

Физиология человека, 2020, T. 46, № 5, стр. 115-125
Влияние тиреоидных гормонов на электрические и механические параметры сердца
И. Х. Джуманиязова 1, *, О. В. Смирнова 1, **
1 Московский государственный университет имени М.В. Ломоносова
Москва, Россия
* E-mail: irisha-dz@mail.ru
** E-mail: smirnova_ov@mail.ru
Поступила в редакцию 28.12.2019
После доработки 04.03.2020
Принята к публикации 19.04.2020
Аннотация
Несмотря на то, что влияние тиреоидных гормонов на работу сердца были замечены еще в 80-х гг. прошлого столетия, молекулярные механизмы, лежащие в его основе, до сих пор выясняются. Данный обзор подводит некоторые итоги изучения молекулярных мишеней действия тиреоидных гормонов, приводящих к изменению электрической и механической активности сердца; затрагивается вопрос изменения гемодинамики, влияющей на работу сердечной мышцы.
Тиреоидные гормоны синтезируются в щитовидной железе и являются необычайно важными для нашего организма на разных этапах онтогенеза. В процессе раннего онтогенеза они участвуют в развитии центральной нервной системы и организации линейного роста [1]. Во взрослом организме тироксин (Т4) и трийодтиронин (Т3) нормализуют обмен веществ и термогенез, а также регулируют деятельность различных органов, в том числе и сердечно-сосудистой системы [2, 3]. Геномный эффект тиреоидных гормонов на клетку-мишень опосредуется ядерными рецепторами к тиреоидным гормонам (TR), среди которых выделяют 2 основных типа, кодируемых разными генами (TRα и TRβ) [4]. Действуя на эти рецепторы, Т3 изменяет экспрессию определенных генов, модулируя активность клетки [2, 4, 5]. Также тиреоидные гормоны обладают негеномным действием и способны напрямую изменять работу митохондрий, активность белков-обменников и каналов плазматической мембраны, строение цитоскелета [4].
Тиреоидные гормоны, обладая широким спектром действия на организм, могут модулировать электрическую и механическую активность сердца [2–5]. Изменение сердечной деятельности может происходить в результате как прямого действия гормонов на клетки органа-мишени, так и опосредованного их влияния на периферическое сопротивление [3, 6], поэтому мы отдельно рассмотрим прямые и опосредованные (непрямые) кардиотропные эффекты Т3 и Т4.
Целью данного обзора является сбор и систематизация информации о молекулярных и интегральных механизмах действия Т3 на структуру и функционирование сердца млекопитающих.
Прямые кардиотропные эффекты тиреоидных гормонов
Сердце – сложный орган, состоящий из кардиомиоцитов и клеток других типов (фибробласты, макрофаги, клетки гладкой мускулатуры, эндотелиальные клетки и пр.) [7, 8], работа которых изменяется под действием обсуждаемых гормонов. Для понимания действия тиреоидных гормонов на активность сердца необходимо рассмотреть влияние Т3 и Т4 на клетки различных типов. Данный обзор посвящен воздействию тиреоидных гормонов на кардиомиоциты, фибробласты и макрофаги, так как совместно они составляют около 90–95% клеточной массы сердца человека [7].
Влияние тиреоидных гормонов на кардиомиоциты. Необходимо отметить, что, в отличие от человеческих кардиомиоцитов, в миоцитах грызунов отсутствует (или присутствует в следовых количествах) деиодиназа 2 типа [9], поэтому в миоцитах сердца крыс и мышей превращение Т4 в активный Т3 незначительно.
Среди изоформ рецепторов тиреоидных гормонов в кардиомиоцитах крыс преобладают TRα1, TRβ1 и TRβ2 [10], у человека же наиболее распространенными ядерными рецепторами являются TRα1, TRα2 и TRβ1 [11].
Тиреоидные гормоны, а именно активный Т3, действуя через ядерные рецепторы, способен изменять экспрессию нескольких генов, активно вовлеченных в нормальное функционирование кардиомиоцита (табл. 1). Кроме того, тиреоидные гормоны обладают негеномными эффектами и способны напрямую взаимодействовать с белками-мишенями в клетке [5, 12, 13], а также с интегрином αVβ3 [14–16].
Таблица 1.
Действие Т3 на экспрессию некоторых белков кардиомиоцита
| Увеличение экспрессии | Уменьшение экспрессии |
|---|---|
| α-тяжелые цепи миозина (α-MHC) [6, 17, 18] | β-тяжелые цепи миозина (β-MHC) [6, 17] |
| Потенциал-зависимые калиевые каналы (Kv1.5, Kv4.2, Kv4.3) [19] | Na+/Ca2+ обменник (NCX) [26] |
| Саркоплазматическая Ca-АТФаза 2 типа (SERCA2) [20] | Фосфоламбан (Plb) [27] |
| Na/K-AТФаза [21] | Рецептор к тиреоидным гормонам α1 [6] |
| Рианодиновые рецепторы саркоплазматического ретикулума (RyR) [22, 23] | |
| Канальные белки фанни-тока HCN2, HCN4 [24] | |
| Na+/H+-антипортер [25] | |
| Актин [18] |
Изменение электрофизиологических и сократительных характеристик миокарда. Показано, что Т3 непосредственно взаимодействует с натриевыми каналами сердца грызунов (кроликов и крыс), вызывая замедление инактивации [28] и увеличение неинактивируемой компоненты Na тока, INa, late. Следствием увеличения INa, late являются угрожающие жизни аритмии [29], которые наблюдаются у больных с повышенным содержанием тиреоидных гормонов [30].
Большинство эффектов Т3 приводит к изменению кальциевой динамики внутри клетки-мишени (↑SERCA2, ↑RyR, ↓NCX, ↓Plb), при этом Т3 не влияет на величину кальциевого тока L-типа [19]. Общий эффект таких изменений заключается в ускорении выброса кальция из саркоплазматического ретикулума (СПР) и его закачивания внутрь него (рис. 1). Кальций – вторичный мессенджер, который, кроме всего прочего, является триггером мышечного сокращения. Итак, изменение кальциевой динамики внутри отдельных миоцитов приводит к развитию быстрых и сильных мышечных сокращений из-за мощного и кратковременного увеличения содержания кальция в цитоплазме (положительный инотроп-ный эффект при введении Т3 грызунам [3]). Тиреоидные гормоны обладают негеномным (возможно, опосредованным через Протеинкиназу C) действием на Ca-АТФазу плазматической мембраны (усиление ее активности [31]) и поэтому могут способствовать выкачиванию кальция из цитоплазмы, компенсируя сниженную экспрессию NCX (рис. 1).
Рис. 1.
Некоторые геномные и негеномные эффекты трийодтиронина, затрагивающие кальциевую динамику рабочего кардиомиоцита. А – работа некоторых каналов и насосов кардиомиоцита. Б – влияние трийодтиронина на каналы и насосы кардиомиоцита. Жирные стрелки обозначают усиление выброса кальция из саркоплазматического ретикулума (или цитоплазматической мембраны) или его накачивания в саркоплазматический ретикулум. Увеличение выброса и накачивания пропорциональны. Под рисунками схематично изображена кривая изменения концентрации кальция в цитоплазме при развитии единичного потенциала действия для отдельного миоцита в норме (А) и при хроническом воздействии Т3 (Б). По [22, 29], с изменениями.
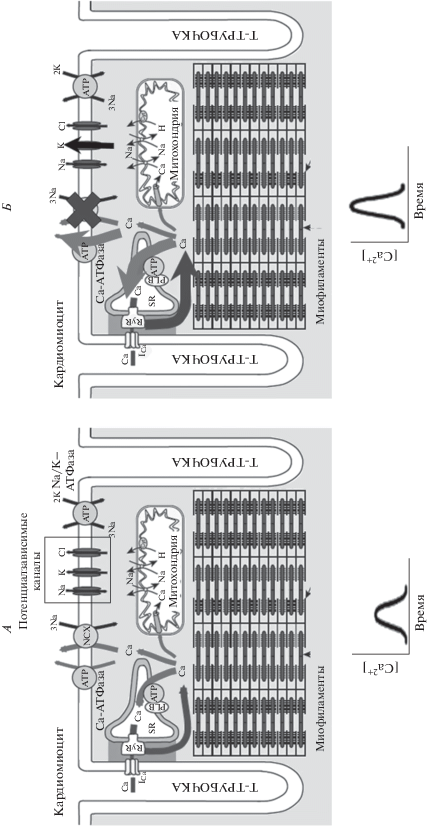
Кальций – ион, динамика которого, наряду с “пейсмекерным” током If, обеспечивает пейсмекерную активность клеток синоатриального узла (САУ) сердца [24, 32, 33]. Внутриклеточный круговорот кальция (“кальциевые часы”) в клетках САУ определяет их способность спонтанной генерации потенциалов действия [32]. Так как оборот кальция ускоряется (такой вывод можно сделать по изменениям экспрессии белков кальциевой динамики, описанным выше), а плотность If увеличивается (из-за увеличения плотности соответствующих канальных белков на мембране клетки САУ, табл. 1), это приводит к ускорению генерации потенциалов действия клетками САУ (положительный хронотропный эффект при введении крысам Т3 [3]).
Отдельное внимание стоит уделить значимому увеличению транскриптов кальциевых каналов L-типа при гипотиреозе [34]. На фоне сниженной экспрессии белков СПР (табл. 1) при гипотиреоидизме особую важность для развития сокращения приобретает внеклеточный кальций. Он попадает в клетку через кальциевые каналы L-типа. Поэтому для развития нормального сокращения клетка при гипотиреоидизме компенсаторно увеличивает экспрессию кальциевых каналов плазматической мембраны.
Сила сокращения кардиомиоцита может изменяться вследствие ярко выраженного геномного действия Т3 на экспрессию разных изоформ тяжелых цепей миозина [3, 6, 17, 35] (табл. 1). α-MHC (тяжелая цепь миозина, изоформа α) называют “быстрой” изоформой миозина. По сравнению с β-MHC, α-MHC обладает более выраженной АТФазной активностью и развивает большую сократительную силу [36]. Таким образом, сдвиг в сторону увеличения экспрессии α-MHC и подавление экспрессии β-MHC под влиянием Т3 позволяет ускорить сокращение и увеличить количество затраченной на него энергии.
Увеличение частоты сердечных сокращений и предшествующее ему увеличение частоты генерации ритма должно приводить к уменьшению длительности потенциалов действия рабочего миокарда (что подтверждается экспериментально на крысах и морских свинках при введении Т3 [12, 19], рис. 2). При невыполнении этого необходимого условия возможно развитие тяжелых предсердных и желудочковых аритмий (иногда наблюдается у пациентов при гипертиреоидизме [30]).
Длительность потенциала действия определяется работой калиевых потенциалзависимых каналов нескольких типов [37, 38]. Качественно калиевые токи в миоцитах основных модельных объектов (крыс и мышей) и человека разительно отличаются друг от друга: у мышей и крыс стадия реполяризации практически полностью обеспечивается транзиторными токами (Ito), обусловленными каналами Kv1.4, Kv4.2, Kv4.3 и ультрабыстрым калиевым током задержанного выпрямления IKur (канальные белки Kv1.5 и др.); у человека же эти токи обеспечивают лишь фазу быстрой реполяризации. Фаза полной реполяризации у человека в первом приближении является результатом взаимодействия IKr и IKs [29, 37].
Увеличение плотности реполяризующих токов приводит к уменьшению длительности потенциала действия. Один из геномных эффектов Т3 заключается в усилении экспрессии калиевых каналов Kv1.5, переносящих ток IKur, ответственный за реполяризацию в миоцитах крыс и мышей. Таким образом, Т3 может способствовать уменьшению длительности потенциала действия. С другой стороны, Т3 уменьшает экспрессию каналов Kv1.4, переносящих ток Ito, что может способствовать увеличению длительности потенциала действия [19, 39, 40]. В то же время у кролика хроническое введение Т3 вызывает значимое увеличение транзиторного тока Ito в желудочковых миоцитах [41]. Последствия таких разнонаправленных и видоспецифичных изменений экспрессии каналов, определяющих токи Ito и IKur, на длительность потенциала действия сложно оценить без контекста регуляции Т3 других калиевых каналов.
KCNQ1 (порообразующая единица) вместе с KCNE1 (регуляторная субъединица) определяют потенциалзависимый ток IKs [29, 37, 38]. Этот же белок уникален своей способностью формировать постоянно открытый канал утечки калия, образуя комплексы с ауксиллярными единицами KCNE2 и/или KCNE3 в клетках невозбудимых тканей [42]. Комплекс KCNQ1 и KCNE2 необходим для нормального накопления йода в клетках щитовидной железы [42, 43], а нокаутные по гену KCNE2 мыши имеют сниженную концентрацию тиреоидных гормонов в крови [44]. К сожалению, в большинстве работ исследуют именно зависимость синтеза тиреоидных гормонов от наличия/нокаута KCNQ1, а не обратную связь, что не позволяет сделать каких-либо выводов о влиянии Т3 на экспрессию или работу комплекса KCNQ1-KCNE1 в сердце. Отсутствие подобных работ можно объяснить малым уровнем экспрессии обсуждаемых белков в кардиомиоцитах крыс и мышей [45], которые являются избранными модельными объектами физиологии.
Действие Т3 на канальные белки, формирующие ток IKr (KCNH2) [29, 38], опосредовано прямым влиянием комплекса TRβ-Т3 на фосфатидилинозитол-3-киназу (PI3K) [46]. Т3 замедляет инактивацию образованных белками каналов KCNH2, что с учетом особенностей развития тока этого типа приводит к увеличению данного тока и ускорению реполяризации.
Ток IK1 (фоновый ток входящего выпрямления), поддерживающий нормальные значения потенциала покоя, определяет морфологию потенциала действия [29, 38, 47]. Увеличение плотности этого тока без компенсации со стороны других токов может приводить к укорочению потенциала действия и влиять на токи фазы плато [29, 47]. Аппликация Т3 в остром эксперименте приводит к увеличению IK1 и укорочению потенциала действия желудочковых миоцитов морской свинки; этот эффект не может быть объяснен взаимодействием с ДНК [12]. Вызванный гипертиреоидизм у крыс вызывает увеличение экспрессии гена KCNJ2, кодирующего канальный белок для IK1 [48], что может способствовать укорочению потенциала действия для предотвращения развития постдеполяризации.
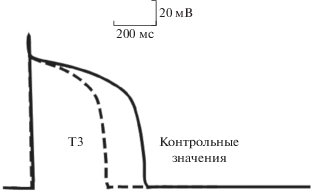
Рис. 2.
Изменение конфигурации потенциала действия при перфузии препарата желудочкового миокарда морской свинки трийодтиронином. По [12].
Липидные рафты “заякоривают” канальные белки в определенных местах плазматической мембраны, что влияет на работу каналов и величину тока, который они определяют. Разрушение липидных рафтов, изменение соотношения жирных кислот в мембране или ее подвижности могут приводить к изменению функционирования канальных белков [29]. Тиреоидные гормоны способны изменять метаболизм липидов [49], это может привести к разнообразным электрофизиологическим эффектам для отдельной клетки и сердца в целом. Вопрос требует дальнейшего изучения.
Отдельные кардиомиоциты сообщаются друг с другом посредством щелевых контактов, образованных в основном (в рабочем миокарде) коннексином 43 (Cх43) [29]. Такие контакты позволяют мышечной ткани формировать функциональный синцитий, способствуют распространению потенциала действия по миокарду с большой скоростью на небольшие расстояния. С другой стороны, нарушение работы щелевых контактов может способствовать появлению аритмогенных очагов и развитию фибрилляций [50]. Т3 не только увеличивает содержание Cх43, но и способствует ускоренному формированию щелевых контактов в культуре крысиных кардиомиоцитов [51, 52]. Так Т3 может способствовать ускорению проведения возбуждения по рабочему миокарду.
Изменение энергетического баланса кардиомиоцита. Усиление транскрипции и трансляции SERCA2 и сопутствующее увеличение количества данного белка на мембране СПР, усиление активности мембранной Ca-АТФазы, ускорение процесса сокращения и увеличение его частоты, переключение на “быструю” изоформу тяжелых цепей миозина может привести к увеличению потребления АТФ клетками. Для этого необходимо ускорить процессы клеточного дыхания, происходящие в митохондриях.
Ускорение процессов клеточного дыхания может происходить несколькими способами. Во-первых, у TRs имеются ядерные гены-мишени, кодирующие ферменты, участвующие в синтезе АТФ. Во-вторых, может увеличиваться синтез белков клеточного дыхания в самих митохондриях. В-третьих, у Т3 могут быть непосредственные мишени на мембране митохондрий [53, 54]. Согласно литературным данным, Т3 увеличивает активность компонентов электрон-транспортной цепи и митохондриальных ферментов [54], ускоряет окисление жиров [55], но усиливает “протонную утечку” за счет увеличения количества и активности унитранспортеров (UCT) мембран митохондрий, способствующих рассеянию большего количества энергии в тепло [53–56].
Кроме того, ускорение работы электрон-транспортной цепи способствует формированию активных форм кислорода, которые в избытке могут приводить к развитию окислительного стресса [57, 58], поэтому содержание основных компонентов антиоксидантной системы зависит от количества тиреоидных гормонов [59].
Влияние тиреоидных гормонов на фибробласты. Резидентные фибробласты составляют большую часть сердца и участвуют в процессах деления и синтеза/разрушения компонентов соединительной ткани (коллаген типа I, III, IV, V, VI, ламинин, эластин) [60], формировании соединительнотканного рубца после инфаркта или при гипертензии (после активации и изменения фенотипа с пролиферативного на миофибробластный MyoFb [61]). Существует несколько источников фибробластов: часть их происходит из эпикардиальных стволовых клеток, часть мигрирует из кровеносного русла [7, 62]. Фибробласты реагируют на большое количество внешних стимулов: механический стресс, циркулирующие сигнальные соединения [62, 63].
Фибробласты (компоненты невозбудимой ткани) могут взаимодействовать с кардиомиоцитами посредством коннексинов (Cx40, Cx43, Cx45), изменяя их электрофизиологические характеристики [7, 64, 65] (рис. 3, А). Количество Cx43 прямо зависит от содержания Т3 [66]. Таким образом, Т3 способствуют усилению взаимодействий фибробласт–миоцит и макрофаг–миоцит.
Рис. 3.
Изменение электрофизиологических характеристик клеток при взаимодействии миоцитов с фибробластами (А) и макрофагами (Б). Показано изменение формы потенциала действия и значений потенциала покоя кардиомиоцита при взаимодействии с фибробластом/макрофагом. На схемах (А) и (Б) показаны результаты математического моделирования. По [8, 92].
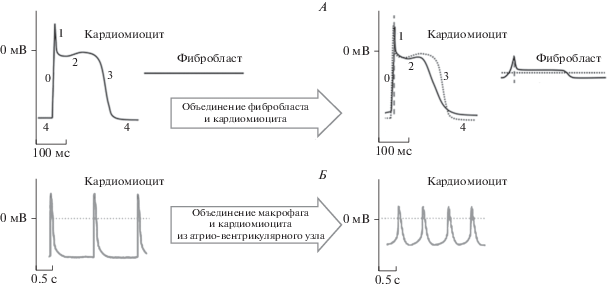
Механическое воздействие на фибробласты сердца приводит к их гиперполяризации, которая может передаваться на миоциты через щелевые контакты. Такое взаимодействие частично препятствует деполяризации миоцитов, вызываемой их растяжением, и последующей индукции аритмий [65]. Таким образом, Т3 может оказывать антиаритмогенное действие на миокард, усиливая коммуникацию между миоцитами и фибробластами посредством увеличения экспрессии Cx43 на мембранах обоих типов клеток.
При гипертиреозе наблюдается физиологическая гипертрофия миокарда [3, 6, 35]. Такой эффект (при исключении влияния циркулирующей крови) может быть вызван несколькими причинами: 1) увеличением объема каждого отдельного миоцита (из-за увеличения содержания сократительных белков (табл. 1); 2) увеличением количества миоцитов и фибробластов, поддержанием их жизни путем активации mTOR и Akt пути [67] (рис. 4).

Несмотря на то, что Т3 подавляет синтез компонентов соединительной ткани сердца [68, 69], он усиливает пролиферацию фибробластов. Таким образом, изменение синтетической активности фибробластов не является основным эффектом тиреоидных гормонов на этот тип клеток, а механические стимулы при изменении кровотока значительно влияют на электрофизиологические параметры системы кардиомиоцит–фибробласт [65].
Влияние тиреоидных гормонов на макрофаги. Макрофаги являются резидентными клетками некоторых отделов сердца, например, атрио-вентрикулярного узла (определяющего проведение возбуждения от предсердий к желудочкам). При этом макрофаги взаимодействуют с кардиомиоцитами посредством Cх43, который экспрессируется в иммунных клетках, в том числе в моноцитах и их клетках-потомках [8, 70], и изменяют их электрическую активность (рис. 3, Б). Также различные популяции активированных макрофагов участвуют в восстановлении миокарда после инфаркта [71].
Как и большинство клеток организма, макрофаги имеют рецепторы тиреоидных гормонов [72]. Аппликация Т3 у грызунов приводит к уменьшению миграции клеток-предшественников макрофагов (моноцитов) из кровеносного русла, изменению активности фагоцитоза, выделению активных форм кислорода; в культурах клеток наблюдается сниженная концентрация провоспалительных цитокинов (IL-1, IL-6) [73–75].
К сожалению, популяция резидентных макрофагов сердца мало изучена, поэтому большинство приведенных данных касается популяции макрофагов кости или выращенных in vitro из резидентной популяции макрофагов кости. Чаще всего активированные популяции макрофагов сердца (М1 или М2) упоминаются в контексте восстановительных процессов после развития инфаркта миокарда, поэтому дальнейшие исследования механизмов влияния тиреоидных гормонов на макрофаги сердца и их взаимодействие с кардиомиоцитами могут иметь терапевтическую направленность (например, использование аналога тиреоидных гормонов с выраженным инотропным эффектом на сердце [76]).
Подводя промежуточный итог, Т3 в различных клетках сердца:
1. Увеличивает частоту генерации потенциалов действия пейсмейкерных клеток.
2. Ускоряет внутриклеточный круговорот кальция.
3. Способствует развитию быстрого и резкого сокращения отдельного миоцита.
4. Разнонаправленно влияет (геномно и негеномно) на калиевые каналы, но все эти изменения приводят к уменьшению длительности потенциала действия возбудимых клеток.
5. Способствует формированию щелевых контактов между кардиомиоцитами и другими типами клеток сердца (фибробластами и макрофагами).
6. Изменяет энергетический баланс кардиомиоцита, увеличивает диссипацию энергии митохондриями.
7. Способствует развитию окислительного стресса в миоците в результате производства большого количества активных форм кислорода быстроработающими ферментами электрон-транспортной цепи митохондрий.
8. Способствует пролиферации фибробластов и их выживанию.
9. Подавляет синтез коллагена фибробластами.
10. Изменяет ответ резидентной популяции макрофагов сердца на воспалительный ответ.
Сочетанный эффект заключается в учащении генерации потенциалов действия, облегчении проведения возбуждения по миокарду, изменении электрофизиологических характеристик кардиомиоцитов и их энергетических потребностей, изменении композиции популяций резидентных фибробластов и макрофагов в сердце.
Непрямое действие тиреоидных гормонов на сердце
Тиреоидные гормоны вызывают изменения объема циркулирующей крови и общего периферического сопротивления, влияющие на функционирование сердца.
Влияние на объем крови и общее периферическое сопротивление. Под действием тиреоидных гормонов изменяется экспрессия некоторых белков-переносчиков клеток канальца нефрона: увеличивается количество белковых молекул Na+/H+-антипортера проксимальных канальцев [77], Na+/Pi-котранспортера [78]. В результате наблюдается снижение концентрации ионов натрия в дистальных канальцах и активация ренин-ангиотензин-альдостероновой системы. Это приводит к задержке соли и воды в организме и увеличению объема крови [3, 5, 35, 79]. Также Т4 и Т3 являются стимуляторами эритропоэза [80], что поддерживает кислород-переносящую способность крови.
Увеличенный объем крови способствует возрастанию преднагрузки на сердечную мышцу – той силы, которая действует на мышечное волокно перед его сокращением. Такое воздействие приводит к увеличению ударного объема и к физиологической гипертрофии миокарда [81].
Существует несколько предположений, позволяющих объяснить снижение общего тонуса сосудов. Во-первых, при воздействии Т3 уменьшается содержание рецепторов к ангиотензину II на мембранах гладкомышечных клеток. Во-вторых, Т3 вызывает увеличение продукции NO, являющегося одним из важных релаксантов гладкой мускулатуры [82]. Возможно, локальное закисление и выделение метаболитов в результате усиленных процессов клеточного дыхания также вносит свой вклад в расширение сосудов [83]. Расширение сосудов и последующее падение общего периферического сопротивления приводит к снижению постнагрузки (того давления, против которого должна работать мышца) на сердечную мышцу. Это способствует изменению архитектуры сердца и увеличению ударного объема.
Изменение архитектуры миокарда при механическом стрессе. Увеличение преднагрузки за счет увеличения объема крови приводит к растяжению миокарда. Такое воздействие может иметь несколько эффектов: во-первых, согласно закону Франка-Старлинга, растяжение сердечной мышцы в физиологических пределах приводит к увеличению силы ее сокращения [84]; во-вторых, механический стресс вызывает повышение содержания цитокинов, способствующих гипертрофии миокарда (например, фактора роста соединительной ткани, CTGF) [85].
Изменение нервной регуляции сердечной деятельности. Вариабельность сердечного ритма – важный показатель, анализ которого позволяет получить информацию о преобладании тонических симпатических или парасимпатических влияний на сердечную деятельность [86]. При гипертиреоидизме наблюдается увеличение мощности низкочастотных волн (определяющих в основном симпатическую регуляцию) при спектральном анализе вариабельности у 30-летних пациентов [87]. Стоит отметить, что такой эффект может наблюдаться не из-за усиления симпатических влияний [88], а из-за ослабления парасимпатических влияний [89]. При гипотиреоидизме наблюдается также усиление симпатических влияний на сердце, имеющее, возможно, компенсаторный характер [90].
Описанные выше эффекты могут объясняться не только модуляцией тонических влияний разных отделов вегетативной нервной системы, но и изменениями в количестве и афинности адренергических и холинергических рецепторов плазматической мембраны кардиомиоцитов. Количество мускариновых рецепторов уменьшается при воздействии на культуру миоцитов Т3 [91], в то время как количество β-адренорецепторов увеличивается [35].
Результатом описанного выше действия Т3 на циркуляторное русло является сниженное диастолическое давление и общее периферическое сопротивление, увеличенная преднагрузка и сниженная постнагрузка на сердце, увеличенный ударный объем, физиологическая гипертрофия миокарда, измененная нервная регуляция сердечной деятельности.
ЗАКЛЮЧЕНИЕ
Была предпринята попытка систематизировать наблюдаемое интегральное действие тиреоидных гормонов на работу и строение сердца с помощью описания их молекулярных белков-мишеней. Рассмотрено влияние гормонов не только на резидентные клетки сердца, но и на кровеносное русло.
Графический результат представлен на рис. 5.

Информированное согласие. Каждый соавтор обзорной статьи представил добровольное письменное информированное согласие, подписанное им после разъяснения ему потенциальных рисков и преимуществ написания данного обзора.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией данной статьи.
Список литературы
Bernal J., Nunez J. Thyroid hormones and brain development // Eur. J. Endocrinol. 1995. V. 133. № 4. P. 390.
Zhang J., Lazar M.A. The Mechanism of Action of Thyroid Hormones // Annu. Rev. Physiol. 2000. V. 62. № 1. P. 439.
Klein I. Thyroid hormone and the cardiovascular system // Am. J. Med. 1990. V. 88. № 6. P. 631.
Cheng S.-Y., Leonard J.L., Davis P.J. Molecular Aspects of Thyroid Hormone Actions // Endocr. Rev. 2010. V. 31. № 2. P. 139.
Brent G.A. Mechanisms of thyroid hormone action // J. Clin. Invest. 2012. V. 122. № 9. P. 3035.
Klein I., Danzi S. Thyroid Disease and the Heart // Curr. Probl. Cardiol. Elsevier. 2016. V. 41. № 2. P. 65.
Porter K.E., Turner N.A. Cardiac fibroblasts: At the heart of myocardial remodeling // Pharmacol. Ther. 2009. V. 123. № 2. P. 255.
Hulsmans M., Clauss S., Xiao L. et al. Macrophages Facilitate Electrical Conduction in the Heart // Cell. 2017. V. 169. № 3. P. 510.
Croteau W., Davey J., Galton V. et al. Cloning of the mammalian type II iodothyronine deiodinase. A selenoprotein differentially expressed and regulated in human and rat brain and other tissues. // J. Clin. Invest. 1996. V. 98. № 2. P. 405.
Schwartz H.L., Lazar M.A., Oppenheimer J.H. Widespread distribution of immunoreactive thyroid hormone β2 receptor (TRβ2) in the nuclei of extrapituitary rat tissues // J. Biol. Chem. 1994. V. 269. № 40. P. 24 777.
d’Amati G. Increased Expression of Thyroid Hormone Receptor Isoforms in End-Stage Human Congestive Heart Failure // J. Clin. Endocrinol. Metab. 2001. V. 86. № 5. P. 2080.
Sakaguchi Y., Cui G., Sen L. Acute effects of thyroid hormone on inward rectifier potassium channel currents in guinea pig ventricular myocytes. // Endocrinology. 1996. V. 137. № 11. P. 4744.
Dillmann W.H. Biochemical basis of thyroid hormone action in the heart // Am. J. Med. 1990. V. 88. № 6. P. 626.
Bergh J., Lin H.-Y., Lansing L. et al. Integrin α V β 3 Contains a Cell Surface Receptor Site for Thyroid Hormone that Is Linked to Activation of Mitogen-Activated Protein Kinase and Induction of Angiogenesis // Endocrinology. 2005. V. 146. № 7. P. 2864.
Sabatino L., Iervasi G., Pingitore A. Thyroid hormone and heart failure: from myocardial protection to systemic regulation // Expert Rev. Cardiovasc. Ther. 2014. V. 12. № 10. P. 1227.
Davis P.J., Leonard J.L., Davis F.B. Mechanisms of nongenomic actions of thyroid hormone // Front. Neuroendocrinol. 2008. V. 29. № 2. P. 211.
Danzi S., Ojamaa K., Klein I. Triiodothyronine-mediated myosin heavy chain gene transcription in the heart // Am. J. Physiol. Circ. Physiol. 2003. V. 284. № 6. P. H2255.
Gustafson T., Bahl J., Markham B. et al. Hormonal regulation of myosin heavy chain and alpha-actin gene expression in cultured fetal rat heart myocytes. // J. Biol. Chem. 1987. V. 262. № 27. P. 13 316.
Watanabe H., Ma M., Washizuka T. et al. Thyroid hormone regulates mRNA expression and currents of ion channels in rat atrium // Biochem. Biophys. Res. Commun. 2003. V. 308. № 3. P. 439.
Rohrer D., Dillmann W.H. Thyroid hormone markedly increases the mRNA coding for sarcoplasmic reticulum Ca2+-ATPase in the rat heart // J. Biol. Chem. 1988. V. 263. № 15. P. 6941.
Ewart H.S., Klip A. Hormonal regulation of the Na(+)-K(+)-ATPase: mechanisms underlying rapid and sustained changes in pump activity // Am. J. Physiol. Physiol. 1995. V. 269. № 2. P. C295.
Carr A.N., Kranias E.G. Thyroid Hormone Regulation of Calcium Cycling Proteins // Thyroid. 2002. V. 12. № 6. P. 453.
Jiang M., Xu A., Tokmakejian S., Narayanan N. Thyroid hormone-induced overexpression of functional ryanodine receptors in the rabbit heart // Am. J. Physiol. Circ. Physiol. 2000. V. 278. № 5. P. H1429.
Biel M. Cardiac HCN Channels Structure, Function, and Modulation // Trends Cardiovasc. Med. 2002. V. 12. № 5. P. 206.
Incerpi S., De Vito P., Luly P. et al. Short-Term Effects of Thyroid Hormones and 3,5-Diiodothyronine on Membrane Transport Systems in Chick Embryo Hepatocytes // Endocrinology. 2002. V. 143. № 5. P. 1660.
Boerth S., Artman M. Thyroid hormone regulates Na+-Ca2+ exchanger expression during postnatal maturation and in adult rabbit ventricular myocardium // Cardiovasc. Res. 1996. V. 31. P. E145.
Kiss E., Jakab G., Kranias E., Edes I. Thyroid hormone-induced alterations in phospholamban protein expression. Regulatory effects on sarcoplasmic reticulum Ca2+ transport and myocardial relaxation. // Circ. Res. 1994. V. 75. № 2. P. 245.
Davis P.J., Davis F.B. Nongenomic Actions of Thyroid Hormone on the Heart // Thyroid. 2002. V. 12. № 6. P. 459.
Zipes D.P., Jaliff J. Cardiac Electrophysiology: From Cell to Bedside. Elsevier, 2014. 1320 p.
Olshausen K., Bischoff S., Kahaly G. et al. Cardiac arrhythmias and heart rate in hyperthyroidism // Am. J. Cardiol. 1989. V. 63. № 13. P. 930.
Davis P.J., Davis F.B. Nongenomic Actions of Thyroid Hormone // Thyroid. 1996. V. 6. № 5. P. 497.
Lakatta E.G., Vinogradova T.M., Maltsev V.A. The Missing Link in the Mystery of Normal Automaticity of Cardiac Pacemaker Cells // Ann. N. Y. Acad. Sci. 2008. V. 1123. № 1. P. 41.
Difrancesco D. The role of the funny current in pacemaker activity // Circ. Res. 2010. V. 106. № 3. P. 434.
Demolombe S., Marionneau C., Le Bouter S. et al. Functional genomics of cardiac ion channel genes // Cardiovasc. Res. 2005. V. 67. № 3. P. 438.
Kahaly G.J., Dillmann W.H. Thyroid Hormone Action in the Heart // Endocr. Rev. 2005. V. 26. № 5. P. 704.
Tardiff J., Hewett T., Factor S. et al. Expression of the β (slow)-isoform of MHC in the adult mouse heart causes dominant-negative functional effects // Am. J. Physiol. Circ. Physiol. 2000. V. 278. № 2. P. H412.
Nerbonne J.M. Molecular basis of functional voltage-gated K+ channel diversity in the mammalian myocardium // J. Physiol. 2000. V. 525. № 2. P. 285.
Grandi E., Sanguinetti M., Bartos D. et al. Potassium channels in the heart: structure, function and regulation // J. Physiol. 2017. V. 595. № 7. P. 2209.
Ma M., Watanabe K., Watanabe H. et al. Different Gene Expression of Potassium Channels by Thyroid Hormone and an Antithyroid Drug Between the Atrium and Ventricle of Rats // Jpn. Heart J. 2003. V. 44. № 1. P. 101.
Nishiyama A. Effects of thyroid status on expression of voltage-gated potassium channels in rat left ventricle // Cardiovasc. Res. 1998. V. 40. № 2. P. 343.
Shimoni Y., Severson D.L. Thyroid status and potassium currents in rat ventricular myocytes // Am. J. Physiol. − Hear. Circ. Physiol. 1995. V. 268. № 2. P. 37.
Purtell K., Roepke T.K., Abbott G.W. Cardiac arrhythmia and thyroid dysfunction: A novel genetic link // Int. J. Biochem. Cell Biol. Elsevier Ltd. 2010. V. 42. № 11. P. 1767.
Roepke T., King E., Reynal-Neyra A. et al. Kcne2 deletion uncovers its crucial role in thyroid hormone biosynthesis // Nat. Med. 2009. V. 15. № 10. P. 1186.
Fröhlich H., Boini K., Seebohm G. et al. Hypothyroidism of gene-targeted mice lacking Kcnq1 // Pflugers Arch. Eur. J. Physiol. 2011. V. 461. № 1. P. 45.
Charpentier F., Merot J., Riochet D. et al. Adult K-CNE1-Knockout mice exhibit a mild cardiac cellular phenotype // Biochem. Biophys. Res. Commun. 1998. V. 251. № 3. P. 806.
Storey N., Gentile S., Ullah H. et al. Rapid signaling at the plasma membrane by a nuclear receptor for thyroid hormone // Proc. Natl. Acad. Sci. 2006. V. 103. № 13. P. 5197.
Hibino H., Inanobe A., Furutani K. et al. Inwardly Rectifying Potassium Channels: Their Structure, Function, and Physiological Roles // Physiol. Rev. 2010. V. 90. № 1. P. 291.
Lozano-Velasco E., Wangensteen R., Quesada A. et al. Hyperthyroidism, but not hypertension, impairs PITX2 expression leading to Wnt-microRNA-ion channel remodeling // PLoS One. 2017. V. 12. № 12. P. e0188473.
Pucci E., Chiovato L., Pinchera A. Thyroid and lipid metabolism // Int. J. Obes. 2000. V. 24. № S2. P. S109.
Kanno S., Saffitz J.E. The role of myocardial gap junctions in electrical conduction and arrhythmogenesis // Cardiovascular Pathology. 2001. V. 10. № 4. P. 169.
Mitašíková M., Lin H., Soukup T. et al. Diabetes and thyroid hormones affect connexin-43 and PKC-ε expression in rat heart atria // Physiol. Res. 2009. V. 58. № 2. P. 211.
Tribulova N., Shneyvays V., Mamedova L. et al. Enhanced connexin-43 and α-sarcomeric actin expression in cultured heart myocytes exposed to triiodo-L-thyronine // J. Mol. Histol. 2004. V. 35. № 5. P. 463.
Harper M.E., Seifert E.L. Thyroid hormone effects on mitochondrial energetics // Thyroid. 2008. V. 18. № 2. P. 145.
Goglia F., Moreno M., Lanni A. Action of thyroid hormones at the cellular level: The mitochondrial target // FEBS Lett. 1999. V. 452. № 3. P. 115.
Sinha R.A., Singh B.K., Yen P.M. Direct effects of thyroid hormones on hepatic lipid metabolism // Nat. Rev. Endocrinol. 2018. V. 14. № 5. P. 259.
Goglia F., Silvestri E., Lanni A. Thyroid hormones and mitochondria // Biosci. Rep. 2002. V. 22. № 1. P. 17.
Birben E., Sahiner U., Sackesen C. et al. Oxidative stress and antioxidant defense // World Allergy Organ. J. 2012. V. 5. № 1. P. 9.
Cesarone M., Belcaro G., Carratelli M. et al. A simple test to monitor oxidative stress // Int. Angiol. 1999. V. 18. № 2. P. 127.
Das K., Chainy G.B. Modulation of rat liver mitochondrial antioxidant defence system by thyroid hormone // Biochim. Biophys. Acta. 2001. V. 1537. № 1. P. 1.
Eghbali M. Localization of types I, III and IV collagen mRNAs in rat heart cells by in situ hybridization // J. Mol. Cell. Cardiol. 1989. V. 21. № 1. P. 103.
Sun Y. Angiotensin Converting Enzyme and Myofibroblasts during Tissue Repair in the Rat Heart // J. Mol. Cell. Cardiol. 1996. V. 28. № 5. P. 851.
Baudino T., Carver W., Giles W., Borg T. Cardiac fibroblasts: friend or foe? // Am. J. Physiol. Circ. Physiol. 2006. V. 291. № 3. P. H1015.
Camelliti P., Borg T.K., Kohl P. Structural and functional characterisation of cardiac fibroblasts // Cardiovasc. Res. 2005. V. 65. № 1. P. 40.
Feld Y., Melamed-Frank M., Kehat I. et al. Electrophysiological Modulation of Cardiomyocytic Tissue by Transfected Fibroblasts Expressing Potassium Channels // Circulation. 2002. V. 105. № 4. P. 522.
Abramochkin D.V., Lozinsky I.T., Kamkin A. Influence of mechanical stress on fibroblast–myocyte interactions in mammalian heart // J. Mol. Cell. Cardiol. Elsevier Ltd. 2014. V. 70. P. 27.
Stock A., Sies H., Stahl W. Enhancement of gap junctional communication and connexin43 expression by thyroid hormones // Biochem. Pharmacol. 1998. V. 55. № 4. P. 475.
Ojamaa K. Signaling mechanisms in thyroid hormone-induced cardiac hypertrophy Thyroid Hormone & Cardiovascular Thyroid Hormone & Cardiovascular // Vascul. Pharmacol. Elsevier Inc. 2010. V. 52. № 3–4. P. 113.
Ciulla M., Paliotti R., Cortelazzi D. et al. Effects of Thyroid Hormones on Cardiac Structure: A Tissue Characterization Study in Patients with Thyroid Disorders Before and After Treatment // Thyroid. 2001. V. 11. № 7. P. 613.
Mikkonen L., Lampiaho K., Kulonen E. Effect of thyroid hormones, somatotrophin, insulin and corticosteroids on synthesis of collagen in granulation tissue both in vivo and in vitro // Acta Endocrinol. (Copenh). 1966. V. 51. № 1. P. 23.
Oviedo-Orta E., Evans W.H. Gap junctions and connexin-mediated communication in the immune system // Biochim. Biophys. Acta − Biomembr. 2004. V. 1662. № 1–2. P. 102.
Frantz S., Nahrendorf M. Cardiac macrophages and their role in ischaemic heart disease // Cardiovasc. Res. 2014. V. 102. № 2. P. 240.
Valledor A.F., Ricote M. Nuclear receptor signaling in macrophages // Biochem. Pharmacol. 2004. V. 67. № 2. P. 201.
Costa Rosa L.F., Cury Y., Curi R. Hormonal control of macrophage function and glutamine metabolism // Biochem. Cell Biol. 1991. V. 69. № 4. P. 309.
Perrotta C., Buldorini M., Assi E. et al. The thyroid hormone triiodothyronine controls macrophage maturation and functions: Protective role during inflammation // Am. J. Pathol. American Society for Investigative Pathology. 2014. V. 184. № 1. P. 230.
Jara E., Munos-Durango N., Llanos C. et al. Modulating the function of the immune system by thyroid hormones and thyrotropin // Immunol. Lett. 2017. V. 184. P. 76.
Abohashem-Aly A., Meng X., Li J. et al. DITPA, a thyroid hormone analog, reduces infarct size and attenuates the inflammatory response following myocardial ischemia // J. Surg. Res. Elsevier Inc. 2011. V. 171. № 2. P. 379.
Baum M., Dwarakanath V., Alpern R., Moe O. Effects of thyroid hormone on the neonatal renal cortical Na+/H+ antiporter // Kidney Int. 1998. V. 53. № 5. P. 1254.
Alcalde A., Sarasa M., Raldula D. et al. Role of Thyroid Hormone in Regulation of Renal Phosphate Transport in Young and Aged Rats // Endocrinology. 1999. V. 140. № 4. P. 1544.
Ibrahim M.M. RAS inhibition in hypertension // J. Hum. Hypertens. 2006. V. 20. № 2. P. 101.
Golde D., Bersch N., Chopra I., Cline M. Thyroid Hormones Stimulate Erythropoiesis in Vitro // Br. J. Haematol. 1977. V. 37. № 2. P. 173.
Syme H.M. Cardiovascular and Renal Manifestations of Hyperthyroidism // Vet. Clin. North Am. – Small Anim. Pract. 2007. V. 37. № 4. P. 723.
Grais I.M., Sowers J.R. Thyroid and the heart // Am. J. Med. Elsevier Ltd. 2014. V. 127. № 8. P. 691.
Clifford P.S. Local control of blood flow // Adv. Physiol. Educ. 2011. V. 35. № 1. P. 5.
John Solaro R. Mechanisms of the Frank-Starling Law of the Heart: The Beat Goes On // Biophys. J. 2007. V. 93. № 12. P. 4095.
Shimizu I., Minamino T. Physiological and pathological cardiac hypertrophy // J. Mol. Cell. Cardiol. Elsevier Ltd. 2016. V. 97. P. 245.
Баевский Р., Иванов Г., Чирейкин Л. и др. Анализ вариабельности сердечного ритма при использовании различных электрокардиографических систем // Вестник аритмологии. 2001. №. 24. С. 65.
Chen J.-L., Chiu H., Tseng Y., Chu W.-C. Hyperthyroidism is characterized by both increased sympathetic and decreased vagal modulation of heart rate: evidence from spectral analysis of heart rate variability // Clin. Endocrinol. (Oxf). 2006. V. 64. № 6. P. 611.
Cairoli V.J., Crout J.R. Role of the autonomic nervous system in the resting tachycardia of experimental hyperthyroidism. // J. Pharmacol. Exp. Ther. 1967. V. 158. № 1. P. 55.
Cacciatori V., Bellavere F., Pezzarossa A. et al. Power spectral analysis of heart rate in hyperthyroidism // J. Clin. Endocrinol. Metab. 1996. V. 81. № 8. P. 2828.
Cacciatori V., Gemma M., Bellavere F. et al. Power spectral analysis of heart rate in hypothyroidism // Eur. J. Endocrinol. 2000. V. 143. № 3. P. 327.
Waisberg M., Shainberg A. Characterization of muscarinic cholinergic receptors in intact myocardial cells in vitro // Biochem. Pharmacol. 1992. V. 43. № 11. P. 2327.
Yue L., Xie J., Nattel S. Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation // Cardiovasc. Res. 2011. V. 89. № 4. P. 744.
Дополнительные материалы отсутствуют.
Инструменты
Физиология человека