

Физиология человека, 2021, T. 47, № 6, стр. 115-124
Молекулярно-клеточные изменения в патогенезе эндометриоза
К. А. Тониян 1, 2, *, О. И. Орлов 1, В. В. Бояринцев 3, И. В. Огнева 1, 4, **
1 ФГБУН ГНЦ РФ – Институт медико-биологических проблем РАН
Москва, Россия
2 ФГБУ Клиническая больница № 1 (Волынская) УДП РФ
Москва, Россия
3 ФГБУ ДПО Центральная государственная медицинская академия УДП РФ
Москва, Россия
4 ФГАОУ ВО Первый Московский государственный медицинский университет
имени И.М. Сеченова (Сеченовский университет)
Москва, Россия
* E-mail: ktoniyan@mail.ru
** E-mail: iogneva@yandex.ru
Поступила в редакцию 22.12.2020
После доработки 26.05.2021
Принята к публикации 01.06.2021
Аннотация
В данном обзоре рассматриваются молекулярно-клеточные изменения в патогенезе эндометриоза. Приведены данные об изменениях врожденного и адаптивного иммунитета, рассмотрена роль нарушений гормонального фона и ядерных рецепторов к стероидным гормонам, обсужден возможный эпителиально-мезенхимальный переход как в клетках самого эндометрия, так и в месте их прикрепления при формировании эктопического очага эндометрия. Обсуждается роль сочетанного действия различных факторов в патогенезе эндометриоза.
В последние годы все большую распространенность среди гинекологических патолгий приобретает эндометриоз, что не позволяет исключить влияния различных экологических факторов на дебют и развитие данного заболевания.
По определению, эндометриоз представляет собой формирование очагов из ткани, подобной эпителию эндометрия, вне полости матки. Такой патогенез противоречит одной из ключевых парадигм эмбрионального развития, где каждый тип клеток после прохождения всех этапов дифференцировки и морфогенеза находится на своем месте в своем микроокружении, взаимодействуя с соседними клетками и типами клеток. Соответственно, формирование очагов ткани в нехарактерной для нее локализации требует с одной стороны, возможности для нее туда попасть, а с другой стороны – изменений в той ткани, куда она встраивается: должна быть возможность для адгезии клеток и иммунологическая толерантность на формирующийся “аутотрансплантат”. Хотя эндометриоз является доброкачественным пролиферативным заболеванием, он имеет общие характеристики с опухолевыми процессами, в частности, воспалительное состояние, инвазия соседних тканей, индукция ангиогенеза и устойчивость к апоптозу [1].
Локализация очагов эндометриоза
Аденомиоз характеризуется наличием в миометрии доброкачественного разрастания эндометриальных желез и стромы эндометрия. Ретроспективные исследования показывают, что дебют заболевания обычно приходится на 30–40 лет, часто сосуществует с лейомиомой, и, хотя, при этом меноррагия является наиболее частым признаком, примерно треть случаев протекает бессимптомно [2].
Локализация эндометриодиного очага в яичнике является самой распространенной среди эктопических очагов. Впервые довольно хорошо описана еще в 1925 г. основоположником теории ретроградной менструации, как причины эндометриоза, J.A. Sampson [3]. Следует отметить, что широкоприменяемый маркер злокачественной опухоли яичников CA-125 может оставаться в пределах нормы, что, вероятно, зависит от степени дифференцировки опухоли [4, 5].
Одной из наиболее подробно описанных локализаций является формирование эндометриодного очага в брюшной стенке. Чаще всего, такую локализацию связывают с предшествующими гинекологическими вмешательствами. Так, считают, что эндометриоз брюшной стенки является редким заболеванием, которое возникает после кесарева сечения или тазовой хирургии и имеет частоту 0.03–1.5% у женщин с подобным анамнезом [6].
Диагностика эндометриоза различных отделов желудочно-кишечного тракта также имеет целый ряд диагностических проблем в связи с его неспецифическими симптомами. В то же время, своевременная и адекватная постановка диагноза позволяет предлагать медикаментозное лечение в качестве терапии первой линии при эндометриозе кишечника, хотя и не во всех случаях [7]. Более того, зафиксированы редкие случаи субмукозной и субэпителиальной опухоли желудка, которые впоследствии были определены как эндометриоз [8, 9].
После брюшины и желудочно-кишечного тракта на втором месте по распространенности очагов локализации эндометриоза вне репродуктивной системы находится мочевыделительная система, в первую очередь, мочевой пузырь и мочеточники, где очаги эндометриоза удаляют хирургически [10].
Наиболее редкими случаями формирования очагов эндометриоза вне органов малого таза являются случаи торакального эндометриоза. Для него характерны катамениальная плевральная боль, одышка, кровохарканье. Так, D.B. Flieder et al. [11] описывают клинико-патологические особенности девяти случаев плевро-легочного эндометриоза и первого случая легочного эктопического децидуоза. На очагах железистого эпителия были выявлены панцитокератин, рецепторы к эстрогену и прогестерону. При этом клетки стромы окрашивались на виментин, актин, десмин, и также, рецепторы к эстрогену и прогестерону [11].
Очень редкими представляются случаи локализации эндометриодиных очагов в носослезном (назолакримальном) канале [12], описанные даже для раннего подросткового возраста, когда у 13-летней девочки наблюдалось катамениальное кровотечение в слезном мениске левого глаза [13].
Таким образом, при эндометриозе манифестное формирование очага эндометрия, представляющего собой эпителиальную ткань (однослойный призматический эпителий), происходит также при его прикреплении к эпителиальной ткани, преимущественно к мезотелию (однослойный плоский эпителий, развивается из мезодермы и способен к регенерации за счет митотических делений), образующему серозные оболочки матки, яичника, брюшной стенки, желудочно-кишечного тракта, мочеточника и мочевого пузыря. Для формирования эндометриоидного очага должно быть сочетанное действие нескольких факторов. С одной стороны, это изменение структуры клеточного слоя в том месте, где возникает очаг, чтобы эндометрийподобная клетка могла прикрепиться и начать пролиферировать, на фоне изменения иммунного статуса. С другой стороны, клетки эндометрия должны претерпевать некоторые изменения, в частности, в способности к миграции, адгезии и последующей пролиферации в нехарактерном окружении. В данном обзоре авторы рассматривают каждый из этих факторов.
Изменения иммунного статуса
Формирование эктопического очага эндометриоидной ткани неизбежно должно сопровождаться изменением иммунологического статуса. Избегание иммунологического надзора и формирование иммунологической толерантности к клеткам эндометрия в месте их нехарактерной локализации могут быть аналогичны таковым при развитии опухоли. Однако, применительно к эндометриозу, эти процессы изучены недостаточно.
Адгезия эндометрийподобных клеток в нехарактерном месте приводит, в первую очередь, к активации резидентых антиген-представляющих клеток в данной ткани и последующему развитию воспаления с вовлечением всех компонентов иммунной системы с регуляцией, в том числе и по цепи обратной связи.
Резидентные антиген-представляющие клетки. Дендритные клетки – это антигенпрезентирующие клетки, происходящие из костного мозга, которые действуют как связующее звено между врожденным и адаптивным иммунитетом и участвуют в формировании иммунной толерантности при воспалительных и опухолевых заболеваниях [14]. В эктопических эндометриодиных очагах, локализованных на брюшине и в окружающей ткани мезотелия, наблюдается значительное увеличение незрелых дендритных клеток по сравнению с эндометрием, локализованным в матке, и с тканью мезотелия, которая находится значительно дальше от места поражения [15]. В связи с этим, авторы исследования полагают, что сохранение эктопического очага связано как раз с тем, что незрелые дендритные клетки не способны адекватно представлять эктопический антиген и это не приводит к его элиминации [15].
Не только дендритные клетки, но и макрофаги презентируют антигены Т-клеткам, помимо целого ряда других, выполняемых ими функций. У женщин с диагностированным эндометриозом были обнаружены значительно более высокие концентрации макрофагов, даже по сравнению со случаями абдоминальных инфекций, однако механизм, определяющий такое распределение клеток, до сих пор неизвестен [14]. Однако известно, что у женщин с эндометриозом перитонеальные макрофаги обладают повышенной способностью секретировать моноцитарный хемотаксический белок-1 моноцитов – МСР-1 (CCL2 – лиганд для хемокиновых рецепторов CC-семейства) [16], играющий роль в рекрутировании моноцитов в места повреждения и воспаления. Кроме того, показано, что вновь рекрутированные макрофаги, экспрессирующие Tie-2 (рецептор ангиопоэтина с собственной тирозинкиназной активностью), проникают в области, окружающие новообразованные эндометриотические кровеносные сосуды [17], что делает эту подгруппу макрофагов возможной мишенью для потенциального лечения эндометриоза.
Изменение уровня простагландина E2 [18] и/или увеличение продукции TNFα [19], отмечаемые при эндометриозе, могут приводить к снижению экспрессии матричных металлопротеиназ и рецепторов CD36 [18], активация которых, необходима для фагоцитоза макрофагами. Уменьшение эффективности фагоцитоза не позволяет очистить клеточный дебрис и элиминировать эктопический очаг эндометриоза.
Однако остается не вполне ясным – эти изменения предшествовали развитию эндометриоза, или развитие эндометриоза привело к изменению активности макрофагов, реркрутизации незрелых дендритных клеток и, как следствие, развитию хронического воспаления и/или иммунологической толерантности.
Воспаление и цитокины. В начале острого воспаления, инициируемого резидентными макрофагами, секретируются медиаторы воспаления – цитокины. Большое количество исследований, свидетельствует об изменении уровней и/или соотношения цитокинов при развитии эндометриоза [20].
Можно предположить, при формировании эктопического очага ткани эндометрия, в первую очередь, синтезируется TNFα и, возможно, IL-1 и IL-6, которые приводят к изменениям в кровотоке и переносу клеток из кровеносных сосудов в ткань. В брюшной полости, у пациенток с эндометриозом были обнаружены повышенные уровни TNFα, IL-1β, IL-6 и IL-8 [21]. В свою очередь, IL-1β стимулирует циклооксигеназную систему 2, приводя к росту эктопического эндометрия путем индукции пролиферации и ангиогенеза в результате стимуляции продукции фактора роста VEGF [22], причем последнее может быть опосредовано и IL-8 [23]. Стромальные клетки растущего эндометрия могут вырабатывать эотаксин, являющийся сильным хемоаттрактантом Т-хелперов Th2 [24], выделяющих IL-4 и, в том числе, таким образом, стимулирующих размножение и созревание В-клеток. Однако в эктопическом эндометрии и у этих же женщин в эутопическом эндометрии была обнаружена повышенная экспрессия IL-22 [25], продуцируемого активированными Т-клетками, основной мишенью действия которого является подавление выработки IL-4 Th2-клетками. При этому у женщин с эндометирозом в сыворотке крови наблюдалось повышение IFNγ, свидетельствующее об активации Th1 клеток, а также MCP-1 и IL-8 (стимулирующего накопление гранулоцитов) [26]. Помимо вышеупомоянутых цитокинов роль в избегании иммунологического надзора при формировании эндометриоидных повреждений может играть IL-15, продуцируемый моноцитами, поскольку он непосредственно стимулировал рост и инвазию стромальных клеток, подавляя при этом активность NK-клеток [27]. Несмотря на все вышесказанное, сравнительное исследование пациентов с ранней и поздней стадиями эндометриоза не обнаружило различий в уровнях IL-2, IL-4, IL-10 и IFN-γ в крови и брюшной жидкости [28].
Однако практически все описанные выше цитокины или продуцируются, или рекрутируют/подавляют различные типы лимфоцитов, таких как NK-клетки и различные виды T-клеток. Тем не менее, данные об участии тех или иных видов лимфоцитов, их содержании и активности при эндометриозе остаются противоречивыми.
Клеточный иммунитет и аутофагия. Активированные NK-клетки способны мигрировать и инфильтрировать эндометриоидные очаги, что может быть использовано даже в терапии [29]. Однако их активация и цитотоксические эффекты снижались наряду с функцией Т-клеток на фоне активации макрофагов при формировании эндометриодиных очагов [30]. Возможно, снижение активации обусловлено увеличением на поверхности NK-клеток рецепторов, участвующих в ингибировании активности (KIR – киллер-ингибирующие рецепторы, такие как KIR2DL1), что было показано у пациентов с развитым эндометриозом [31]. Другая возможность для снижения активации NK-клеток может быть связана с аберрантной продукцией цитокинов другими клетками (например, вышеописанных IL-15 или IL-6 за счет модуляции экспрессии протеин-фосфатазы-2 (SHP-2), содержащей область гомологии Src-2 [32]). Более того, некоторые авторы предполагают, что именно функционирование NK-клеток является ключевым в патогенезе эндометриоза [33].
Существенный вклад в патогенез эндометриоидного поражения вносят Т-лимфоциты, причем показано, что при минимальном или легком эндометриотическом поражении в перитонеальной жидкости преобладают цитокины, производимые Th1-клетками (взаимодействующими с мононуклеарными фагоцитами), а на более поздних и тяжелых стадиях эндометриоза – цитокины, производимые Th2-клетками (взаимодействующими с В-клетками) [34], иными словами меняется соотношение Th1/Th2 [35, 36]. Кроме соотношения между этими двумя классическими подтипами Т-лимфоцитов, по-видимому, в прогрессировании эндометриоза может играть роль соотношение Th17/Treg [37]. Содержание Treg в перитонеальной жидкости, которые могут быть активированы высоким уровнем эстрадиола, что характерно для эндометриоза [38], увеличивается [39] и может приводить к локальному снижению иммунологического надзора [40].
Практически ничего не известно о роли В-клеток в патогенезе эндометриоза, хотя, некоторое количество данных, которые суммировали в своем обзоре L.G.C. Riccio et al. [41], свидетельствует о повышении их количества [42]. Последнее может иметь ключевое значение при обсуждении аутоиммунной природы эндометриоза. Кроме того, накопление Treg в эндометриоидных очагах может привести к истощению пула этих клеток и, как следствие, к мультисистемному аутоиммунному ответу у мышей и людей [43]. Действительно, у эндометриоза достаточно много общего с аутоиммунными заболеваниями, в частности, аберрантный иммунологический надзор с аномальной выработкой цитокинов и соотношением клеток иммунной системы, повышенный гуморальный ответ и воспалительное повреждение [44]. Действительно, показано, что в сыворотке, цервикальном и вагинальном секрете у женщин с эндометриозом возрастает концентрация аутоантител [44, 45].
Однако дисбаланс в производстве цитокинов может играть также важную роль в регуляции аутофагии. Вероятно, эстроген, обычно повышенный при эндометриозе опосредует активацию CXCL12/CXCR4-опосредованного пути ингибирования аутофагии, снижая ее уровень в эктопическом эндометрии [46, 47]. Однако, в некоторых случаях, а именно при эндометриоидном поражении яичников показано, что снижение содержания p53 – основного индуктора апоптоза, может стимулировать аутофагию [48]. Кроме того, в этом случае клетки эндометрия подвергаются окислительному стрессу на фоне повышения ионов железа, что может действовать как активатор аутофагии [49]. Причиной возникновения вышеупомянутого окислительного стресса может быть гипоксия. Матка является хорошо васкуляризированным органом, что обеспечивает высокую оксигенацию эутопического эндометрия, попадание которого в эктопические места (яичники, брюшину и т.п.) неизбежно приводит к снижению получения кислорода и тканевой гипоксии [50]. В эндометриотических поражениях яичников увеличивается экспрессия индуцируемого гипоксией фактора-1α HIF-1α – транскрипционного фактора, опосредующего клеточный ответ на гипоксию, что приводит к активации аутофагии и ускорению миграции и инвазии клеток эндометрия [47].
Изменения структуры клеточного слоя в месте прикрепления и ангиогенез
Возникновение очагов эктопического эндометрия невозможно без адгезии эндометриальных клеток на поверхности, чаще всего сформированной мезотелием. В последнее время появляется все больше доказательств того, что ведущую роль в адгезии эндометриальных клеток на мезотелии брюшины играет эпителиально-мезенхимальный переход, которому подверглись клетки мезотелия [51]. В этом случае, мезотелиальный барьер более не обеспечивает защиту базального слоя, и клетки эндометрия могут легко прикрепляться к строме, формируя далее эктопические очаги. Свидетельством такого перехода является снижение эпителиальных (E-кадгерин, десмоплакин, муцин-1, окклюдин и клаудин) и повышение мезенхимальных (N-кадгерин, гладкомышечный актин, виментин, фибронектин и др.) маркеров, что отмечалось при поражении брюшины и яичников [52].
Разнонаправленные изменения активности матриксных металлопротеиназ (внеклеточные цинк-зависимые эндопептидазы), которые наблюдаются при эндометриозе [18, 53], могут приводить к изменениям внеклеточного матрикса, что способствует прикреплению клеток эндометрия, обладающих повышенной адгезионной способностью к различным компонентам внеклеточного матрикса, включая коллаген типа IV, ламинин, витронектин и фибронектин [54].
Полученные на культуре клеток данные свидетельствуют о том, что адгезия стромальных и эпителиальных клеток эндометрия к мезотелию брюшины приводит к изменениям экспрессии различных тирозин-киназ, а также колониестимулирующего фактора и его рецептора, причем как в мезотелиальных клетках, так и в эндометриальных, что может усиливать инвазию [55] и индуцировать дальнейшие изменения.
Далее, после адгезии и инвазии, пролиферация клеток эндометрия и рост эктопического очага будет в значительной степени зависеть от ангиогенеза, который опосредуется цитокинами, приводящими, в первую очередь, к увеличению содержания фактора роста эндотелия сосудов VEGF [23, 56], причем его уровни коррелируют с тяжестью заболевания [57]. По-видимому, экспрессия VEGF контролируется трансформирующим фактором роста-бета 1 (TGF-β1), экспрессирующимся в эндометрии, скорее всего, под гормональным контролем, причем в эндометриотических поражениях его экспрессия увеличивается [58]. TGF-β1 увеличивает концентрацию мРНК ID1 (белок-ингибитор связывания с ДНК транскрипционных факторов), что, в свою очередь, приводит к увеличению экспрессии VEGF, как было показано в экспериментах с использованием siРНК [59], и делает ингибиторы ID1 хорошими кандидатами для терапии эндометриоза [60].
Предполагаемое сходство эндометриоза и аутоиммунных заболеваний, а также необходимость васкуляризации для роста эктопических очагов эндометрия позволяет предположить участие молекул клеточной адгезии эндотелия сосудов в патогенезе эндометриоза. Проведенное A.K. Schutt et al. [61] исследование показало, что экспрессия молекул клеточной адгезии сосудов VCAM-1 на мезотелии брюшины больных эндометриозом увеличивается по сравнению с контрольной группой, что может быть ответом на увеличение содержания TNFα, IL-1, IL-4.
Изменение клеток эндометрия
Ядерные рецепторы к стероидным гормонам. Большинство клинических фактов и многочисленные клеточно-молекулярные исследования позволяют считать эндометриоз гормон-зависимым воспалительным заболеванием [62]. Эстроген способствует выживаемости эндометриальных клеток в эктопических очагах, воспалительному ответу и прогрессированию заболевания [63].
Каноническим путем действия стероидных гормонов является их связывание с ядерными рецепторами. Рецепторы эстрогена α и β (ESR1 и ESR2) и рецептор прогестерона (PGR) являются ключевыми стероидными рецепторами, участвующими в патогенезе эндометриоза [62, 64]. Кроме того, поскольку ядерные рецепторы NR5A1 и NR2F2 играют роль в регуляции локальных концентраций эстрогена (например, при превращении холестерина в эстрадиол), то они также могут быть задействованы в развитии эндометриоза [65].
Показано, что в эутопическом эндометрии женщин с подтвержденным диагнозом эндометриоза содержание мРНК гена, кодирующего рецептор NR5A1 примерно в 12 000 раз выше, нежели в контрольной группе [66]. Такое исключительное повышение должно поддерживаться несколькими механизмами, в частности изменением метилирования ДНК, модификацией гистонов и посттранскрипционной регуляцией [63] (рис. 1). Во-первых, наблюдается гипометилирование CpG-островка в промоторной области гена NR5A1 в эндометриальных сторомальных клетках при эндометриозе яичников [66, 67]. Во-вторых, гистоны H2 и H3 в промоторной области гена NR5A1 обогащены ацетильными группами [68]. Хотя в целом, активность деацетилаз HDAC1 и HDAC2 в стромальных клетках эндометрия в эктопических очагах повышается [69], что позволяет предлагать использование их ингибиторов в качестве таргетной терапии, как было показано в экспериментах на мышах [70]. Однако почему гистоны промоторной области гена NR5A1 избегают глобального деацетилирования пока неясно. В-третьих, можно предположить и роль посттранскрипционных модификаций в поддержании такого уровня мРНК, в первую очередь, микроРНК [71].
Рис. 1.
Схематичное представление роли ядерных рецепторов к стероидным гормонам в патогенезе эндометриоза. meCpG – метилирование CpG-островков в промоторных областях соответствующих генов; acH2, acH3 – ацетилирование коровых гистонов H2 и H3; miРНК – микроРНК; NR5A1 – ядерный рецептор к стероидным гормонам; ESR – рецептор к эстрогену; PGR – рецептор к прогестерону; HSD17B2 – 17β-гидроксистероиддегидрогеназа типа 2. Стрелки вверх/вниз показывают увеличение/уменьшение содержания.
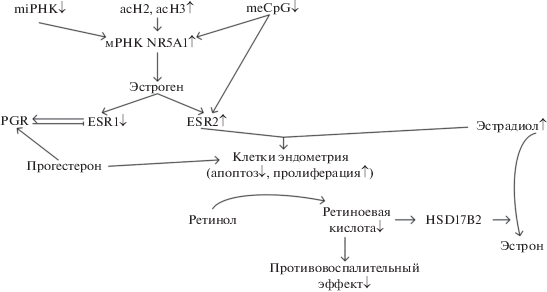
Эстроген регулирует пролиферацию ткани эндометрия путем связывания с ядерными рецепторами ESR1 и ESR2, однако в норме доминирующий вклад в этот процесс вносит ESR1 [72], который после активации индуцирует экспрессию гена, кодирующего рецептор прогестерона PGR [73]. Однако, при эндометриозе в клетках, полученных in vivo, а также в экспериментах in vitro – в культивируемых стромальных клетках эндометрия, содержание мРНК ESR1 примерно в 7 раз ниже [74, 75] (рис. 1). При этом содержание мРНК ESR2 в 40–140 раз выше [74–76]. В связи с этим, предполагают, что такой паттерн экспрессии у здоровых женщин может быть существенным фактором риска развития эндометриоза [77]. Увеличение содержания ESR2 на фоне повышения эстрадиола может приводить к пролиферации первичных эндометриотических клеток и способствовать их выживанию путем ингибирования апоптоза [77]. Причиной увеличения экспрессии ESR2, вероятно, является снижение уровня метилирования CpG-островка в промоторной области гена, обнаруженное в эндометриотических стромальных клетках по сравнению с эутопическим эндометрием [75].
Тем не менее, несмотря на убедительные доказательства роли изменения уровня метилирования в регуляции экспрессии NR5A, и рецепторов эстрогена, остается пока не вполне ясным причина этого изменения in vivo. ДНК-метилтрансфераза 1 (DNMT1) и DNMT3B по-разному экспрессируются в эктопическом эндометрии и в норме [78]. Учитывая, что DNMT3B – метилаза de novo, то требуется ее направленная рекрутизация к промоторной области определенного гена, например, ESR1. Однако, что может служить таким направляющим фактором – пока неясно.
Мишенями действия прогестерона в клетке являются две гомологичные изоформы его рецептора, которые, в свою очередь, проявляют разные уровни активности в разных тканях [79, 80]. В эндометрии только PGR-A оказывает репрессорное действие на ESR1 [81], ингибируя эстроген-индуцированный рост и пролиферацию [82]. Помимо действия на рецептор эстрогена, в ответ на прогестерон стромальные клетки эндометрия поглощают ретинол и продуцируют ретиноевую кислоту, которая индуцирует фермент 17β-гидроксистероиддегидрогеназу типа 2 (HSD17B2) в эпителиальных клетках эндометрия [83], превращающую эстрадиол в менее мощный эстрон [84] (рис. 1). Однако при эндометриозе развивается резистентность к прогестерону [85], что приводит к снижению продукции ретиноевой кислоты, обладающей противовоспалительным действием [86] и, соответственно, к развитию воспаления.
Кроме того, изменения содержания гормонов и активности их рецепторов, наряду с гипоксическим сигналом, могут приводить к индукции эпителиально-мезенхимального перехода не только в мезотелии, но и в самом эндометрии. Отрыв клеток эндометрия от внеклеточного матрикса должен был бы приводить к аноикису, устойчивость к которому наблюдается при эндометриотических поражениях [87]. Однако, имея исходно мезенхимальное происхождение, клетки эндометрия могут иметь тенденцию к такой своеобразной “обратной дифференцировке” [88]. Показано, что рецептор к эстрогену может напрямую связываться с промотором фактора роста гепатоцитов (HGF), что далее приводит к индукции эпителиально-мезенхимального перехода в эпителиальных клетках эндометрия человека [89]. Кроме того, к такому же эффекту приводит и оверэкспрессия индуцируемого гипоксией фактора HIF-1α [90].
Цитоскелет. Ведущую роль в миграции, адгезии, инвазии и пролиферации клеток играет цитоскелет, в том числе и в эпителиальных и стромальных клетках эндометрия, однако экспериментальные данные о его участии в патогенезе эндометриоза очень немногочисленны. Однако, даже скрининговое исследование, целью которого было выявление дифференциально экспрессируемых генов у пациентов с эндометриозом, показывает, что большинство из них кодируют белки, участвующие в формировании фокально-адгезивных комплексов, актинового цитоскелета и в сигнальном пути митоген-активируемых фосфокиназ [91].
Показано, что эстрогены и селективные модуляторы его рецепторов через внеядерные сигнальные каскады (G-белки и к Rho-ассоциированную киназу) регуляруют ремоделирование актинового цитоскелета, меняя адгезионную и инвазивную способность клеток эндометрия, причем особым эффектом обладает 17β-эстрадиол [92]. Миграция эндометриальных стромальных клеток в ответ на 17β-эстрадиол и прогестерон также менялась у пациентов с эндометриозом по сравнению с контрольной группой: 17β-эстрадиол стимулировал миграцию эндометриальных стромальных клеток и в контроле, и, с более сильным эффектом, в группе с эндометриозом, а прогестерон – только в группе с эндометриозом [93].
Альтернативную гипотезу предлагают H.M. Albertsen и K. Ward [94], которые, проведя также скрининговое исследование, уделяют внимание 4 генам (WNT4, CDC42, ID4, VEZT), которые контролируют актиновый цитоскелет, однако делают предположение, что нарушение целостности мезотелиального барьера в результате аберрантной экспрессии этих генов может быть причиной развития эндометриоза. Однако данные гены широко экспрессируются в различных клетках, и наравне с клетками мезотелия, могут быть дисрегулированы и в эндометриальных клетках.
ЗАКЛЮЧЕНИЕ
Наибольшее количество представленных в литературе данных посвящено роли нарушений гормонального фона и иммунологического статуса, однако о влиянии различных факторов на изменение самих клеток эндометрия, например, на цитоскелет, являющийся ключевым фактором миграции, адгезии, инвазии и пролиферации, на сегодняшний день известно очень мало. Тем не менее, можно полагать, что исследование изменений организации цитоскелета, регуляции экспрессии генов, в том числе и эпигенетической, кодирующих основные белки цитоскелета, может быть полезным при разработке новых терапевтических подходов к лечению эндометриоза.
На основании вышесказанного, можно полагать, что для формирования эндометриоза необходимо сочетанное действие нескольких факторов:
1) клетка эндометрия должна избежать аноикиса, отрываясь от внеклеточного матрикса в матке. Если это происходит случайным образом, то таких клеток будет очень незначительное количество, но и его может оказаться достаточно для формирования эктопического очага, в случае попадания в “благоприятные условия”. Если есть предпосылки для направленного избегания апоптоза (например, индуцирован эпителиально-мезенхимальный переход), то таких клеток будет гораздо больше и, следовательно, при прочих равных условиях, существенно выше вероятность образования эндометриоидного очага;
2) клетка эндометрия должна прикрепиться в нехарактерном окружении, что, в норме, должно приводить к активации резидентных клеток иммунной системы, активации продукции цитокинов, рекрутизации различных механизмов адаптивной иммунной системы. Однако, в случае нарушения целостности клеточного слоя в месте прикрепления, изменения иммунологического статуса, как локального, так и системного, клетки эндометрия могут избегать иммунологического надзора и/или может формироваться иммунологическая толерантность;
3) после адгезии и инвазии, клетка эндометрия должна начать пролиферировать для формирования популяции эндометрийподобных клеток и, как следствие, очага эктопического эндометрия. Для этого требуется активация ангиогенеза и все более устойчивое подавление иммунного ответа;
4) вклад любого из этих процессов в каждом конкретном случае может быть различным, но суммарный эффект должен достигать, вероятно, некоторого порогового значения, превышение которого приводит к развитию заболевания.
Финансирование работы. Работа поддержана программой фундаментальных исследований ГНЦ РФ – ИМБП РАН (Москва) 65.4, Программой стратегического академического лидерства (Сеченовский универститет, Москва).
Благодарности. Авторы выражают благодарность Марии Александровне Усик за неоценимую помощь при подготовке обзора.
Список литературы
Guo S.W. Endometriosis and ovarian cancer: potential benefits and harms of screening and risk-reducing surgery // Fertil. Steril. 2015. V. 104. № 4. P. 813.
Emmanuel I., Ochigbo A., Philip A., Nyam E.Y. Adenomyosis: A Clinico-pathological Study // West Afr. J. Med. 2019. V. 36. № 1. P. 88.
Sampson J.A. Peritoneal endometriosis due to the menstrual dissemination of endometrial tissue into the peritoneal cavity // Am. J. Obstet. Gynecol. 1927. V. 14. P. 422.
Nahar K., Ferdous B., Akhter N. et al. Ovarian Endometrioid Adenocarcinoma Arising in Endometriosis: A Case Report // Mymensingh Med. J. 2018. V. 27. № 2. P. 420.
Tsai C., Huang S.H., Huang C.Y. Polypoid endometriosis – A rare entity of endometriosis mimicking ovarian cancer // Taiwan. J. Obstet. Gynecol. 2019. V. 58. № 3. P. 328.
Saliba C., Jaafoury H., El Hajj M. et al. Abdominal Wall Endometriosis: A Case Report // Cureus. 2019. V. 11. № 2. P. e4061.
Andres M.P., Mendes R.F.P., Hernandes C. et al. Hormone treatment as first line therapy is safe and relieves pelvic pain in women with bowel endometriosis // Einstein (Sao Paulo). 2019. V. 17. № 2. P. eAO4583.
Kashyap P., Medeiros F., Levy M., Larson M. Unusual submucosal tumor in the stomach. Diagnosis: Endometriosis // Gastroenterology. 2011. V. 140. № 7. P. e7.
Ha J.K., Choi C.W., Kim H.W. et al. An extremely rare case of gastric subepithelial tumor: gastric endometriosis // Clin. Endosc. 2015. V. 48. № 1. P. 74.
Fernandes R.P., Centini G., Afors K. et al. Standard Approach to Urinary Bladder Endometriosis // J. Minim. Invasive Gynecol. 2018. V. 25. № 6. P. 955.
Flieder D.B., Moran C.A., Travis W.D. et al. Pleuro-pulmonary endometriosis and pulmonary ectopic deciduosis: a clinicopathologic and immunohistochemical study of 10 cases with emphasis on diagnostic pitfalls // Hum. Pathol. 1998. V. 29. № 12. P. 1495.
Oner A., Karakucuk S., Serin S. Nasolacrimal endometriosis. A case report // Ophthalmic Res. 2006. V. 38. № 5. P. 313.
Türkçüoğlu I., Türkçüoğlu P., Kurt J., Yildirim H. Presumed nasolacrimal endometriosis // Ophthalmic Plast Reconstr. Surg. 2008. V. 24. № 1. P. 47.
Zhang T., De Carolis C., Man G.C.W., Wang C.C. The link between immunity, autoimmunity and endometriosis: a literature update // Autoimmun. Rev. 2018. V. 17. № 10. P. 945.
Schulke L., Berbic M., Manconi F. et al. Dendritic cellpopulations in the eutopic and ectopic endometrium of women with endometriosis // Hum. Reprod. 2009. V. 24. № 7. P. 1695.
Akoum A., Kong J., Metz C., Beaumont M.C. Spontaneous and stimulated secretion of monocyte chemotactic protein-1 and macrophage migration inhibitory factor by peritoneal macrophages in women with and without endometriosis // Fertil. Steril. 2002. V. 77. № 5. P. 989.
Capobianco A., Monno A., Cottone L. et al. Proangiogenic Tie2(+) macrophages infiltrate human and murine endometriotic lesions and dictate their growth in a mouse model of the disease // Am. J. Pathol. 2011. V. 179. № 5. P. 2651.
Wu M.H., Shoji Y., Wu M.C. et al. Suppression of matrix metalloproteinase-9 by prostaglandin E(2) in peritoneal macrophage is associated with severity of endometriosis // Am. J. Pathol. 2005. V. 167. № 4. P. 1061.
Zhang R.J., Wild R.A., Ojago J.M. Effect of tumor necrosis factor-alpha on adhesion of human endometrial stromal cells to peritoneal mesothelial cells: an in vitro system // Fertil. Steril. 1993. V. 59. P. 1196.
Wu M.Y., Ho H.N. The role of cytokines in endometriosis // Am. J. Reprod. Immunol. 2003. V. 49. № 5. P. 285.
De Barros I.B.L., Malvezzi H., Gueuvoghlanian-Silva B.Y. “What do we know about regulatory T cells and endometriosis? A systematic review” // J. Reprod. Immunol. 2017. V. 120. P. 48.
Kao A.P., Wang K.H., Long C.Y. et al. Interleukin-1β induces cyclooxygenase-2 expression and promotes the invasive ability of human mesenchymal stem cells derived from ovarian endometrioma // Fertil. Steril. 2011. V. 96. № 3. P. 678.e1.
Sikora J., Smycz-Kubańska M., Mielczarek-Palacz A., Kondera-Anasz Z. Abnormal peritoneal regulation of chemokine activation-The role of IL-8 in pathogenesis of endometriosis // Am. J. Reprod. Immunol. 2017. V. 77. № 4. https://doi.org/10.1111/aji.12622
Osuga Y., Koga K., Hirota Y. et al. Lymphocytes in endometriosis // Am. J. Reprod. Immunol. 2011. V. 65. № 1. P. 1.
Guo Y., Chen Y., Liu L.B. et al. IL-22 in the endometriotic milieu promotes the proliferation of endometrial stromal cells via stimulating the secretion of CCL2 and IL-8 // Int. J. Clin. Exp. Pathol. 2013. V. 6. № 10. P. 2011.
Măluțan A.M., Drugan T., Ciortea R. et al. Endometriosis-associated changes in serum levels of interferons and chemokines // Turk. J. Med. Sci. 2017. V. 47. № 1. P. 115.
Yu J.J., Sun H.T., Zhang Z.F. et al. IL15 promotes growth and invasion of endometrial stromal cells and inhibits killing activity of NK cells in endometriosis // Reproduction. 2016. V. 152. № 2. P. 151.
Hassa H., Tanir H.M., Tekin B. et al. Cytokine and immune cell levels in peritoneal fluid and peripheral blood of women with early- and late-staged endometriosis // Arch. Gynecol. Obstet. 2009. V. 279. № 6. P. 891.
Montenegro M.L., Ferriani R.A., Basse P.H. Exogenous activated NK cells enhance trafficking of endogenous NK cells to endometriotic lesions // BMC Immunol. 2015. V. 16. P. 51.
Gogacz M., Gałczyński K., Wojtaś M. Fas-related apoptosis of peritoneal fluid macrophages in endometriosis patients: understanding the disease // J. Immunol. Res. 2017. V. 2017. P. 3175394.
Wu M.Y., Yang J.H., Chao K.H. et al. Increase in the expression of killer cell inhibitory receptors on peritoneal natural killer cells in women with endometriosis // Fertil. Steril. 2000. V. 74. № 6. P. 1187.
Kang Y.J., Jeung I.C., Park A. et al. An increased level of IL-6 suppresses NK cell activity in peritoneal fluid of patients with endometriosis via regulation of SHP-2 expression // Hum. Reprod. 2014. V. 29. № 10. P. 2176.
Thiruchelvam U., Wingfield M., O’Farrelly C. Natural Killer Cells: Key Players in Endometriosis // Am. J. Reprod. Immunol. 2015. V. 74. № 4. P. 291.
Andreoli C.G., Genro V.K., Souza C.A. et al. T helper (Th)1, Th2, and Th17 interleukin pathways in infertile patients with minimal/mild endometriosis // Fertil. Steril. 2011. V. 95. № 8. P. 2477.
Podgaec S., Dias Junior J.A., Chapron C. et al. Th1 and Th2 ummune responses related to pelvic endometriosis // Rev. Assoc. Med. Bras. (1992). 2010. V. 56. № 1. P. 92.
Takamura M., Koga K., Izumi G. et al. Simultaneous Detection and Evaluation of Four Subsets of CD4+ T Lymphocyte in Lesions and Peripheral Blood in Endometriosis // Am. J. Reprod. Immunol. 2015. V. 74. № 6. P. 480.
Berbic M., Fraser I.S. Regulatory T cells and other leukocytes in the pathogenesis of endometriosis // J. Reprod. Immunol. 2011. V. 88. № 2. P. 149.
Wei C., Mei J., Tang L. et al. 1-Methyl-tryptophan attenuates regulatory T cells differentiation due to the inhibition of estrogen-IDO1-MRC2 axis in endometriosis // Cell Death Dis. 2016. V. 7. № 12. P. e2489.
Králíčková M., Fiala L., Losan P. et al. Altered Immunity in Endometriosis: What Came First? // Immunol Invest. 2018. V. 47. № 6. P. 569.
Polanczyk M.J., Hopke C., Vandenbark A.A., Offner H. Treg suppressive activity involves estrogen-dependent expression of programmed death-1 (PD-1) // Int. Immunol. 2007. V. 19. № 3. P. 337.
Riccio L.G.C., Baracat E.C., Chapron C. et al. The role of the B lymphocytes in endometriosis: A systematic review // J. Reprod. Immunol. 2017. V. 123. P. 29.
Gleicher N., el-Roeiy A., Confino E., Friberg J. Is endometriosis an autoimmune disease? // Obstet. Gynecol. 1987. V. 70. № 1. P. 115.
Buckner J.H. Mechanisms of impaired regulation by CD4+CD25+FOXP3+ regulatory T cells in human autoimmune diseases // Nat. Rev. Immunol. 2010. V. 10. № 12. P. 849.
Eisenberg V.H., Zolti M., Soriano D. Is there an association between autoimmunity and endometriosis? // Autoimmun. Rev. 2012. V. 11. № 11. P. 806.
Fernández-Shaw S., Hicks B.R., Yudkin P.L. et al. Anti-endometrial and anti-endothelial auto-antibodies in women with endometriosis // Hum. Reprod. 1993. V. 8. № 2. P. 310.
Mizushima N., Levine B., Cuervo A.M., Klionsky D.J. Autophagy fights disease through cellular self-digestion // Nature. 2008. V. 451. № 7182. P. 1069.
Yang H.L., Mei J., Chang K.K. et al. Autophagy in endometriosis // Am. J. Transl. Res. 2017. V. 9. № 11. P. 4707.
Vousden K.H., Prives C. Blinded by the light: the growing complexity of p53 // Cell. 2009. V. 137. № 3. P. 413.
Soares M.P., Bach F.H. Heme oxygenase-1: from biology to therapeutic potential // Trends Mol. Med. 2009. V. 15. № 2. P. 50.
Wu M.H., Hsiao K.Y., Tsai S.J. Hypoxia: The force of endometriosis // J. Obstet. Gynaecol. Res. 2019. V. 45. № 3. P. 532.
Albertsen H.M., Ward K. Genes Linked to Endometriosis by GWAS Are Integral to Cytoskeleton Regulation and Suggests That Mesothelial Barrier Homeostasis Is a Factor in the Pathogenesis of Endometriosis // Reprod. Sci. 2017. V. 24. № 6. P. 803.
Proestling K., Birner P., Gamperl S. et al. Enhanced epithelial to mesenchymal transition (EMT) and upregulated MYC in ectopic lesions contribute independently to endometriosis // Reprod. Biol. Endocrinol. 2015. V. 13. P. 75.
Bostanci Durmus A., Dincer Cengiz S., Yılmaz H. et al. The levels of matrix metalloproteinase-9 and neutrophil gelatinase-associated lipocalin in different stages of endometriosis // J. Obstet. Gynaecol. 2019. V. 39. № 7. P. 991.
Christodoulakos G., Augoulea A., Lambrinoudaki I. et al. Pathogenesis of endometriosis: the role of defective “immunosurveillance” // Eur. J. Contracept. Reprod. Health Care. 2007. V. 12. № 3. P. 194.
Nair A.S., Nair H.B., Lucidi R.S. et al. Modeling the early endometriotic lesion: mesothelium-endometrial cell co-culture increases endometrial invasion and alters mesothelial and endometrial gene transcription // Fertil. Steril. 2008. V. 90. № 4(Suppl). P. 1487.
McLaren J. Vascular endothelial growth factor and endometriotic angiogenesis // Hum. Reprod. Update. 2000. V. 6. № 1. P. 45.
Shifren J.L., Tseng J.F., Zaloudek C.J. et al. Ovarian steroid regulation of vascular endothelial growth factor in the human endometrium: implications for angiogenesis during the menstrual cycle and in the pathogenesis of endometriosis // J. Clin. Endocrinol. Metab. 1996. V. 81. № 8. P. 3112.
Omwandho C.O., Konrad L., Halis G. et al. Role of TGF-betas in normal human endometrium and endometriosis // Hum. Reprod. 2010. V. 25. № 1. P. 101.
Young V.J., Ahmad S.F., Brown J.K. et al. Peritoneal VEGF-A expression is regulated by TGF-β1 through an ID1 pathway in women with endometriosis // Sci. Rep. 2015. V. 5. P. 16859.
Fong S., Debs R.J., Desprez P.Y. Id genes and proteins as promising targets in cancer therapy // Trends Mol. Med. 2004. V. 10. № 8. P. 387.
Schutt A.K., Atkins K.A., Slack-Davis J.K., Stovall D.W. VCAM-1 on peritoneum and α4β1 integrin in endometrium and their implications in endometriosis // Int. J. Gynecol. Pathol. 2015. V. 34. № 1. P. 85.
Yilmaz B.D., Bulun S.E. Endometriosis and nuclear receptors // Hum. Reprod. Update. 2019. V. 25. № 4. P. 473.
Bulun S.E. Endometriosis // N. Engl. J. Med. 2009. V. 360. № 3. P. 268.
Attia G.R., Zeitoun K., Edwards D. et al. Progesterone receptor isoform A but not B is expressed in endometriosis // J. Clin. Endocrinol. Metab. 2000. V. 85. № 8. P. 2897.
Bernardi L.A., Dyson M.T., Tokunaga H. et al. The Essential Role of GATA6 in the Activation of Estrogen Synthesis in Endometriosis // Reprod. Sci. 2019. V. 26. № 1. P. 60.
Xue Q., Lin Z., Yin P. et al. Transcriptional activation of steroidogenic factor-1 by hypomethylation of the 5' CpG island in endometriosis // J. Clin. Endocrinol. Metab. 2007. V. 92. № 8. P. 3261.
Yamagata Y., Nishino K., Takaki E. et al. Genome-wide DNA methylation profiling in cultured eutopic and ectopic endometrial stromal cells // PLoS One. 2014. V. 9. № 1. P. e83612.
Monteiro J.B., Colón-Díaz M., García M. et al. Endometriosis is characterized by a distinct pattern of histone 3 and histone 4 lysine modifications // Reprod. Sci. 2014. V. 21. № 3. P. 305.
Samartzis E.P., Noske A., Samartzis N. et al. The expression of histone deacetylase 1, but not other class I histone deacetylases, is significantly increased in endometriosis // Reprod. Sci. 2013. V. 20. № 12. P. 1416.
Imesch P., Fink D., Fedier A. Romidepsin reduces histone deacetylase activity, induces acetylation of histones, inhibits proliferation, and activates apoptosis in immortalized epithelial endometriotic cells // Fertil. Steril. 2010. V. 94. № 7. P. 2838.
Bartel D.P. MicroRNAs: target recognition and regulatory functions // Cell. 2009. V. 136. № 2. P. 215.
Huhtinen K., Ståhle M., Perheentupa A., Poutanen M. Estrogen biosynthesis and signaling in endometriosis // Mol. Cell. Endocrinol. 2012. V. 358. № 2. P. 146.
Lin Z., Reierstad S., Huang C.C., Bulun S.E. Novel estrogen receptor-alpha binding sites and estradiol target genes identified by chromatin immunoprecipitation cloning in breast cancer // Cancer Res. 2007. V. 67. № 10. P. 5017.
Smuc T., Pucelj M.R., Sinkovec J. et al. Expression analysis of the genes involved in estradiol and progesterone action in human ovarian endometriosis // Gynecol. Endocrinol. 2007. V. 23. № 2. P. 105.
Xue Q., Lin Z., Cheng Y.H. et al. Promoter methylation regulates estrogen receptor 2 in human endometrium and endometriosis // Biol. Reprod. 2007. V. 77. № 4. P. 681.
Yang H., Kang K., Cheng C. et al. Integrative Analysis Reveals Regulatory Programs in Endometriosis // Reprod. Sci. 2015. V. 22. № 9. P. 1060.
Monsivais D., Dyson M.T., Yin P. et al. ERβ- and prostaglandin E2-regulated pathways integrate cell proliferation via Ras-like and estrogen-regulated growth inhibitor in endometriosis // Mol. Endocrinol. 2014. V. 28. № 8. P. 1304.
Hsiao K.Y., Wu M.H., Chang N. et al. Coordination of AUF1 and miR-148a destabilizes DNA methyltransferase 1 mRNA under hypoxia in endometriosis // Mol. Hum. Reprod. 2015. V. 21. № 12. P. 894.
Mulac-Jericevic B., Mullinax R.A., DeMayo F.J. et al. Subgroup of reproductive functions of progesterone mediated by progesterone receptor-B isoform // Science. 2000. V. 289. № 5485. P. 1751.
Mulac-Jericevic B., Lydon J.P., DeMayo F.J., Conneely O.M. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform // Proc. Natl. Acad. Sci. USA. 2003. V. 100. № 17. P. 9744.
Vegeto E., Shahbaz M.M., Wen D.X. et al. Human progesterone receptor A form is a cell- and promoter-specific repressor of human progesterone receptor B function // Mol. Endocrinol. 1993. V. 7. № 10. P. 1244.
Kim J.J., Kurita T., Bulun S.E. Progesterone action in endometrial cancer, endometriosis, uterine fibroids, and breast cancer // Endocr. Rev. 2013. V. 34. № 1. P. 130.
Cheng Y.H., Yin P., Xue Q. et al. Retinoic acid (RA) regulates 17beta-hydroxysteroid dehydrogenase type 2 expression in endometrium: interaction of RA receptors with specificity protein (SP) 1/SP3 for estradiol metabolism // J. Clin. Endocrinol. Metab. 2008. V. 93. № 5. P. 1915.
Yang S., Fang Z., Gurates B. et al. Stromal PRs mediate induction of 17beta-hydroxysteroid dehydrogenase type 2 expression in human endometrial epithelium: a paracrine mechanism for inactivation of E2 // Mol. Endocrinol. 2001. V. 15. № 12. P. 2093.
Kao L.C., Germeyer A., Tulac S. et al. Expression profiling of endometrium from women with endometriosis reveals candidate genes for disease-based implantation failure and infertility // Endocrinology. 2003. V. 144. № 7. P. 2870.
Schug T.T., Berry D.C., Shaw N.S. et al. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors // Cell. 2007. V. 129. № 4. P. 723.
Jia J., Zhang W., Liu J.Y. et al. Epithelial mesenchymal transition is required for acquisition of anoikis resistance and metastatic potential in adenoid cystic carcinoma // PLoS One. 2012. V. 7. № 12. P. e51549.
Matsuzaki S., Darcha C. Epithelial to mesenchymal transition-like and mesenchymal to epithelial transition-like processes might be involved in the pathogenesis of pelvic endometriosis // Hum. Reprod. 2012. V. 27. № 3. P. 712.
Ono Y.J., Hayashi M., Tanabe A. et al. Estradiol-mediated hepatocyte growth factor is involved in the implantation of endometriotic cells via the mesothelial-to-mesenchymal transition in the peritoneum // Am. J. Physiol. Endocrinol. Metab. 2015. V. 308. № 11. P. E950.
Wu M.H., Chen K.F., Lin S.C. et al. Aberrant expression of leptin in human endometriotic stromal cells is induced by elevated levels of hypoxia inducible factor-1alpha // Am. J. Pathol. 2007. V. 170. №2. P. 590.
Ping S., Ma C., Liu P. et al. Molecular mechanisms underlying endometriosis pathogenesis revealed by bioinformatics analysis of microarray data // Arch. Gynecol. Obstet. 2016. V. 293. № 4. P. 797.
Flamini M.I., Sanchez A.M., Goglia L. et al. Differential actions of estrogen and SERMs in regulation of the actin cytoskeleton of endometrial cells // Mol. Hum. Reprod. 2009. V. 15. № 10. P. 675.
Gentilini D., Vigano P., Somigliana E. et al. Endometrial stromal cells from women with endometriosis reveal peculiar migratory behavior in response to ovarian steroids // Fertil. Steril. 2010. V. 93. № 3. P. 706.
Albertsen H.M., Ward K. Genes Linked to Endometriosis by GWAS Are Integral to Cytoskeleton Regulation and Suggests That Mesothelial Barrier Homeostasis Is a Factor in the Pathogenesis of Endometriosis // Reprod. Sci. 2017. V. 24. № 6. P. 803.
Дополнительные материалы отсутствуют.
Инструменты
Физиология человека