

Цитология, 2020, T. 62, № 3, стр. 151-159
Роль р53-зависимой аутофагии в регуляции поведения плюрипотентных клеток
Г. И. Сутула 1, М. Л. Воробьев 1, И. И. Суворова 1, *
1 Институт цитологии РАН
194064 Санкт-Петербург, Россия
* E-mail: irsuvorov@yandex.ru
Поступила в редакцию 25.11.2019
После доработки 16.12.2019
Принята к публикации 18.12.2019
Аннотация
Эмбриональные стволовые клетки (ЭСК), а также их искусственные аналоги индуцированные плюрипотентные стволовые клетки (ИПСК) являются предшественниками всех типов клеток взрослого организма. По этой причине они представляют собой неиссякаемый клеточный источник для регенеративной медицины. Однако успешное применение ЭСК и ИПСК в клинике сопряжено с риском образования тератомы после трансплантации их дифференцированных продуктов. Как правило, онкогенный потенциал связан с тем, что в популяции зрелых клеток сохраняются плюрипотентные клетки, устойчивые к дифференцировке. Эти дефектные клетки по каким-то причинам не смогли выйти из состояния плюрипотентности in vitro при действии митогенных стимулов и остались недифференцированными. В эмбриогенезе существуют особые механизмы устранения непригодных для развития зародыша клеток, которые массивно запускаются перед гаструляцией – начальным этапом дифференцировки клеток в зародышевые листки. Известно, что до момента имплантации критическую роль в формировании эмбриона играет аутофагия, которую можно рассматривать как одну из главных клеточных стратегий, направленных на масштабную перестройку внутриклеточного материала после оплодотворения. Если предположить, что процесс внутриклеточной реорганизации зародышевых клеток прошел неэффективно, то такие клетки будут иметь дефектный протеостаз, что отразится на их дифференцировочном потенциале. По этой причине высокий уровень апоптоза, наблюдаемый перед гаструляцией в эмбриогенезе, связан с устранением мутантных клеток, не пригодных для дифференцировки. Поврежденные клетки маркируются активированным белком р53, что свидетельствует о р53-зависимых механизмах элиминации. И, по всей видимости, механизм активации р53 связан с нарушением клеточного протеостаза, в основе которого лежит аутофагия. Таким образом, целью настоящего обзора является исследование роли р53-зависимой аутофагии в определении дальнейшей судьбы плюрипотентных клеток: индукция клеточной гибели и (или) резистентность к дифференцировке. Мы показали, что белок р53 находится в очень тесной взаимосвязи с аутофагией и при наличии дефектного протеостаза достаточно эффективно переводит процесс аутофагии на путь индуцируемой клеточной гибели в плюрипотентных клетках.
Механизмы, обеспечивающие качественный контроль внутриклеточного содержимого, являются критичными в процессе эмбрионального развития и, соответственно, должны быть высокоэффективными в эмбриональных стволовых клетках (ЭСК), из которых образуются все типы тканей взрослого организма. Недавно было показано, что дефектные зародышевые стволовые клетки в эмбриогенезе устраняются из популяции по механизму “конкурентного преимущества” (Sancho et al., 2013). Клеточная конкуренция – это тип межклеточного взаимодействия, впервые изученный у Drosophila, у которых сосуществование двух клеточных популяций с различными метаболическими свойствами или уровнем пролиферации приводит к экспансии более сильной популяции за счет слабой (Moreno et al., 2002). Предполагается, что процесс распознавания и устранения дефектных, неправильно сформированных или аномальных клеток играет важную роль в гомеостазе тканей, контроле размеров органов и в поддержании популяции стволовых клеток (Levayer, Moreno, 2013).
У эмбрионов мыши перед началом этапа гаструляции наблюдается высокая апоптотическая гибель клеток (Manova et al., 1998; Spruce et al., 2010). К тому же с началом дифференцировки эмбрион становится гиперчувствительным к повреждению ДНК, вызванному облучением в низких дозах (Heyer et al., 2000). Все это свидетельствует о том, что на данном этапе эмбриогенеза может осуществляться тщательный контроль пригодности клеток, направляющихся на дифференцировку (Sancho et al., 2013). Так, например, в процессе развития зародыша эмбриональные клетки, имеющие дефекты в сигнальном пути BMP, в процессе аутофагии или являющиеся полиплоидными, элиминируются из популяции на стадии гаструляции по механизму программируемой клеточной гибели и только в присутствии нормальных клеток (Sancho et al., 2013).
Основной механизм элиминации дефектных клеток, входящих в дифференцировку – ингибирование сигнального пути киназы mTOR (Bowling et al., 2018). Путь mTOR активируется при действии ростовых факторов, а также в условиях достаточного количества аминокислот в среде, и, соответственно, стимулирует анаболические процессы в клетке – белковый синтез и клеточный рост. Делеция гена mtor приводит к гибели эмбриона сразу же после имплантации в стенку матки, что свидетельствует о критической роли пути mTOR в процессе дифференцировки (Gangloff et al., 2004; Murakami et al., 2004). До стадии имплантации эмбриона, когда начинается дифференцировка в направлении трех зародышевых листков, активность mTOR не является критичной, и образование ЭСК (клеток, выделенных из внутренней клеточной массы бластоцисты) происходит в отсутствии функциональной активности этого пути. В это время в клетках высоко активна аутофагия – антагонистичный по отношению к mTOR сигнальный путь, позволяющий осуществлять масштабную реорганизацию внутриклеточных компонентов оплодотворенного ооцита при переходе к эмбриогенезу (Tsukamoto et al., 2008). Учитывая, что время раннего эмбрионального развития является достаточно быстрым и деградация белков с помощью убиквитинпротеасомной системы за столь короткое время неэффективна, то именно аутофагия играет на этом этапе эмбрионального развития ключевую роль. Если процесс аутофагии будет не полным, то недеградированные внутриклеточные компоненты могут препятствовать дальнейшему развитию эмбриона.
Известно, что различные нарушения во внутриклеточных процессах неизбежно сопровождаются активацией белка р53 – главного “защитника генома” клеток, так как он эффективно удаляет аномальные клетки из популяции. Следовательно, дефектные стволовые клетки, возникающие в эмбриогенезе, маркируются функционально-активным белком р53, что приводит к их последующей элиминации и, таким образом, предотвращается их дифференцировка и включение в будущую ткань (Bowling et al., 2018). Тем не менее, не совсем ясен механизм активации белка р53 в мутантных клетках и его согласование с сигнальным каскадом mTOR. По всей видимости, природа этой активации может быть связана с нарушениями в процессе аутофагии, когда деградация внутриклеточных компонентов при переходе от материнского типа экспрессии генов к зиготическому полностью не завершена, и путь mTOR в ответ на дифференцировочные стимулы активироваться не может, так как активная аутофагия его супрессирует. В этом случае происходит стимуляция белка р53, который устраняет мутантные клетки. Однако каким образом этапы аутофагии функционально скоординированы с активностью р53 в плюрипотентных клетках, до сих пор до конца неясно.
Чтобы исследовать этот вопрос, в настоящем обзоре были поставлены следующие задачи: рассмотреть общие клеточные механизмы, вовлекающие р53 в регуляцию аутофагии; определить, каким образом они реализуются в клетках, обладающих плюрипотентными свойствами и высоким уровнем пролиферации; показать, что тесная сопряженность белка р53 с процессом аутофагии может быть основой для использования ее в качестве фармакологической мишени для элиминации дефектных плюрипотентных клеток из популяции.
ИЗВЕСТНЫЕ МЕХАНИЗМЫ ВОВЛЕЧЕННОСТИ p53 В РЕГУЛЯЦИЮ АУТОФАГИИ В КЛЕТКАХ
Белок p53 является мультифункциональным, так как он играет ключевую роль в клеточном ответе на повреждение ДНК при действии различных стресс-факторов (гипоксия, ионизирующее излучение, высокая концентрация NO и др.) и, соответственно, участвует в координации множества сигнальных путей для формирования общего ответа клеток на неблагоприятный стимул. Как транскрипционный фактор, р53 активирует и репрессирует многочисленные гены-мишени, что в результате приводит к остановке прогрессии клеток в контрольных точках клеточного цикла (G1/S, S, G2/M) и позволяет устранить генотоксические нарушения с помощью репарации, или индуцировать программируемую клеточную гибель при наличии обширных повреждений (Speidel, 2015).
По этой причине сигнальные р53-зависимые механизмы являются одной из главных клеточных стратегий подавления опухолевой трансформации, однако до сих пор так и остаются до конца не раскрытыми. Для глубокого понимания биологии функционирования р53 был проведен глобальный анализ транскрипционных сетей, регулируемых этим белком, в эмбриональных фибробластах мыши в ответ на повреждение ДНК (Kenzelmann et al., 2013). В результате среди прямых мишеней р53 было идентифицировано множество генов, белковые продукты которых участвуют в аутофагии.
Аутофагия – консервативный механизм, направленный на избавление от дисфункциональных органелл и внутриклеточных компонентов, а также на сохранение жизнеспособности клеток при неблагоприятных энергетических условиях (Parzych, Klionsky, 2014). Соответственно к основным факторам, инициирующим аутофагию в клетках, можно отнести голодание, наличие в цитоплазме поврежденных органелл, частично денатурированных белков и их агрегатов. В результате предназначенный для утилизации внутриклеточный материал секвестрируется двумембранным компартментом – аутофагосомой. Далее аутофагосома сливается с лизосомой и формируется аутофаголизосома, в которой происходит деградация содержимого. Как правило, запуск аутофагии сопровождается активацией аденозинмонофосфат-зависимой киназой (АМФК), которая функционирует в виде гетеродимеров, состоящих из каталитических α-субъединиц и регуляторных субъединиц β и γ. В условиях недостаточного количества питательных веществ молекула АТФ не может конкурентно ингибировать связывание АМФ и АДФ с γ-субъединицей, что приводит к аллостерической активации киназы, которая функционирует, соответственно, в виде внутриклеточного сенсора уровней аденозинфосфатов (АМФ/АТФ и АДФ/АТФ). Дополнительно активность АМФК регулируется ключевым фосфорилированием по треонину 172 α-субъединицы, осуществляемым, по крайней мере, тремя киназами, среди которых стоит отметить хорошо изученное взаимодействие с белком LKB (Hardie et al., 2016). При активации АМФК сигнальный путь mTOR ингибируется, в свою очередь при активации mTOR происходит подавление аутофагии. Главный механизм взаимного антагонизма этих сигнальных путей осуществляется через киназу Ulk1, на которой сходятся сигналы, идущие от белков АМФК и mTOR, и в результате формируется общий метаболический ответ клетки. Белок mTOR прямо ингибирует киназу Ulk1 и запускает анаболические процессы в клетке, в то время как АМФК активирует Ulk1, что сопровождается диссоциацией Ulk1 от ее комплекса с mTOR, и индуцируется клеточный катаболизм.
Таким образом, канонический путь запуска аутофагии происходит через активацию сигнального пути АМФК/Ulk1, что сопровождается ингибированием mTOR и подавлением процессов клеточного синтеза. Соответственно можно предположить, что остальные регуляторы аутофагии, которые вовлекаются в этот процесс в ответ на различные стресс-стимулы, должны находиться в тесном взаимодействии с сигнальным модулем АМФК/Ulk1/mTOR. Согласно многочисленным данным литературы, добавление активаторов АМФК в среду для культивирования приводит к фосфорилированию р53 по серину 15, что свидетельствует о наличие прямого или опосредованного взаимодействия между АМФК и р53 (Kim et al., 2016). Как оказалось, АМФК может прямо фосфорилировать р53 по серину 15 в эмбриональных фибробластах мыши, что приводит к его стабилизации и активации и последующему формированию блока клеточного цикла в условиях пониженного содержания глюкозы в среде (Jones et al., 2005). Таким образом, можно предположить, что АМФК-зависимая активация р53 является ключевым условием, при котором р53 вовлекается в процесс аутофагии в клетках.
В настоящее время известно, что р53 регулирует аутофагию посредством двух механизмов в зависимости от своей локализации в клетке: ядерной или цитоплазматической. Ядерный пул р53 функционирует в виде транскрипционного фактора и стимулирует аутофагию через запуск экспрессии своих генов-мишеней. Так, р53 активирует транскрипцию субъединиц β1 и β2 киназы АМФК, что свидетельствует о существовании положительной обратной связи между р53 и АМФК (Feng et al., 2007). Во-вторых, р53 индуцирует экспрессию генов Sestrin1 и Sestrin2, гиперэкспрессия которых понижает уровень фосфорилированной формы белка p70S6K – одной из главных мишеней киназы mTOR, и, следовательно, ингибирует путь mTOR (Budanov, Karin, 2008). В-третьих, p53 активирует транскрипцию гена dram1, который кодирует лизосомальный белок, участвующий в процессе слияния аутофагосом с лизосомами на этапе образования аутофаголизосом (Crighton, 2006). Недавно киназа Ulk1 и модулятор аутофагии ISG20L1 были также идентифицированы как прямые мишени p53 в клетках человека (Eby et al., 2010; Gao et al. 2011). Таким образом, р53 вовлекается в регуляцию аутофагию посредством АМФК-зависимой активации и в виде транскрипционного фактора индуцирует экспрессию коровых генов аутофагии. Поскольку основная деятельность р53 в клетке заключается в формировании апоптотического ответа, то логично предположить, что участие р53 в регуляции аутофагии напрямую связано с этой функцией. Так, уровень апоптоза в ответ на действие доксорубицина в фибробластах мыши с делецией аутофагического гена ATG5 был значительно слабее чем в контрольных клетках (Kenzelmann Broz et al., 2013). Авторы утверждают, что в контексте активации белка p53 аутофагия способствует не выживанию клетки, а эффективному p53-зависимому апоптозу и, как следствие, подавлению клеточной трансформации.
Второй механизм, с помощью которого р53 вовлекается в регуляцию аутофагии, обусловлен цитоплазматическим характером локализации р53 в клетках и функционально противоположен первому. Не совсем понятна природа активации р53 в этом случае, но цитоплазматический р53 ингибирует аутофагию, что было косвенно продемонстрировано на различных модельных системах. В частности, было показано, что инактивация p53 в клетках мыши, нематоды и человека путем нокаута, РНК-интерференции или фармакологического воздействия индуцирует аутофагию (Tasdemir, 2008). Кроме того, было показано, что мутантный р53, который не способен взаимодействовать с ДНК и локализован в цитоплазме, ингибирует аутофагию в раковых клетках (Morselli, 2008). Несмотря на установленную роль цитоплазматического пула р53 в ингибировании аутофагии, сам механизм еще недостаточно изучен и требует дальнейших исследований.
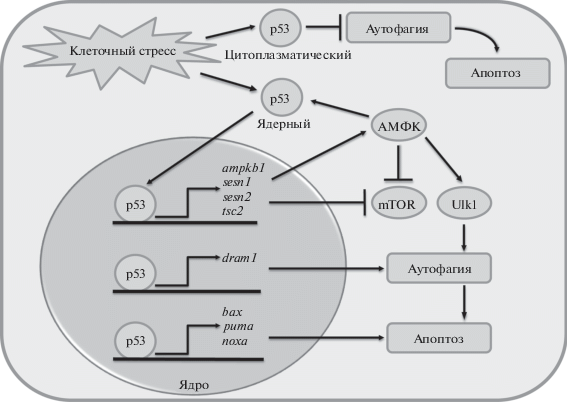
Таким образом, аутофагия как стратегия клеточного выживания при неблагоприятных условиях может быть использована как один из путей клеточной гибели, который требует участия белка р53. Соответственно киназа АМФК функционирует как метаболический сенсор в определении дальнейшей судьбы мутантных клеток посредством активации p53, связывая сигналы анти- и про-клеточной гибели. Следовательно, регуляция активностей белков р53 и АМФК обеспечивает баланс между жизнеспособностью и гибелью клеток посредством активации аутофагии (рис. 1).
Рис. 1.
Общая схема р53-зависимой регуляции аутофагии в клетках. При действии стресс-факторов (ДНК-повреждающие агенты, окислительный стресс, гипоксия и др.) активируется белок р53, который посредством АМФК-зависимого фосфорилирования вовлекается в регуляцию аутофагии: р53 транслоцируется в ядро, запуская экспрессию генов, белковые продукты которых участвуют в аутофагии. С одной стороны, р53 активирует аутофагические гены ampkb1, sesn1, sesn2, tsc2, которые усиливают аутофагический ответ клетки, поддерживая активацию сигнального пути АМФK/Ulk1. С другой стороны, р53 активирует аутофагический ген dram1, белковый продукт которого совместно с белками bax, puma, noxa, может быть главным переключателем аутофагического ответа клетки с выживания на апоптоз.
АУТОФАГИЯ В ПЛЮРИПОТЕНТНЫХ КЛЕТКАХ
Аутофагия массивно индуцируется практически сразу же после оплодотворения (Tsukamoto et al., 2008). В это время материнские белки и органеллы быстро разлагаются и замещаются белками, происходящими из зародышевого генома, и оплодотворенный ооцит претерпевает крупномасштабную внутриклеточную перестройку или процесс так называемого перехода транскриптов от материнских к зиготическим (Schier, 2007). Соответственно, аутофагия позволяет провести обширную реорганизацию внутреннего содержимого клетки, образуя достаточное количество аминокислот для вновь синтезированных белков. В процессе эмбриогенеза аутофагия временно подавляется на поздней стадии одноклеточного бластомера и далее вновь активируется. В результате клетки ВКМ (внутренняя клеточная масса), из которой получают ЭСК и которые являются предшественниками всех типов клеток взрослого организма, имеют высокий уровень аутофагической активности, которая тесно сопряженa с недифференцированным состоянием клеток. Ключевая роль аутофагии в поддержании плюрипотентности подтверждена результатами исследований по репрограммированию соматических клеток в индуцированные плюрипотентные стволовые клетки (ИПСК). Так, значительная активация аутофагии необходима на ранних стадиях получения ИПСК, что позволяет утилизовать внутриклеточные компоненты взрослых клеток и запустить синтез протеома, характерного для недифференцированного состояния (Wang et al., 2013).
Как уже было сказано выше, в основе канонической индукции аутофагии лежит сигнальный каскад АМФК/Ulk1/mTOR. В процессе эмбриогенеза активация пути mTOR является критичной на стадии гаструляции, соответственно, до этого момента активность АМФК/Ulk1 имеет ключевое значение в клетках эмбриона и, таким образом, превалирует в плюрипотентных клетках. Например, белок Ulk1, а также белки ATG13, FIP200, и ATG101, составляющие с киназой Ulk1 комплекс и участвующие в инициации аутофагии, транскрибируются на высоком уровне в плюрипотентных клетках по сравнению с фибробластами (Gong et al., 2018). Нокаут Ulk1 в ЭСК мыши резко снижает уровень аутофагической активности и приводит к ослаблению недифференцированного состояния клеток, уменьшая экспрессию маркеров плюрипотентности Oct4, Sox2, Nanog (Gong et al., 2018). Активность Ulk1 инициируется и поддерживается рядом АМФК-зависимых фосфорилирований.
Таким образом, конститутивная активация Ulk1 с помощью АМФК может быть одним из ключевых внутриклеточных механизмов, регулирующих клеточную идентичность плюрипотентных клеток. Это предположение было подтверждено результатами, полученными на линии ЭСК мыши с эндогенной активацией локуса ulk1 (Suvorova et al., 2019). Индуцибельная транскрипция гена ulk1 приводит к последующему накоплению белка и его фосфорилированию по серинам 555 и 317 киназой АМФК, что сопровождается увеличением уровня экспрессии маркеров плюрипотентности Oct4, Sox2, Nanog, Klf4. Следовательно, активация сигнального пути АМФК/Ulk1 и ингибирование пути mTOR являются чувствительными точками во внутриклеточной сигнализации ЭСК, поддерживающей их плюрипотентность и индуцирующей дифференцировку.
Последние данные, полученные на основе геномного скрининга нокаутных ЭСК человека с помощью технологии CRISPR/Cas9, выявили ключевую роль белка FLCN в комитированности недифференцированных клеток к дифференцировке (Mathieu et al., 2019). Было показано, что нокаут гена flcn приводит к резистентности клеток к дифференцировке, так как в этих условиях путь mTOR, критичный для дифференцировки, не активируется. Известно, что FLCN является эволюционно консервативным негативным регулятором киназы АМФК (Possik et al., 2014). Поэтому нокаут flcn приводит к конститутивной активации АМФК, активность которой является ключевой в поддержании плюрипотентности клеток (Possik et al., 2014). По этой причине ЭСК человека, нокаутные по flcn, могут оставаться плюрипотентными, поскольку они сохраняют высокий уровень аутофагии из-за персистентной активности АМФК/Ulk1-зависимой аутофагии.
МЕХАНИЗМЫ ВОВЛЕЧЕННОСТИ БЕЛКА p53 В РЕГУЛЯЦИЮ АУТОФАГИИ В ПЛЮРИПОТЕНТНЫХ КЛЕТКАХ
Известно, что процесс дифференцировки ЭСК сопровождается активацией белка р53 (Lin, Lin, 2017). Было показано, что р53 прямо регулирует экспрессию генов, ответственных за дифференцировку, плюрипотентность и самообновление (Li et al., 2012). Несмотря на непосредственное участие в дифференцировке, мыши с делецией по этому белку являются жизнеспособными в отличие от мышей, у которых отсутствует активность киназы mTOR. Наличие у мышей с делецией р53 многочисленных опухолей свидетельствует о том, что первостепенная роль р53 в эмбриогенезе – поддержание стабильности генома дифференцирующихся клеток.
Как уже было сказано выше, функционирование сигнального пути АМФК/Ulk1 тесно связано с активностью белка р53, который играет важную роль в элиминации мутантных клеток из популяции. Можно предположить, что дефектные эмбриональные стволовые клетки в эмбриогенезе имеют высокий уровень активации белка р53 вследствие персистирующей аутофагии, опосредованной сигнальным путем АМФК/Ulk1 (Bowling et al., 2018). В пользу данного предположения можно привести результаты, демонстрирующие, что прямой активатор киназы АМФК синтетическое соединение AICAR, который имитирует повышение уровня АМФ, приводит к фосфорилированию белка р53 по серину 15 в ЭСК мыши (Григораш и др., 2018). Кроме того, обработка ЭСК мыши ресвератролом, индуктором аутофагии, приводит к совместной активации АМФК и р53, что свидетельствует о наличии прямого или опосредованного взаимодействия между этими белками в недифференцированных клетках (Suvorova et al., 2018). При действии ресвератрола АМФК стимулирует р53, который далее транслоцируется в ядро и участвует в активации аутофагии транскрипционно-зависимым способом. Так, р53 запускает экспрессию гена dram1, который кодирует лизосомальный белок, требуемый для формирования аутолизосом в процессе аутофагии. Но при этом белок Dram1 также является критическим компонентом р53-зависимого апоптотического ответа, однако сам по себе он не может запустить апоптоз. Чтобы индуцировать программируемую клеточную гибель, р53 должен активировать не только транскрипцию dram1, а также экспрессию одного или нескольких проапоптотических генов, чтобы в результате эти сигнальные пути конвергировались в единый механизм индукции апоптоза (рис. 1).
СИГНАЛЬНЫЙ МОДУЛЬ АМФК/Ulk1/p53 КАК ПЕРСПЕКТИВНАЯ ФАРМАКОЛОГИЧЕСКАЯ МИШЕНЬ В ПОДДЕРЖАНИИ СТАБИЛЬНОЙ ПОПУЛЯЦИИ ПЛЮРИПОТЕНТНЫХ КЛЕТОК IN VITRO
Известно, что в процессе длительного культивирования в популяции ЭСК накапливаются генетические изменения (Draper et al., 2004; Baker et al., 2007; Spits et al., 2008). Возникающие мутации являются основной проблемой использования ЭСК на практике из-за их потенциальной онкогенности. Генетические аберрации могут появляться одновременно в разных хромосомных локусах в течение одного пассажа и постепенно накапливаться при длительном культивировании клеток. В то же время возникающие мутации влияют на фенотип ЭСК, что сопровождается ослаблением плюрипотентности и индукцией спонтанной дифференцировки или, наоборот, ослаблением потенциала дифференцировки и устойчивой экспрессией маркеров плюрипотентности. Эта проблема также относится к индуцированным плюрипотентным клеткам человека (ИПСК), привлекающим большое внимание своим потенциалом для использования в регенеративной медицине. Следовательно, для успешного использования ЭСК на практике необходимо решить две задачи: поддержание устойчивой экспансии ЭСК in vitro и предотвращение возникающих мутаций.
Различные мутации, наблюдаемые в плюрипотентных клетках in vitro, а также связанные с ними молекулярные механизмы и фенотипические характеристики, уже во многом описаны. Например, генетические аберрации, часто возникающие в культивируемых ЭСК человека, связаны с повышенной экспрессией гена Bcl2l1 и (или) мутацией в гене p53 (Amir et al., 2017). Нарушения в сигнальных путях, связанных с этими генами, делают клетки более устойчивыми к стрессу и приводят к преимущественному выживанию мутантных ЭСК и последующей замене нормальных клеток во время последующих пассажей. Стоит отметить, что те же самые механизмы используются опухолевыми клетками для выживания.
Белки семейства Bcl2 являются ключевыми медиаторами клеточного ответа на стресс. Они подразделяются на три группы в зависимости от состава домена гомологии Bcl-2 (BH) и их функции: антиапоптотические многодоменные белки (домены BH1–BH4), проапоптотические многодоменные белки (домены BH1–BH3) и проапоптотические однодоменные (только BH3). Члены семейства Bcl-2, способствующие клеточному выживанию, противодействуют апоптозу путем секвестрирования доменов BH3 проапоптотических белков. Таким образом, относительное количество тех или иных про- и антиапоптотических белков Bcl-2 определяет судьбу клеток: выживание или гибель. Белок р53 связывается с членами семейства Bcl-2 и запускает независимый от транскрипции апоптоз. Соответственно при мутации р53 активность антиапоптотических белков семейства Bcl-2 является высокой и способствует выживаемости дефектных клеток. Чтобы элиминировать из популяции клетки, несущие мутации белка р53, были разработаны такие фармакологические препараты как миметики BH3. Миметики BH3 связываются с мотивом BH3 антиапоптотических белков семейства Bcl-2 и ингибируют таким образом их активность, восстанавливая апоптотический ответ в клетке.
Так как ЭСК имеют высокий уровень аутофагии, то логично предположить, что манипулирование этим чувствительным процессом является хорошим инструментом в регуляции клеточного поведения. В настоящее время использование миметиков BH3, вызывающих гибель клеток в результате активации аутофагии, является одной из основных стратегий элиминации мутантных ЭСК из популяции in vitro (Cho et al., 2018). Дело в том, что если р53 как ключевой активатор аутофагии играет критичную роль в поддержании плюрипотентности, то клетки с мутантным р53 будут терять свой недифференцированный статус. Согласно последним данным плюрипотентные клетки в отличие от соматических клеток демонстрируют различную степень восприимчивости к использованию миметиков BH3, что создает возможность элиминировать уходящие в дифференцировку р53-мутантные клетки из популяции, не затрагивая нормальные ЭСК (Garcia et al., 2016). Предполагают, что эффективность действия того или иного миметика BH3 определяется уровнем аутофагии в клетках. Так как ЭСК имеют высокий уровень аутофагии, то логично предположить, что манипулирование этим чувствительным процессом через миметики BH3 является хорошим инструментом в регуляции клеточного поведения.
Один из механизмов такой элиминации – воздействие на комплекс между аутофагическим белком Beclin1 и антиапоптотическим белком семейства Bcl-2. При нормальных условиях Bcl-2 ингибирует Beclin1. Так как Beclin1 является мишенью киназы Ulk1, и, соответственно, без Ulk1-зависимого фосфорилирующего белка Beclin1 невозможна сборка пре-аутофагических мембран – затравки для формирования аутофагических вакуолей аутофагосом. Поскольку миметики BH3 действуют на комплекс Bcl-2/Beclin1, то, соответственно, затрагиваются механизмы, связанные с аутофагией. Например, миметики BH3 (ABT-737 и HA14-1) разрушают комплекс Bcl-2/Beclin1 и активируют различные проаутофагические пути посредством активации АМФК и ингибирования mTOR (Malik et al., 2011). Активация АМФК/Ulk1 и ингибирование пути mTOR являются чувствительными точками в поддержании клеточной плюрипотентности и тесно связаны с активностью р53, которая играет важную роль в элиминации мутантных клеток. Как уже было сказано выше, активность пути АМФК/Ulk1 также поддерживается р53-зависимыми механизмами, поэтому уровень аутофагии в ЭСК мыши значительно снижается после ингибирования транскрипционной активности р53 (Suvorova et al., 2018). Таким образом, важно иметь в виду, что активность пути АМФК/Ulk1 может быть снижена в ЭСК с дисфункциональной активностью p53, что необходимо учитывать при выборе миметиков BH3 для элиминации мутантных ЭСК из популяции.
ЗАКЛЮЧЕНИЕ
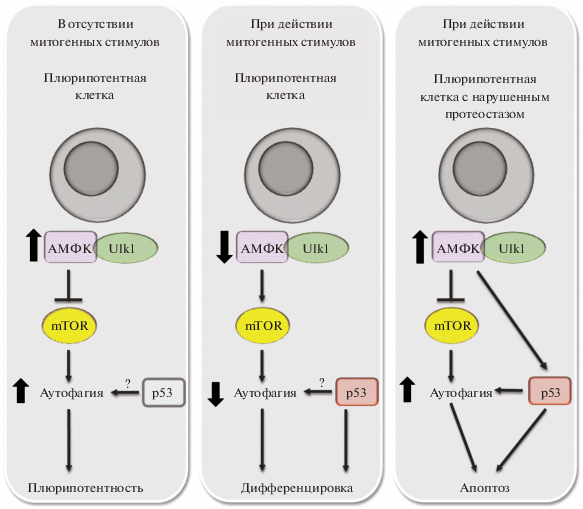
В настоящее время понимание роли белка р53 в регуляции аутофагии в клетках неоднозначно. С одной стороны, ядерный р53 активирует аутофагию, с другой, цитоплазматически локализованный р53 ингибирует этот процесс. Кроме того, сам механизм интеграции р53 в механизм аутофагии и его роль в индукции клеточной гибели в контексте аутофагической активации в клетках до конца не изучен. А между тем этот механизм может играть главную роль в элиминации мутантных плюрипотентных клеток в процессе эмбриогенеза перед этапом гаструляции. На рис. 2 представлена схема, демонстрирующая механизм вовлеченности р53 в элиминацию мутантных клеток из эмбриона до момента имплантации. В отсутствии митогенных стимулов, когда образуются клетки внутренней массы бластоцисты, обладающие высоким уровнем аутофагии, степень вовлеченности р53 в поддержание катаболических процессов еще не известно. В этом случае аутофагия за счет активации сигнального пути АМФК/Ulk1 поддерживает жизнеспособность клеток и участвует в формировании их плюрипотентного профиля. В процессе дифференцировки митогенные стимулы снижают уровень аутофагической активности в клетках за счет активации mTOR-зависимых анаболических процессов, необходимых для клеточной специализации. Известно, что р53 вовлечен в регуляцию дифференцировки клеток, и можно предположить, что контроль этого белка в этом процессе осуществляется через активацию или ингибирование аутофагии. При нарушении протеостаза, когда в клетках наблюдается высокий уровень катаболических процессов, не прекращающихся на действие митогенных факторов, активированный белок 53 переключает аутофагию на путь апоптоза и, соответственно, такие клетки элиминируются из эмбриогенеза (рис. 2).
Рис. 2.
Механизмы вовлеченности р53 в регуляцию аутофагии в клетках в отсутствие митогенных стимулов и при их действии, а также в условиях нарушения клеточнго протеостаза. Комментарии в тексте (раздел “Заключение”).
Таким образом, выяснение роли p53 в регуляции аутофагии в ЭСК является необходимым условием для разработки безопасной клеточной терапии на основе плюрипотентных клеток.
Список литературы
Григораш Б.Б., Суворова И.И., Поспелов В.А. 2018. AICAR-зависимая активация киназы АМФК не сопровождается блоком G1/S в эмбриональных стволовых клетках мыши. Молекулярная биология. Т. 52. № 3. С. 489. (Grigorash B.B., Suvorova I.I., Pospelov V.A. 2018. AICAR-dependent activation of АМФК kinase is not accompanied by G1/S block in mouse embryonic stem cells. Mol. Biol. V. 52. P. 489.)
Amir H., Touboul T., Sabatini K., Chhabra D., Garitaonandia I., Loring J.F., Morey R., Laurent L.C. 2017. Spontaneous single-copy loss of TP53 in human embryonic stem cells markedly increases cell proliferation and survival. Stem Cells. V. 35. P. 872.
Baker D.E.C., Harrison N.J., Maltby E., Smith K., Moore H.D., Shaw P.J., Heath P.R., Holden H., Andrews P.W. 2007. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat. Biotechnol. V. 25. P. 207.
Bowling S., Di Gregorio A., Sancho M., Pozzi S., Aarts M., Signore M., Schneider M.D., Martinez-Barbera J.P., Gil J., Rodríguez T.A. 2018. P53 and mTOR signalling determine fitness selection through cell competition during early mouse embryonic development. Nat. Commun. V. 9. P. 1763.
Budanov A.V., Karin M. 2008. The p53-regulated sestrin gene products inhibit mTOR signaling. Cell. V. 134. P. 451.
Cho S.J., Kim K.T., Jeong H.C., Park J.C., Kwon O.S., Song Y.H., Shin J.G., Kang S., Kim W., Shin H.D., Lee M.O., Moon S.H., Cha H.J. 2018. Selective elimination of culture-adapted human embryonic stem cells with BH3 mimetics. Stem Cell Rep. V.11. P. 1244.
Crighton D., Wilkinson S., O’Prey J., Syed N., Smith P., Harrison P.R., Gasco M., Garrone O., Crook T., Ryan K.M. 2006. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. V. 126. P. 121.
Draper J.S., Smith K., Gokhale P., Moore H.D., Maltby E., Johnson J., Meisner L., Zwaka T. P., Thomson J.A., Andrews P.W. 2004. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat. Biotechnol. V. 22. P. 53.
Eby K.G., Rosenbluth J.M., Mays D.J., Marshall C.B., Barton C.E., Sinha S., Johnson K.N., Tang L., Pietenpol J.A. 2010. ISG20L1 is a p53 family target gene that modulates genotoxic stress-induced autophagy. Mol. Cancer. V. 9. P. 95.
Feng Z., Hu W., de Stanchina E., Teresky A.K., Jin S., Lowe S., Levine A.J. 2007. The regulation of AMФK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. V. 67. P. 3043.
Gangloff Y.-G., Mueller M., Dann S.G., Svoboda P., Sticker M., Spetz J.-F., Um S.H., Brown E.J., Cereghini S., Thomas G., Kozma S.C. 2004. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol. Cell. Biol. V. 24. P. 9508.
Gao W., Shen Z., Shang L., Wang X. 2011. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ. V. 18. P. 1598.
García C.P., Videla Richardson G.A., Dimopoulos N.A., Fernandez Espinosa D.D., Miriuka S.G., Sevlever G.E., Romorini L., Scassa M.E. 2016. Human pluripotent stem cells and derived neuroprogenitors display differential degrees of susceptibility to BH3 mimetics ABT-263, WEHI-539 and ABT-199. PloS One. V. 11. e0152607. https://doi.org/10.1371/journal.pone.0152607
Gong J., Gu H., Zhao L., Wang L., Liu P., Wang F., Xu H., Zhao T. 2018. Phosphorylation of ULK1 by AMФK is essential for mouse embryonic stem cell self-renewal and pluripotency. Cell Death Dis. V. 9. P. 38.
Hardie D.G., Schaffer B.E., Brunet A. 2016. AMФK: An energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. V. 26. P. 190.
Heyer B.S., MacAuley A., Behrendtsen O., Werb Z. 2000. Hypersensitivity to DNA damage leads to increased apoptosis during early mouse development. Genes Dev. V. 14. P. 2072.
Jones R.G., Plas D.R., Kubek S., Buzzai M., Mu J., Xu Y., Birnbaum M.J., Thompson C.B. 2005. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell. V. 18. P. 283.
Kenzelmann Broz D., Spano Mello S., Bieging K.T., Jiang D., Dusek R.L., Brady C.A., Sidow A., Attardi L.D. 2013. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. V. 27. P. 1016.
Kim J., Yang G., Kim Y., Kim J., Ha J. 2016. AMФK activators: mechanisms of action and physiological activities. Exp. Mol. Med. V. 48. e224. https://doi.org/10.1038/emm.2016.16
Levayer R., Moreno E. 2013. Mechanisms of cell competition: Themes and variations. J. Cell Biol. V. 200. P. 689.
Li M., He Y., Dubois W., Wu X., Shi J., Huang J. 2012. Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol. Cell. V. 46. P. 30.
Lin T., Lin Y. 2017. p53 switches off pluripotency on differentiation. Stem Cell Res. Therapy. V. 8. P. 44.
Malik S.A., Orhon I., Morselli E., Criollo A., Shen S., Mariño G., BenYounes A., Bénit P., Rustin P., Maiuri M.C., Kroemer G. 2011. BH3 mimetics activate multiple pro-autophagic pathways. Oncogene. V. 30. P. 3918.
Manova K., Tomihara-Newberger C., Wang S., Godelman A., Kalantry S., Witty-Blease K., De Leon V., Chen W.S., Lacy E., Bachvarova R.F. 1998. Apoptosis in mouse embryos: elevated levels in pregastrulae and in the distal anterior region of gastrulae of normal and mutant mice. Dev. Dynam. An Official Publication of the American Association of Anatomists. V. 213. P. 293.
Mathieu J., Detraux D., Kuppers D., Wang Y., Cavanaugh C., Sidhu S., Levy S., Robitaille A.M., Ferreccio A., Bottorff T., McAlister A., Somasundaram L., Artoni F., Battle S., D. Hawkins R., Moon R.T., Ware C.B., Paddison P.J., Ruohola-Baker H. 2019. Folliculin regulates mTORC1/2 and WNT pathways in early human pluripotency. Nat. Commun. V. 10. P. 632.
Moreno E., Basler K., Morata G. 2002. Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature. V. 416. P. 755.
Morselli E., Tasdemir E., Maiuri M.C., Galluzzi L., Kepp O., Criollo A., Vicencio J.M., Soussi T., Kroemer G. 2008. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle (Georgetown, Tex.). V. 7. P. 3056.
Murakami M., Ichisaka T., Maeda M., Oshiro N., Hara K., Edenhofer F., Kiyama H., Yonezawa K., Yamanaka S. 2004. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol. Cell. Biol. V. 24. P. 6710.
Parzych K.R., Klionsky D.J. 2014. An overview of autophagy: morphology, mechanism, and regulation. Antioxid. Redox Signal. V. 20. P. 460.
Possik E., Jalali Z., Nouët Y., Yan M., Gingras M.-C., Schmeisser K., Panaite L., Dupuy F., Kharitidi D., Chotard L., Jones R.G., Hall D.H., Pause A. 2014. Folliculin regulates AMФK-dependent autophagy and metabolic stress survival. PLoS Genetics. V. 10. e1004273. https://doi.org/10.1371/journal.pgen.1004273
Sancho M., Di-Gregorio A., George N., Pozzi S., Sánchez J.M., Pernaute B., Rodríguez T.A. 2013. Competitive interactions eliminate unfit embryonic stem cells at the onset of differentiation. Dev. Cell. V. 26. P. 19.
Schier A.F. 2007. The maternal-zygotic transition: death and birth of RNAs. Science. V. 316. P. 406.
Speidel D. 2015. The role of DNA damage responses in p53 biology. Arch. Toxicol. V. 89. P. 501.
Spits C., Mateizel I., Geens M., Mertzanidou A., Staessen C., Vandeskelde Y., Van der Elst J., Liebaers I., Sermon K. 2008. Recurrent chromosomal abnormalities in human embryonic stem cells. Nat. Biotechnol. V. 26. P. 1361.
Spruce T., Pernaute B., Di-Gregorio A., Cobb B.S., Merkenschlager M., Manzanares M., Rodriguez T.A. 2010. An early developmental role for miRNAs in the maintenance of extraembryonic stem cells in the mouse embryo. Dev. Cell. V. 19. P. 207.
Suvorova, I.I., Knyazeva A.R., Pospelov V.A. 2018. Resveratrol-induced p53 activation is associated with autophagy in mouse embryonic stem cells. Biochem. Biophys. Res. Commun. V. 503. P. 2180.
Suvorova, I.I., Knyazeva A.R., Petukhov A.V., Aksenov N.D., Pospelov V.A. 2019. Resveratrol enhances pluripotency of mouse embryonic stem cells by activating AMФK/Ulk1 pathway. Cell Death Dis. V. 5. P. 61.
Tasdemir E., Maiuri M.C., Galluzzi L., Vitale I., Djavaheri-Mergny M., D’Amelio M., Criollo A., Morselli E., Zhu C., Harper F., Nannmark U., Samara C., Pinton P., Vicencio J.M., Carnuccio R., Moll U.M., Madeo F., Paterlini-Brechot P., Rizzuto R., Szabadkai G., Pierron G., Blomgren K., Tavernarakis N., Codogno P., Cecconi F., Kroemer G. 2008. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. V. 10. P. 676.
Tsukamoto S., Kuma A., Murakami M., Kishi C., Yamamoto A., Mizushima N. 2008. Autophagy is essential for preimplantation development of mouse embryos. Science. V. 321. P. 117.
Wang S., Xia P., Ye B., Huang G., Liu J., Fan Z. 2013. Transient activation of autophagy via Sox2-mediated suppression of mTOR is an important early step in reprogramming to pluripotency. Cell Stem Cell. V. 13. P. 617.
Дополнительные материалы отсутствуют.
Инструменты
Цитология