

Доклады Российской академии наук. Физика, технические науки, 2020, T. 492, № 1, стр. 68-72
МОЛЕКУЛЯРНО-ДИНАМИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ОБЪЕМНОГО МОДУЛЯ УПРУГОСТИ ДЛЯ КРЕМНИЯ И КАРБИДА КРЕМНИЯ
А. В. Уткин 1, *, академик РАН В. М. Фомин 1, 2
1 Институт теоретической и прикладной механики
им. С.А. Христиановича Сибирского отделения Российской академии наук
Новосибирск, Россия
2 Новосибирский государственный университет
Новосибирск, Россия
* E-mail: utkin@itam.nsc.ru
Поступила в редакцию 23.12.2019
После доработки 16.03.2020
Принята к публикации 23.03.2020
Аннотация
Предложена методология нахождения объемного модуля упругости на основании молекулярно-динамического моделирования. При помощи этой методологии было исследовано влияние размера сферического кластера на объемный модуль упругости. Было показано, что, начиная с некоторого критического размера кластера, модуль упругости начинает возрастать по мере уменьшения объема структуры. В качестве объекта исследования были использованы кремний Si и кубическая форма карбида кремния 3C-SiC.
Нанокластеры кремния и карбида кремния широко используются для улучшения механических свойств металлов и керамики. Кроме этого, нанокластеры являются прекрасной добавкой для улучшения механических и термических свойств композитных материалов на основе эпоксидной смолы. Они заполняют полости композита, а также усиливают связи между полимерными цепями. Однако механические свойства композиционных материалов определяются не только объемной концентрацией нанокластеров, но также и их характерным размером [1]. При этом изменение характерного размера нанокластеров может приводить к изменению механических характеристик в десятки раз. Необходимо отметить, что экспериментальное исследование вопроса влияния размера нанокластера на характер физических процессов осложняется масштабами явлений во времени и в пространстве. Таким образом, актуальным становится проведение численных экспериментов, в частности, моделирование методом молекулярной динамики [2, 3].
МЕТОДОЛОГИЯ И ФИЗИЧЕСКАЯ СИСТЕМА
В качестве физической системы рассматривались сферический кластер кремния Si и карбида кремния, имеющего структуру цинковой обманки (кубическая форма 3C-SiC). Диаметр кластеров изменялся в широком диапазоне значений (от 3 до 22 нм для карбида кремния и от 4 до 33 нм для кремния).
Кристаллический кластер, вписанный в сферу определенного радиуса, вырезался из идеального кубического образца, при этом диагональ этого куба, проходящая через начало координат, совпадала с направлением [1, 1, 1 ]. Полученная система имеет не совсем сферическую форму, но для простоты в дальнейшем мы будем использовать термин сферический кластер.
Интегрирование уравнения движения проводилось с использованием скоростной модификации схемы Верле второго порядка точности по временному шагу, а сам шаг по времени в численном расчете составлял 10–16 с. Для численного моделирования был разработан и программно реализован параллельный алгоритм, позволяющий проводить расчеты с использованием графических процессоров (ГПУ) общего назначения компании NVIDIA, поддерживающих технологию CUDA (Compute Unified Device Architecture) [4].
Основным преимуществом метода молекулярной динамики (ММД) при моделировании процессов на микроуровне является то, что ММД не требует привлечения дополнительных моделей, как это делается в механике деформированного твердого тела. Таким образом, в основе предлагаемого подхода лежит закон Ньютона (или теория Гамильтона), при этом термодинамические и физические свойства материалов определяются потенциалом межатомного взаимодействия. Система охлаждалась при помощи метода искусственной вязкости, в результате чего ансамбль атомов в конце процесса охлаждения находился в минимуме потенциальной энергии [5]. При этом начальное состояние системы было близким к нулю (10–7 К). В рамках классической механики при температуре системы, стремящейся к нулю, импульс также стремится к нулю. Метод молекулярной динамики не учитывает нулевые колебания системы.
В представленной работе для моделирования процесса деформирования кремния был использован потенциал Стиллинжера–Вебера [6], а для исследования свойств карбида кремния был использован потенциал, предложенный группой Вашишты [7]. Оба этих потенциала хорошо апробированы для моделирования различных ударно-волновых явлений в рассматриваемых веществах и корректно описывают процессы разрушения на микроуровне. И для кремния, и для карбида кремния потенциал межатомного взаимодействия состоит из суммы двухчастичной и трехчастичной частей:
(1)
$U = \sum\limits_{i < j} {U_{{ij}}^{2}({{r}_{{ij}}})} + \sum\limits_{i,j < k} {U_{{ijk}}^{3}({{r}_{{ij}}},{{r}_{{ik}}})} ,$В случае карбида кремния двухчастичная часть (1) включает в себя члены, отвечающие за пространственное влияние ионов, кулоновские взаимодействия, заряд диполя и ван-дер-ваальсовы взаимодействия:
(2)
$\begin{gathered} U_{{ij}}^{2}\left( r \right) = \frac{{{{H}_{{ij}}}}}{{{{r}^{{{{\eta }_{{ij}}}}}}}} + \frac{{{{Z}_{i}}{{Z}_{j}}}}{r}\exp \left( { - \frac{r}{\lambda }} \right) - \\ \, - \frac{{{{D}_{{ij}}}}}{{2{{r}^{4}}}}\exp \left( { - \frac{r}{\xi }} \right) - \frac{{{{W}_{{ij}}}}}{{{{r}^{6}}}}, \\ \end{gathered} $Трехчастичная часть (1) ответственна за структурные трансформации под действием давления и процесс плавления:
(3)
$U_{{ijk}}^{3}\left( {{{r}_{{ij}}},{{r}_{{ik}}}} \right) = {{R}^{3}}\left( {{{r}_{{ij}}},{{r}_{{ik}}}} \right){{P}^{3}}\left( {{{\theta }_{{ijk}}}} \right),$(4)
$\begin{gathered} {{R}^{3}}\left( {{{r}_{{ij}}},{{r}_{{ik}}}} \right) = {{B}_{{ijk}}}\exp \left( {\frac{\gamma }{{{{r}_{{ij}}} - {{r}_{0}}}} + \frac{\gamma }{{{{r}_{{ik}}} - {{r}_{0}}}}} \right) \times \\ \, \times \Theta \left( {{{r}_{0}} - {{r}_{{ij}}}} \right)\Theta \left( {{{r}_{0}} - {{r}_{{ik}}}} \right), \\ \end{gathered} $(5)
${{P}^{3}}\left( {{{\theta }_{{ij}}}_{k}} \right) = \frac{{{{{\left( {\cos {{\theta }_{{ij}}}_{k} - \cos {{{\bar {\theta }}}_{{ijk}}}} \right)}}^{2}}}}{{1 + {{C}_{{ijk}}}{{{\left( {\cos {{\theta }_{{ij}}}_{k} - \cos {{{\bar {\theta }}}_{{ijk}}}} \right)}}^{2}}}}.$Здесь Bijk определяет прочность связи, ${{\theta }_{{ij}}}_{k}$ – угол, образованный векторами rij и rik, Cijk ${{\bar {\theta }}_{{ijk}}},$ γ – константы, $\Theta \left( {{{r}_{0}} - r} \right)$ – функция Хевисайда, а r0 = 2.9 Å. Значения констант, входящих в трехчастичную часть потенциала межатомного взаимодействия, приведены в статье [7].
С целью экономии вычислительных ресурсов была выполнена модификация двухчастичной части потенциала взаимодействия (2). Потенциал был обрезан на расстоянии rc = 7.35 Å и сдвинут таким образом, чтобы и потенциал, и его производная обращались в нуль на расстоянии радиуса обрезания:
(6)
$\begin{gathered} U_{{ij}}^{{{\text{2shifted}}}}\left( r \right) = \\ \, = \left\{ \begin{gathered} U_{{ij}}^{2}\left( r \right) - U_{{ij}}^{2}\left( {{{r}_{c}}} \right) - \left( {r - {{r}_{c}}} \right){{\left( {\frac{{dU_{{ij}}^{2}}}{{dr}}} \right)}_{{r = {{r}_{C}}}}},\quad r \leqslant {{r}_{c}} \hfill \\ 0,\quad r > {{r}_{c}}. \hfill \\ \end{gathered} \right. \\ \end{gathered} $Для кремния двухчастичная часть потенциала межатомного взаимодействия (1) определяется следующим выражением:
(7)
$\begin{gathered} U_{{ij}}^{2}\left( r \right) = A\left( {B{{{\left( {\frac{\sigma }{{{{r}_{{ij}}}}}} \right)}}^{p}} - {{{\left( {\frac{\sigma }{{{{r}_{{ij}}}}}} \right)}}^{q}}} \right) \times \\ \, \times \exp \left( {\frac{\sigma }{{{{r}_{{ij}}} - r}}} \right)\Theta \left( {{{r}_{0}} - {{r}_{{ij}}}} \right), \\ \end{gathered} $Трехчастичная часть потенциала (1) для кремния определяется как
(8)
$U_{{ijk}}^{3}\left( {{{r}_{{ij}}},{{r}_{{ik}}}} \right) = {{R}^{3}}\left( {{{r}_{{ij}}},{{r}_{{ik}}}} \right){{P}^{3}}\left( {{{\theta }_{{ijk}}}} \right),$(9)
$\begin{gathered} {{R}^{3}}\left( {{{r}_{{ij}}},{{r}_{{ik}}}} \right) = \lambda \exp \left( {\frac{\gamma }{{{{r}_{{ij}}} - {{r}_{0}}}} + \frac{\gamma }{{{{r}_{{ik}}} - {{r}_{0}}}}} \right) \times \\ \, \times \Theta \left( {{{r}_{0}} - {{r}_{{ij}}}} \right)\Theta \left( {{{r}_{0}} - {{r}_{{ik}}}} \right), \\ \end{gathered} $(10)
${{P}^{3}}\left( {{{\theta }_{{ij}}}_{k}} \right) = {{\left( {\cos {{\theta }_{{ij}}}_{k} + \frac{1}{3}} \right)}^{2}}.$Здесь ${{\theta }_{{ij}}}_{k}$ – угол, образованный векторами rij и rik, радиус обрезания r0 = 3.77 Å, $\lambda ,$ ${{\bar {\theta }}_{{ijk}}},$ γ – константы [6].
Объемный модуль упругости является характеристикой, описывающей упругие свойства вещества под действием всестороннего сжатия, и определяется как
(11)
$K = - \frac{{\Delta P}}{{{{\Delta V} \mathord{\left/ {\vphantom {{\Delta V} {{{V}_{0}}}}} \right. \kern-0em} {{{V}_{0}}}}}},$В численном эксперименте проводилось квазистатическое сжатие первоначально охлажденного сферического кластера [4]. В качестве внешних параметров задавалось конечное сжимающее давление и величина приращения давления 0.0001 ГПа на каждом шаге (линейный рост внешнего давления).
При моделировании в рамках ММД необходимо оперировать такими характеристиками, как силы, действующие на атомы системы. Поэтому на каждом шаге интегрирования определялись силы, соответствующие прикладываемому сжимающему давлению и действующие на поверхностные атомы в направлении к центру масс кластера:
где S – площадь поверхности кластера, а NS – число поверхностных атомов.Поверхностные атомы кластера определялись при помощи анализа координационного числа всех атомов системы (у поверхностных атомов кремния и карбида кремния координационное число меньше четырех).
Во время всего процесса сжатия проводилось дополнительное охлаждение кластера при помощи метода искусственной вязкости. В проводимых численных экспериментах в качестве верхнего предельного значения была использована величина давления 1 ГПа. Выбор такой величины позволяет гарантированно вести речь об исследовании процессов в области упругих деформаций.
РАСЧЕТ ОБЪЕМНОГО МОДУЛЯ УПРУГОСТИ
На рис. 1 представлена временная динамика изменения объема кластера для кремния и для карбида кремния. Несмотря на то, что временной интервал приложения внешнего давления был всего 1 пс, последующее изменение объема кластера продолжалось несоизмеримо больший временной интервал.
Рис. 1.
Зависимость изменения объема кластера от времени. а – кремний, начальный диаметр 16 нм, б – кубическая форма карбида кремния, начальный диаметр 14 нм.
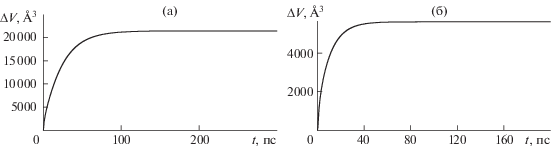
Видно, что объем кластера перестает изменяться и выходит на постоянные асимптотические значения к 80 пс для карбида кремния и в районе 200 пс для кремния. Значительную роль в установлении квазиравновесного состояния сжатого кластера играет его размер. Чем больше размер, тем больше времени требуется сжатой системе для установления конечного объема.
При определении объемного модуля упругости для маленьких кластеров принципиальное значение приобретает корректное определение объема рассматриваемой структуры. При определении объема условно больших кластеров (диаметр превышает 10 нм для карбида кремния и 15 нм для кремния) можно использовать обычное определение объема сферы. При этом радиус определяется как расстояние от центра масс системы до максимально удаленного атома, который, тем не менее, находится в пределах радиуса действия других атомов системы.
Однако по мере уменьшения диаметра кластера использование приближения сферы для определения его объема вносит значительные погрешности в определение объемного модуля упругости. По этой причине необходимо использовать более сложные подходы для определения объема кластера. В представленной работе был использован алгоритм, основанный на триангуляции Делоне и реализованный в свободно распространяемом пакете-анализаторе OVITO [9, 10 ] .
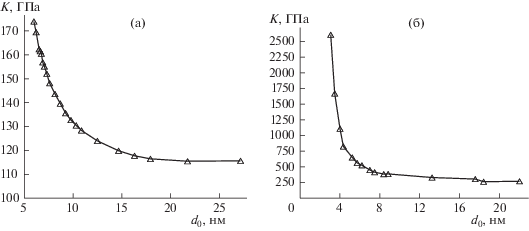
На рис. 2 представлены зависимости объемного модуля упругости от диаметра нанокластера. Зависимости для кремния и для карбида кремния показывают, что объемный модуль упругости K зависит от размера кластера и начиная с некоторого критического значения резко возрастает по мере уменьшения диаметра. Соответственно при увеличении размера значение K выходит на постоянные асимптотические значения, которые составляют: для кремния $K \approx 110$ ГПа, а для карбида кремния $K \approx 260$ ГПа. Полученные в результате молекулярно-динамического моделирования величины несколько превышают экспериментальные значения: для кремния K = 95 ГПа, а для карбида кремния K = 225 ГПа. Это связано с тем, что в качестве физической системы рассматривается идеальная бездефектная структура [6, 7].
ЗАКЛЮЧЕНИЕ
В работе предложена методология нахождения объемного модуля упругости на основании молекулярно-динамического моделирования. С помощью данной методологии были определены объемные модули упругости для кремния Si и для кубической формы карбида кремния 3C-SiC. Для обоих веществ было показано, что начиная с некоторого критического размера кластера объемный модуль упругости начинает возрастать по мере уменьшения объема структуры. Соответственно при увеличении размера структуры объемный модуль упругости стремится к постоянному асимптотическому значению, при этом отличие от экспериментальных данных находится в пределах 15–17%.
Список литературы
Фомин В.М., Филиппов А.А. Теоретико-экспериментальный метод определения упругих характеристик наноматериалов // ДАН. 2019. Т. 489. № 5. С. 469–472. https://doi.org/10.31857/S0869-56524895469-472
Fan J., Chu P K. Silicon Carbide Nanostructures Fabrication, Structure, and Properties, B.: Springer International Publishing, 2014.
Frenkel D., Smit B. Understanding Molecular Simulation: From Algorithms to Applications. Amsterdam: Elsevier Academic Press, 2001.
Utkin A.V. Analysis of Parallel Molecular Dynamics for MPI, CUDA and CUDA-MPI Implementation // Mathematica Montisnigri 2017. V. 39. P. 101–109. WoS: 000419276200008
Golovnev I.F., Golovneva E.I., Fomin V.M. Simulation of Quasistatic Processes in Crystals by a Molecular Dynamics Method // Phys. Mesomech. 2003. V. 6. P. 41–45.
Stillinger F.H., Weber T.A. Computer Simulation of Local Order in Condensed Phases of Silicon // Phys. Rev. B. 1985. V. 31. P. 5262–5271. https://doi.org/10.1103/PhysRevB.31.5262
Vashishta P., Kalia R.K., Nakano A., Rino J.P. Interaction Potential for Silicon Carbide: A Molecular Dynamics Study of Elastic Constants and Vibrational Density of States for Crystalline and Amorphous Silicon Carbide // J. Applied Physics. 2007. V. 101. P. 103515: 1-12. https://doi.org/10.1063/1.2724570
Stukowski A., Albe K. Extracting Dislocations and Non-Dislocation Crystal Defects from Atomistic Simulation Data // Modelling Simul. Mater. Sci. Eng. 2010. V. 18. P. 085001. https://doi.org/10.1088/0965-0393/18/8/085001
Stukowski A. Computational Analysis Methods in Atomistic Modeling of Crystals // J. Minerals, Metals and Materials Society. 2014. V. 66. P. 399–407. https://doi.org/10.1007/s11837-013-0827-5
Дополнительные материалы отсутствуют.
Инструменты
Доклады Российской академии наук. Физика, технические науки