.
Takahashi K., Ginis I., Nishioka R., Klimanis D., Barone F.C., White R.F., Chen Y., Hallenbeck J.M. 2004. Glucosylceramide synthase activity and ceramide levels are modulated during cerebral ischemia after ischemic preconditioning. J. Cereb. Blood Flow Metab. 24 (6), 623–627. https://doi.org/10.1097/01.WCB.0000119990.06999.A9
Cutler R.G., Kelly J., Storie K., Pedersen W.A., Tammara A., Hatanpaa K., Troncoso J.C., Mattson M.P. 2004. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA. 101, 2070–2075.
Frisardi V., Panza F., Seripa D., Farooqui T., Farooqui A.A. 2011. Glycerophospholipids and glycerophospholipid-derived lipid mediators: A complex meshwork in Alzheimer’s disease pathology. Prog. Lipid Res. 50, 313–330.
Akiyama H., Barger S., Barnum S., Bradt B., Bauer J., Cole G.M., Cooper N.R., Eikelenboom P., Emmerling M., Fiebich B.L., Finch C.E., Frautschy S., Griffin W.S., Hampel H., Hull M., Landreth G., Lue L., Mrak R., Mackenzie I.R., McGeer P.L., O’Banion M.K., Pachter J., Pasinetti G., Plata-Salaman C., Rogers J., Rydel R., Shen Y., Streit W., Strohmeyer R., Tooyoma I., Van Muiswinkel F.L., Veerhuis R., Walker D., Webster S., Wegrzyniak B., Wenk G., Wyss-Coray T. 2000. Inflammation and Alzheimer’s disease. Neurobiol. Aging. 21, 383–421.
Halder N., Lal G. 2021. Cholinergic system and its therapeutic importance in inflammation and autoimmunity. Front. Immunol. 12, 660342.
Barrantes F.J., Borroni V., Vallés S. 2010. Neuronal nicotinic acetylcholine receptor-cholesterol crosstalk in Alzheimer’s disease. FEBS Lett. 584, 1856–1863.
Zhu D., Xiong W.C., Mei L. 2006. Lipid rafts serve as a signaling platform for nicotinic acetylcholine receptor clustering. J. Neurosci. 26 (18), 4841–4851. https://doi.org/10.1523/JNEUROSCI.2807-05.2006
Coyle J.T., Price D.L., DeLong M.R. 1983. Alzheimer’s disease: A disorder of cortical cholinergic innervation. Science. 219, 1184–1190.
Auld D.S., Kornecook T.J., Bastianetto S., Quirion R. 2002. Alzheimer’s disease and the basal forebrain cholinergic system: Relations to beta-amyloid peptides, cognition, and treatment strategies. Prog. Neurobiol. 68, 209–245.
Schliebs R., Arendt T. 2006. The significance of the cholinergic system in the brain during aging and in Alzheimer’s disease. J. Neural Transm. (Vienna). 113 (11), 1625–1644. https://doi.org/10.1007/s00702-006-0579-2
Vonsattel J.P., Myers R.H., Stevens T.J., Ferrante R.J., Bird E.D., Richardson E.P. 1985. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 44, 559–577.
The Huntington’s Disease Collaborative Research Group. 1993. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 72, 971–983.
Valencia A., Reeves P.B., Sapp E., Li X., Alexander J., Kegel K.B., Chase K., Aronin N., DiFiglia M. 2010. Mutant huntingtin and glycogen synthase kinase 3-beta accumulate in neuronal lipid rafts of a presymptomatic knock-in mouse model of Huntington’s disease. J. Neurosci. Res. 88, 179–190.
Hanagasi H.A., Tufekcioglu Z., Emre M. 2017. Dementia in Parkinson’s disease. J. Neurol. Sci. 374, 26–31.
Bonifati V. 2006. Parkinson’s disease: The LRRK2-G2019S mutation: Opening a novel era in Parkinson’s disease genetics. Eur. J. Hum. Genet. EJHG. 14 (10), 1061–1062. https://doi.org/10.1038/sj.ejhg.5201695
Hatano T., Kubo S.-I., Imai S., Maeda M., Ishikawa K., Mizuno Y., Hattori N. 2007. Leucine-rich repeat kinase 2 associates with lipid rafts. Hum. Mol. Genet. 16, 678–690.
West A.B., Moore D.J., Biskup S., Bugayenko A., Smith W.W., Ross C.A., Dawson V.L., Dawson T.M. 2005. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. USA. 102, 16842–16847.
Krüger R., Kuhn W., Müller T., Woitalla D., Graeber M., Kösel S., Przuntek H., Epplen J.T., Schöls L., Riess O. 1998. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108.
Singleton A.B., Farrer M., Johnson J., Singleton A., Hague S., Kachergus J., Hulihan M., Peuralinna T., Dutra A., Nussbaum R., Lincoln S., Crawley A., Hanson M., Maraganore D., Adler C., Cookson M.R., Muenter M., Baptista M., Miller D., Blancato J., Hardy J., Gwinn-Hardy K. 2003. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 302, 841. https://doi.org/10.1126/science.1090278
Fortin D.L., Troyer M.D., Nakamura K., Kubo S., Anthony M.D., Edwards R.H. 2004. Lipid rafts mediate the synaptic localization of alpha-synuclein. J. Neurosci. 24, 6715–6723. https://doi.org/10.1523/JNEUROSCI.1594-04.2004
Martinez Z., Zhu M., Han S., Fink A.L. 2007. GM1 specifically interacts with alpha-synuclein and inhibits fibrillation. Biochemistry. 46, 1868–1877.
Schneider J.S. 1998. GM1 ganglioside in the treatment of Parkinson’s disease. Ann. N.Y. Acad. Sci. 845, 363–373.
Pasinelli P., Brown R.H. 2006. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 7, 710–723.
Küst B.M., Copray J.C.V.M., Brouwer N., Troost D., Boddeke H.W. 2002. Elevated levels of neurotrophins in human biceps brachii tissue of amyotrophic lateral sclerosis. Exp. Neurol. 177, 419–427. Doi https://doi.org/10.1006/exnr.2002.8011
Annunziata P., Maimone D., Guazzi G.C. 1995. Association of polyclonal anti-GM1 IgM and anti-neurofilament antibodies with CSF oligoclonal bands in a young with amyotrophic lateral sclerosis. Acta Neurol. Scand. 92, 387–393.
Pestronk A., Choksi R. 1997. Multifocal motor neuropathy. Serum IgM anti-GM1 ganglioside antibodies in most patients detected using covalent linkage of GM1 to ELISA plates. Neurology. 49, 1289–1292.
Stevens A., Weller M., Wiethölter H. 1993. A characteristic ganglioside antibody pattern in the CSF of patients with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry. 56, 361–364.
Söderberg M., Edlund C., Alafuzoff I., Kristensson K., Dallner G. 1992. Lipid composition in different regions of the brain in Alzheimer’s disease/senile dementia of Alzheimer’s type. J. Neurochem. 59, 1646–1653.
Fabelo N., Martín V., Marín R., Moreno D., Ferrer I., Díaz M. 2014. Altered lipid composition in cortical lipid rafts occurs at early stages of sporadic Alzheimer’s disease and facilitates APP/BACE1 interactions. Neurobiol. Aging. 35, 1801–1812.
Wood P.L., Tippireddy S., Feriante J., Woltjer R.L. 2018. Augmented frontal cortex diacylglycerol levels in Parkinson’s disease and Lewy Body Disease. PLoS One. 13 (3), e0191815. https://doi.org/10.1371/journal.pone.0191815
Cheng D., Jenner A.M., Shui G., Cheong W.F., Mitchell T.W., Nealon J.R., Kim W.S., McCann H., Wenk M.R., Halliday G.M., Garner B. 2011. Lipid pathway alterations in Parkinson’s disease primary visual cortex. PLoS One. 6 (2), e17299. https://doi.org/10.1371/journal.pone.0017299
Fanning S., Selkoe D., Dettmer U. 2020. Parkinson’s disease: Proteinopathy or lipidopathy? NPJ Park. Dis. 6, 1–9. https://doi.org/10.1038/s41531-019-0103-7
Stok R., Ashkenazi A. 2020. Lipids as the key to understanding α-synuclein behaviour in Parkinson disease. Nat. Rev. Mol. Cell Biol. 21, 357–358.
Chaves-Filho A.B., Pinto I.F.D., Dantas L.S., Xavier A.M., Inague A., Faria R.L., Medeiros M.H.G., Glezer I., Yoshinaga M.Y., Miyamoto S. 2019. Alterations in lipid metabolism of spinal cord linked to amyotrophic lateral sclerosis. Sci. Rep. 9, 11642.
Dodge J.C., Jensen E.H., Yu J., Sardi S.P., Bialas A.R., Taksir T.V., Bangari D.S., Shihabuddin L.S. 2020. Neutral lipid cacostasis contributes to disease pathogenesis in amyotrophic lateral sclerosis. J. Neurosci. 40, 9137–9147.
Mapstone M., Cheema A.K., Fiandaca M.S., Zhong X., Mhyre T.R., MacArthur L.H., Hall W.J., Fisher S.G., Peterson D.R., Haley J.M., Nazar M.D., Rich S.A., Berlau D.J., Peltz C.B., Tan M.T., Kawas C.H., Federoff H.J. 2014. Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 20, 415–418.
Goodenowe D.B., Cook L.L., Liu J., Lu Y., Jayasinghe D.A., Ahiahonu P.W.K., Heath D., Yamazaki Y., Flax J., Krenitsky K.F., Sparks D.L., Lerner A., Friedland R.P., Kudo T., Kamino K., Morihara T., Takeda M., Wood P.L. 2007. Peripheral ethanolamine plasmalogen deficiency: A logical causative factor in Alzheimer’s disease and dementia. J. Lipid Res. 48, 2485–2498.
González-Domínguez R., García-Barrera T., Gómez-Ariza J.L. 2014. Combination of metabolomic and phospholipid-profiling approaches for the study of Alzheimer’s disease. J. Proteomics. 104, 37–47.
Dorninger F., Moser A.B., Kou J., Wiesinger C., Forss-Petter S., Gleiss A., Hinterberger M., Jungwirth S., Fischer P., Berger J. 2018. Alterations in the plasma levels of specific choline phospholipids in Alzheimer’s disease mimic accelerated aging. J. Alzheimers Dis. 62, 841–854. https://doi.org/10.3233/JAD-171036
Girotti A.W., Kriska T. 2004. Role of lipid hydroperoxides in photo-oxidative stress signaling. Antioxid. Redox Signal. 6, 301–310.
Scholte B.J., Horati H., Veltman M., Vreeken R.J., Garratt L.W., Tiddens H.A.W.M., Janssens H.M., Stick S.M. 2019. Oxidative stress and abnormal bioactive lipids in early cystic fibrosis lung disease. J. Cyst. Fibros. 18, 781–789.
Chernomordik L., Chanturiya A., Green J., Zimmerberg J. 1995. The hemifusion intermediate and its conversion to complete fusion: Regulation by membrane composition. Biophys. J. 69, 922–929.
Khandelia H., Loubet B., Olzyńska A., Jurkiewicz P., Hof M. 2014. Pairing of cholesterol with oxidized phospholipid species in lipid bilayers. Soft Matter. 10, 639–647.
Aureli M., Grassi S., Prioni S., Sonnino S., Prinetti A. 2015. Lipid membrane domains in the brain. Biochim. Biophys. Acta. 1851, 1006–1016.
Fantini J., Yahi N. 2011. Molecular basis for the glycosphingolipid-binding specificity of α-synuclein: Key role of tyrosine 39 in membrane insertion. J. Mol. Biol. 408, 654–669.
Di Scala C., Yahi N., Boutemeur S., Flores A., Rodriguez L., Chahinian H., Fantini J. 2016. Common molecular mechanism of amyloid pore formation by Alzheimer’s β-amyloid peptide and α-synuclein. Sci. Rep. 6, 28781.
Yu R.K., Nakatani Y., Yanagisawa M. 2009. The role of glycosphingolipid metabolism in the developing brain. J. Lipid Res. 50 Suppl, S440–S445.
Segler-Stahl K., Webster J.C., Brunngraber E.G. 1983. Changes in the concentration and composition of human brain gangliosides with aging. Gerontology. 29, 161–168.
Svennerholm L., Boström K., Helander C.G., Jungbjer B. 1991. Membrane lipids in the aging human brain. J. Neurochem. 56, 2051–2059.
Svennerholm L., Boström K., Jungbjer B., Olsson L. 1994. Membrane lipids of adult human brain: Lipid composition of frontal and temporal lobe in subjects of age 20 to 100 years. J. Neurochem. 63, 1802–1811.
Chapman J., Sela B.A., Wertman E., Michaelson D.M. 1988. Antibodies to ganglioside GM1 in patients with Alzheimer’s disease. Neurosci. Lett. 86, 235–240.
Brooksbank B.W., McGovern J. 1989. Gangliosides in the brain in adult Down’s syndrome and Alzheimer’s disease. Mol. Chem. Neuropathol. 11, 143–156.
Crino P.B., Ullman M.D., Vogt B.A., Bird E.D., Volicer L. 1989. Brain gangliosides in dementia of the Alzheimer type. Arch. Neurol. 46, 398–401.
Das T., Sa G., Hilston C., Kudo D., Rayman P., Biswas K., Molto L., Bukowski R., Rini B., Finke J.H., Tannenbaum C. 2008. GM1 and tumor necrosis factor-alpha, overexpressed in renal cell carcinoma, synergize to induce T-cell apoptosis. Cancer Res. 68, 2014–2023.
Hayashi H., Kimura N., Yamaguchi H., Hasegawa K., Yokoseki T., Shibata M., Yamamoto N., Michikawa M., Yoshikawa Y., Terao K., Matsuzaki K., Lemere C.A., Selkoe D.J., Naiki H., Yanagisawa K. 2004. A seed for Alzheimer amyloid in the brain. J. Neurosci. 24, 4894–4902.
Ariga T., McDonald M.P., Yu R.K. 2008. Role of ganglioside metabolism in the pathogenesis of Alzheimer’s disease–a review. J. Lipid Res. 49, 1157–1175.https://doi.org/10.1194/jlr.R800007-JLR200
Yamamoto N., Matsubara T., Sato T., Yanagisawa K. 2008. Age-dependent high-density clustering of GM1 ganglioside at presynaptic neuritic terminals promotes amyloid β-protein fibrillogenesis. Biochim. Biophys. Acta, Biomembr. 1778, 2717–2726. https://doi.org/10.1016/j.bbamem.2008.07.028
Di Scala C., Yahi N., Lelièvre C., Garmy N., Chahinian H., Fantini J. 2013. Biochemical identification of a linear cholesterol-binding domain within Alzheimer’s β amyloid peptide. ACS Chem. Neurosci. 4, 509–517.
Yu X., Zheng J. 2012. Cholesterol promotes the interaction of Alzheimer β-amyloid monomer with lipid bilayer. J. Mol. Biol. 421, 561–571.
Bao R., Li L., Qiu F., Yang Y. 2011. Atomic force microscopy study of ganglioside GM1 concentration effect on lateral phase separation of sphingomyelin/dioleoylphosphatidylcholine/cholesterol bilayers. J. Phys. Chem. B. 115, 5923–5929.
Galimzyanov T.R., Lyushnyak A.S., Aleksandrova V.V., Shilova L.A., Mikhalyov I.I., Molotkovskaya I.M., Akimov S.A., Batishchev O.V. 2017. Line activity of ganglioside GM1 regulates the raft size distribution in a cholesterol-dependent manner. Langmuir. 33, 3517–3524.
Ladisch S., Kitada S., Hays E.F. 1987. Gangliosides shed by tumor cells enhance tumor formation in mice. J. Clin. Invest. 79, 1879–1882.
Heitger A., Ladisch S. 1996. Gangliosides block antigen presentation by human monocytes. Biochim. Biophys. Acta. 1303, 161–168.
Staneva G., Chachaty C., Wolf C., Quinn P.J. 2010. Comparison of the liquid-ordered bilayer phases containing cholesterol or 7-dehydrocholesterol in modeling Smith–Lemli–Opitz syndrome. J. Lipid Res. 51, 1810–1822. https://doi.org/10.1194/jlr.M003467
Staneva G., Osipenko D.S., Galimzyanov T.R., Pavlov K.V., Akimov S.A. 2016. Metabolic precursor of cholesterol causes formation of chained aggregates of liquid-ordered domains. Langmuir. 32, 1591–1600.
Heftberger P., Kollmitzer B., Rieder A.A., Amenitsch H., Pabst G. 2015. In situ determination of structure and fluctuations of coexisting fluid membrane domains. Biophys. J. 108, 854–862.
Marsh D. 2009. Cholesterol-induced fluid membrane domains: A compendium of lipid-raft ternary phase diagrams. Biochim. Biophys. Acta. 1788, 2114–2123.
Rinia H.A., Snel M.M.E., van der Eerden J.P.J.M., de Kruijff B. 2001. Visualizing detergent resistant domains in model membranes with atomic force microscopy. FEBS Lett. 501, 92–96.
García-Sáez A.J., Chiantia S., Schwille P. 2007. Effect of line tension on the lateral organization of lipid membranes. J. Biol. Chem. 282, 33 537–33 544. https://doi.org/10.1074/jbc.M706162200
Khadka N.K., Ho C.S., Pan J. 2015. Macroscopic and nanoscopic heterogeneous structures in a three-component lipid bilayer mixtures determined by atomic force microscopy. Langmuir. 31, 12417–12425.
Trabelsi S., Zhang S., Lee T.R., Schwartz D.K. 2008. Linactants: Surfactant analogues in two dimensions. Phys. Rev. Lett. 100, 037802.
Brewster R., Safran S.A. 2010. Line active hybrid lipids determine domain size in phase separation of saturated and unsaturated lipids. Biophys. J. 98, L21–L23.
Galimzyanov T.R., Molotkovsky R.J., Bozdaganyan M.E., Cohen F.S., Pohl P., Akimov S.A. 2015. Elastic membrane deformations govern interleaflet coupling of lipid-ordered domains. Phys. Rev. Lett. 115, 088101.
Pinigin K.V., Kondrashov O.V., Jiménez-Munguía I., Alexandrova V.V., Batishchev O.V., Galimzyanov T.R., Akimov S.A. 2020. Elastic deformations mediate interaction of the raft boundary with membrane inclusions leading to their effective lateral sorting. Sci. Rep. 10, 4087.
O’Brien J.S., Sampson E.L. 1965. Lipid composition of the normal human brain: Gray matter, white matter, and myelin. J. Lipid Res. 6, 537–544.
Yanagisawa K., Odaka A., Suzuki N., Ihara Y. 1995. GM1 ganglioside-bound amyloid beta-protein (A beta): A possible form of preamyloid in Alzheimer’s disease. Nat. Med. 1, 1062–1066.
Amaro M., Šachl R., Aydogan G., Mikhalyov I.I., Vácha R., Hof M. 2016. GM1 Ganglioside inhibits β-amyloid oligomerization induced by sphingomyelin. Angew. Chem. Int. Ed. Engl. 55 (32), 9411–9415. https://doi.org/10.1002/anie.201603178
Cebecauer M., Hof M., Amaro M. 2017. Impact of GM1 on membrane-mediated aggregation/oligomerization of β-amyloid: Unifying view. Biophys. J. 113, 1194–1199.
Šachl R., Amaro M., Aydogan G., Koukalová A., Mikhalyov I.I., Boldyrev I.A., Humpolíčková J., Hof M. 2015. On multivalent receptor activity of GM1 in cholesterol containing membranes. Biochim. Biophys. Acta. 1853, 850–857.
Knuplez E., Curcic S., Theiler A., Bärnthaler T., Trakaki A., Trieb M., Holzer M., Heinemann A., Zimmermann R., Sturm E.M., Marsche G. 2020. Lysophosphatidylcholines inhibit human eosinophil activation and suppress eosinophil migration in vivo. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids. 1865, 158686. https://doi.org/10.1016/j.bbalip.2020.158686
Ma M.-T., Yeo J.-F., Farooqui A.A., Zhang J., Chen P., Ong W.-Y. 2010. Differential effects of lysophospholipids on exocytosis in rat PC12 cells. J. Neural. Transm. 117, 301–308.
Wang D., Zheng W. 2015. Dietary cholesterol concentration affects synaptic plasticity and dendrite spine morphology of rabbit hippocampal neurons. Brain Res. 1622, 350–360.
Jose M., Sivanand A., Channakeshava C. 2021. Membrane cholesterol is a critical determinant for hippocampal neuronal polarity. Front. Mol. Neurosci. 14, 746211.
Furukawa K., Ohmi Y., Tajima O., Ohkawa Y., Kondo Y., Shuting J., Hashimoto N., Furukawa K. 2018. Gangliosides in inflammation and neurodegeneration. Prog. Mol. Biol. Transl. Sci. 156, 265–287.
Sandhoff K., Harzer K. 2013. Gangliosides and gangliosidoses: Principles of molecular and metabolic pathogenesis. J. Neurosci. 33, 10195–101208.
Fusco M., Vantini G., Schiavo N., Zanotti A., Zanoni R., Facci L., Skaper S.D. 1993. Gangliosides and neurotrophic factors in neurodegenerative diseases: From experimental findings to clinical perspectives. Ann. N.Y. Acad. Sci. 695, 314–317. https://doi.org/10.1111/j.1749-6632.1993.tb23074.x
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии