

Доклады Российской академии наук. Химия, науки о материалах , 2021, T. 499, № 1, стр. 3-39
Окислительная С–Н-функционализация аренов: инструмент “зеленой химии” XXI века. Обзор
А. В. Щепочкин 1, Ф. В. Антипин 1, 2, академик РАН В. Н. Чарушин 1, 2, *, академик РАН О. Н. Чупахин 1, 2
1 Институт органического синтеза
им. И.Я. Постовского Уральского отделения Российской академии наук
620219 Екатеринбург, Россия
2 ФГАОУ ВО “Уральский федеральный университет
им. первого Президента России Б.Н. Ельцина”
620002 Екатеринбург, Россия
* E-mail: charushin@ios.uran.ru
Поступила в редакцию 16.03.2021
После доработки 13.08.2021
Принята к публикации 20.08.2021
Аннотация
Впервые предпринята попытка осветить современные окислительные, в том числе каталитические, методологии прямой функционализации аренов, неактивированных к нуклеофильной атаке, за счет замещения атома водорода С–Н-связи, как одного из основных инструментов “зеленой химии”. Приведены примеры построения связей углерод–углерод, углерод–кислород, углерод–азот и углерод–сера с использованием окислителей разного типа, включая соединения гипервалентного иода, а также путем электрохимических и фотохимических превращений.
1. ВВЕДЕНИЕ
Ароматические соединения являются одним из самых обширных классов органических веществ; они широко распространены в природе, входят в состав нефти [1] и каменного угля [2]. Многие соединения ароматического ряда являются крупнотоннажными продуктами нефтехимического синтеза, а также могут быть получены в результате переработки каменноугольной смолы [2]. Они важны для фармацевтической промышленности, синтеза органических красителей, полупроводников, светодиодов и других материалов [3].
Химия ароматических соединений в течение многих десятилетий традиционно основывалась на процессах электрофильного замещения атомов водорода в аренах, позволяющих ввести в ароматическое кольцо атомы галогена, нитро- и сульфогруппы, ацильный и другие электрофильные фрагменты, с дальнейшей функционализацией аренов за счет замещения этих группировок (схема 1) [4, 5]. Важно отметить, что генерирование электрофильных частиц часто требует применения агрессивных реагентов (азотная и серная кислоты, галогены, ацилхлориды, ангидриды кислот и т.д.) и довольно жестких условий, сопровождается образованием большого количества отходов и противоречит принципам “зеленой химии”, включая основополагающий: “лучше предотвратить потери, чем перерабатывать и чистить остатки” [6].
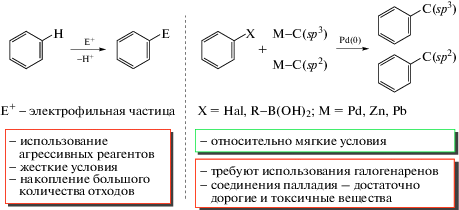
Реакции кросс-сочетания, катализируемые комплексами переходных металлов (именные реакции Хека, Кумады, Стилле, Судзуки–Миуры, Соногаширы и др.) значительно расширили возможности функционализации ароматических соединений, став новым, эффективным инструментом модификации связей С–Х (где Х – галоген) и С–Н в аренах [4, 7]. Следует отметить, в большинстве случаев эти реакции применимы к галогенпроизводным аренов, а палладий и его соединения, используемые в качестве катализаторов, являются дорогими и токсичными, что ограничивает использование данных реакций при создании лекарственных препаратов, компонентов органической электроники и других материалов (схема 1).
Альтернативным подходом к модификации С–Н-связей в аренах являются реакции нуклеофильного ароматического замещения водорода, реализуемые по окислительному или элиминационному механизмам [8]. Сегодня их также квалифицируют как metal-free-процессы нуклеофильной С–Н-функционализации аренов, подчеркивая тем самым возможность протекания реакций в отсутствие переходных металлов в качестве катализаторов [9]. К той же группе химических превращений относятся окислительные кросс-сочетания, сокращенно называемые OCDC-процессами (Oxidative Cross Dehydrogenative Coupling). Все эти методы стремительно развиваются в последние десятилетия и обладают заметными преимуществами в сравнении с классическими реакциями кросс-сочетания с участием галогенаренов. Окислительные кросс-сочетания позволяют создавать связи углерод–углерод и углерод–гетероатом непосредственно из С–Н-производных аренов, без их предварительной функционализации [8, 9]. В качестве окислителей в данных реакциях могут выступать пероксиды, соли меди, серебра, соединения гипервалентного иода, кислород и другие вещества (схема 2) [8–10].

Разновидностью окислительных кросс-сочетаний являются каталитические реакции, которые протекают под действием комплексов переходных металлов и ведут к образованию связи углерод–углерод между двумя арильными остатками за счет окисления атомов водорода двух С–Н-связей сочетаемых аренов. В этих реакциях комплексы металлов выступают как в качестве катализаторов, так и в качестве окислителей, что позволяет относить эти процессы к каталитическим дегидрогенизационным кросс-сочетаниям (Catalytic Dehydrogenative Cross-Coupling) (схема 2) [11].
Важнейшим условием протекания OCDC-реакций является участие окислителя, причем в стехиометрических количествах, а сами CDC-процессы (Cross Dehydrogenative Coupling) имеют много разновидностей, среди которых можно выделить два основных типа превращений.
Первый тип реакций OCDC реализуется при наличии в трехкомпонентной системе “А + В + + окислитель” высокоактивных реакционных партнеров (арена и нуклеофила), которые в результате взаимодействия дают σH-аддукты, с последующим окислением последних внешним окислителем. Классическим примером таких превращений, прочно вошедшим в теорию и практику химии π-дефицитных аренов и гетероаренов, являются реакции нуклеофильного ароматического замещения водорода ${\text{S}}_{{\text{N}}}^{{\text{H}}}$(AO), механизм которых включает две стадии: присоединение нуклеофила и окислительное элиминирование атома водорода в виде катиона (схема 3) [8, 9]. Следует отметить, что нуклеофильные функционализации С–Н-связей в активированных аренах, протекающие в отсутствие катализа металлами, отличаются высокой атомной эффективностью, поскольку они позволяют исключить стадии предварительного введения функциональных групп в исходные реагенты, что отвечает принципу PASE (Pot, Atom, Step Economic) – одному из основных требований “зеленой химии” XXI века.

Второй тип окислительной С–Н-фунционализации аренов – OCDC-процесс в трехкомпонентной системе “А + В + окислитель” с участием партнеров, у которых реакционная способность по отношению друг к другу выражена слабо и, как следствие, образования ковалентной связи между реакционными партнерами на первой стадии не происходит. Для этой ситуации характерны процессы одноэлектронного переноса (SET, single electron transfer) с участием окислителя. Активация арена достигается, как правило, через формирование арильного катион-радикала, который впоследствии и реагирует с нуклеофилом. Для обозначения этого способа активации Зеебахом (Seebach) в 1979 г. был предложен термин “umpolung”, подчеркивающий изменение электронного состояния аренов при формировании из них катион-радикалов [12]. Прием довольно активно используется в органическом синтезе, однако сегодня вместо термина “umpolung” используется более широкое понятие “cation pool” [13], причем генерация катион-радикалов может достигаться как с помощью химических реагентов, так и путем фотохимической или электрохимической активации.
Как уже отмечалось выше, характерной особенностью OCDC-реакций является то, что химический процесс реализуется как трехкомпонентный синтез, поскольку окислитель вводится в систему одновременно с реакционными партнерами. Вполне естественно, что при этом возникает вероятность развития многих, в том числе нежелательных процессов, связанных с окислением как арена, так и нуклеофильного агента, формированием катион-радикалов и радикальных частиц и, как следствие, возможно протекание побочных реакций. Поскольку разные типы трехкомпонентных реакционных систем предполагают принципиально разные механизмы кросс-сочетаний, выбор окислительного агента является исключительно важным [8, 9].
Одной из тенденций развития химии является переход от традиционных методов органического синтеза к электрохимическим [14], применение которых позволяет количественно определить окислительно-восстановительные потенциалы реагентов и промежуточных соединений, что дает возможность осмысленного и рационального выбора окислителя.
Использование электросинтеза позволяет повысить атомную эффективность и минимизировать материальные потери. Так, при анодной активации происходит одноэлектронное окисление арена в катион-радикал, который активно реагирует с нуклеофилом, с последующим окислением радикальных интермедиатов.
Другим способом активации ароматических субстратов для окислительного кросс-сочетания является использование фотохимической активации. Активированный светом фотокатализатор инициирует процессы одноэлектронного переноса, генерируя катион-радикалы аренов, которые участвуют в дальнейших трансформациях [15–17] (схема 4).
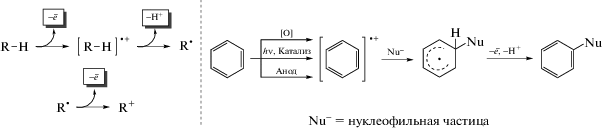
Активными интермедиатами, способными участвовать в реакциях сочетания, являются радикальные частицы, образующиеся в результате элиминирования катиона атома водорода от катион-радикалов; последние способны вступать в реакции радикального присоединения и радикал-радикальные кросс-сочетания.
Обзоры, посвященные нуклеофильной С–Н-функционализации ароматических соединений (т.е. реакциям ${\text{S}}_{{\text{N}}}^{{\text{H}}}$), уделяют значительное внимание OCDC-реакциям первого типа, т.е. превращениям аренов, содержащих электроноакцепторные группировки, а также их гетероароматическим аналогам, в которых за счет влияния гетероатомов в ароматическом кольце создается дефицит электронной плотности [8, 9, 18–21]. И только малая часть этого материала посвящена OCDC-реакциям второго типа, т.е. прямой С–Н-функционализации неактивированных аренов, таких как бензол, нафталин, антрацен и другие полициклические ароматические соединения. При этом в последние годы в литературе появляется все больше данных о прямом синтезе таких промышленно важных соединений, как фенол и анилин, непосредственно из бензола в одну стадию. В качестве примера хотели бы обратить внимание на работу [22], которая посвящена фотоиндуцированному гидроксилированию бензола под действием воды (схема 5).
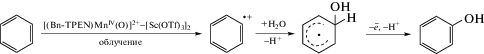
Условия реализации этого превращения являются действительно необычными – реакция протекает при возбуждении комплекса оксида марганца, скоординированного со сложным органическим лигандом и связанного с ионами скандия [(Bn-TPEN)MnIV(O)]2+-[Sc(OTf)3]2, где символом (Bn-TPEN) обозначен N-бензил-N,N,N-трис(2-пиридинил-метил)-1,2-диаминоэтан. При наносекундном облучении лазером в деаэрированной смеси растворителей CF3CH2OH/CH3CN (1 : 1 по объему) комплекс переходит в возбужденное состояние. Далее катион-радикал бензола взаимодействует с водой и трансформируется в фенол с выходом 24%.
Цель настоящего обзора – обобщить современные данные, касающиеся методов окислительной С–Н-функционализации неактивированных к взаимодействию с нуклеофилами аренов (бензол, нафталин, антрацен и другие ароматические системы), ведущих к построению связей С(sp2)–C(sp3), C(sp2)–C(sp2), C(sp2)–гетероатом (O-, N-, S- и др.).
2. ОКИСЛИТЕЛЬНАЯ СН-ФУНКЦИОНАЛИЗАЦИЯ НЕАКТИВИРОВАННЫХ АРЕНОВ: ПОСТРОЕНИЕ СВЯЗЕЙ “УГЛЕРОД–УГЛЕРОД”
2.1. Нуклеофильное алкилирование аренов
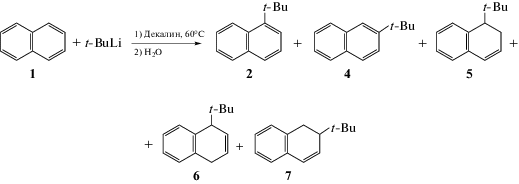
Карбанионы алкиллитиевых, натриевых или калиевых солей в реакциях с ароматическими соединениями вызывают, прежде всего, депротонирование и образование продуктов металлирования аренов. Тем не менее нуклеофильное алкилирование родоначальников ряда ароматических систем, таких как бензол, нафталин и фенантрен, удается реализовать посредством их длительного нагревания с алкиллитиевыми реагентами (t-BuLi, s-BuLi, n-BuLi) в углеводородных растворителях (декалин) (табл. 1). Так, к примеру, реакция нафталина 1 с t-BuLi в декалине протекает при температуре 165°С в течение более 40 ч и приводит к получению моно- и диалкильных производных нафталина 2 и 3 с выходами 30 и 50% соответственно [23] (схема 6).
Таблица 1.
Выходы продуктов нуклеофильного алкилирования аренов
| R–Li | Арен | Выходы продуктов, % | |
|---|---|---|---|
| Моно- | Ди- | ||
| t-Bu– | Бензол | 15 | – |
| n-Bu– | Нафталин | 15 | – |
| s-Bu– | Нафталин | 20 | – |
| t-Bu– | Нафталин | 30–45 | 50–30 |
| t-Bu– | Фенантрен | 50 | – |

Авторы, к сожалению, не дают в своих работах ответов на вопросы, касающиеся причин образования и строения диалкильных производных, а также механизма реакций: какова судьба замещаемых алкильным анионом атомов водорода и за счет чего реализуется окислительный процесс, позволяющий вернуть ароматичность соединениям 2 и 3. Любопытно, что при проведении той же реакции при температуре 60°С, помимо α- и β-трет-бутилнафталинов, образуется смесь трет-бутилдигидронафталинов с общим выходом дигидросоединений примерно 17% (схема 7). Образование дигидросоединений указывает на возможное участие исходного нафталина в окислительных превращениях промежуточных С-аддуктов, но механизм этих превращений до конца не ясен, хотя данные кинетического исследования этой реакции указывают на образование комплекса состава [(RLi)2(ArH)] с последующим присоединением алкиллитиевой соли к нафталину. К сожалению, роль окислителей, а также механизм окислительно-восстановительных превращений обстоятельно не изучались [24].
Реакции нафталина с реактивом Гриньяра также позволяют получить продукты нуклеофильного алкилирования. Например, н-бутилмагний бромид реагирует с нафталином при нагревании до 200°С в декалине. Единственным выделенным продуктом реакции является 1-бутилнафталин 8 (9%), получаемый обработкой интермедиата 9 хлоранилом, который играет роль окислителя [25] (схема 8).

Нуклеофильное метилирование антрацена 10 в реакции с MeLi (5.3 экв.) в ТГФ/Et2O протекает при температуре 50–55°С и выдержке в течение 7 ч. Последующая обработка реакционной массы Pd/C в ксилоле приводит к получению смеси 1-, 2- и 9-метилантраценов 11 : 12 : 13 в процентном соотношении 7 : 1 : 92. При проведении метилирования в диэтиловом эфире при УФ-облучении в присутствии избытка MeLi (70 экв.) в продуктах реакции наряду с метилантраценами 11–13 были обнаружены 9,10-дигидроантрацен 14, а также 1-, 2- и 9-метилдигидроантрацены 15–17, дегидрирование которых в присутствии Pd/C приводит к образованию соединений 11 (3%), 12 (12%) и 13 (19%) [23] (схема 9).

Изучение фотолиза реакционных смесей n-BuLi с антраценом, фенантреном и нафталином показало, что реакционная способность аренов в этих реакциях снижается в ряду антрацен > фенантрен > > нафталин. Бутилирование фенантрена 18 приводит к получению 9-n-бутил-9,10-дигидрофенантрена 19 (27%) вместе с ароматическим производным 20 (30%). Аналогично протекает и реакция n-бутиллития с нафталином 1, ведущая к образованию смеси 1-бутил-1,2-дигидронафталина 21 (20%) и 2-бутилнафталина 22 (10%) [26] (схема 10).

Изопропил-, t-Bu-, бензил- и циклооктади-ениллитиевые соединения были успешно введены в реакцию нуклеофильного присоединения к антрацену в ТГФ. При подкислении реакционной массы были получены дигидросоединения 23–26 с выходами от 27 до 97%. При облучении анионного интермедиата видимым светом или длинноволновым УФ-излучением при температуре –80°С наблюдались элиминирование LiH и образование ароматических продуктов 27, 28 [27] (схема 11). Сам факт нуклеофильного алкилирования антрацена крайне интересен, хотя детально механизм реакций в работах того времени не обсуждался.

Ясно, что в реакциях антрацена и нафталина с алкиллитиевыми производными окислительно-восстановительные процессы, в том числе реакции одноэлектронного переноса, играют важную роль и конкурируют с нуклеофильным присоединением.
Одним из способов инициировать одноэлектронный перенос в реакциях аренов с литий- и магнийорганическими реагентами является использование гексаметилфосфамида (ГМФА) в качестве растворителя. Так, исследования реакции n-BuLi с бифенилом и антраценом в различных растворителях показали, что концентрация ГМФА напрямую влияет на образование радикал-анионных частиц (максимальный выход в диэтиловом эфире достигается при концентрации ГМФА 5.07 М) [28, 29] (схема 12).

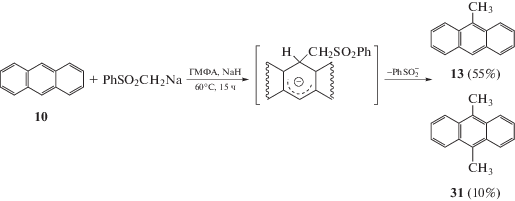
Интересным случаем нуклеофильного метилирования является реакция метилфенилсульфона с антраценом, протекающая в ГМФА в присутствии гидрида натрия [30]. Нагревание реагентов при температуре 60°С в течение 15 ч позволяет получить 9-метилантрацен с выходом 55%, а побочным продуктом является 9,10-диметилантрацен. Авторы полагают, что механизм реакции включает в себя присоединение карбаниона к ароматическому кольцу с последующим гидридным сдвигом и удалением фенилсульфонильного аниона [31] (схема 13).
2.2. Арилирование аренов
Существует большое количество примеров реакций образования связей С(sp2)–C(sp2) между двумя арильными фрагментами, протекающих при катализе переходными металлами. Каталитические системы, которые используются для С–Н-функционализации аренов, очень разнообразны и включают производные таких металлов, как палладий, родий, иридий, кобальт, никель, железо, платина и др., что нашло свое отражение в отдельных обзорах, рассматривающих те или иные каталитические процессы трансформации С–Н-связи в аренах [32–41]. В данном обзоре мы решили сконцентрироваться на окислительных кросс-сочетаниях неактивированных аренов, которые реализуются как в присутствии химических окислителей, так и в электрохимическом варианте.
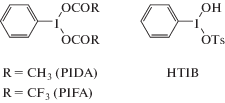
2.2.1. Арилирование с участием соединений гипервалентного иода. Альтернативой использованию катализаторов на основе переходных металлов является применение органических окислителей – производных гипервалентного иода, таких как фенилиодоний диацетат [PhI(OAc)2 – phenyliodine (III) diacetate, PIDA], фенилиодоний бис(трифторацетат) (PhI(OCOCF3)2 – [bis(trifluoroacetoxy)iodo]benzene, PIFA), [гидрокси(тозилокси)иодо]бензол [PhI(OH)OTs – [hydroxy(tosyloxy)iodo]benzene, HTIB) (рис. 1). Использование в качестве растворителей фторированных спиртов – гексафторизопропанола (1,1,1,3,3,3-hexafluoropropan-2-ol, HFIP) и 2,2,2-трифторэтанола (2,2,2-trifluoroethanol, TFE) – значительно увеличивает выходы продуктов окислительных С–Н/С–Н-сочетаний [42].
Гипервалентные производные иода могут выступать в качестве селективных окислителей, осуществляющих одноэлектронный перенос от обогащенных электронами ароматических соединений. Механизм данных реакций включает в себя образование комплекса переноса заряда (КПЗ) между ароматическим субстратом и иодсодержащим реагентом с последующим одноэлектронным переносом и образованием катион-радикала. Дальнейшее взаимодействие катион-радикала с нуклеофилом ароматической природы приводит к образованию продукта С–Н/С–Н-кросс-сочетания (схема 14) [43].

Примером окислительного С–Н/С–Н-сочетания аренов может служить внутримолекулярное сочетание двух арильных фрагментов в 1,3-диарилпропане под действием PIFA. Реакция протекает в трифторэтаноле (TFE), ацетонитриле или дихлорметане при температурах около –40°С, позволяя получать циклические биарильные производные с выходами 25–65%. Использование BF3 ∙ Et2O в тех же условиях позволяет значительно увеличить выходы реакции. Действительно, при циклизации 32 в дихлорметане при температуре –40°С образуется продукт 33 с выходом 91%. Это объясняется тем, что BF3 ∙ Et2O увеличивает способность фенилиодония вызывать одноэлектронный перенос с участием ароматического субстрата за счет координации с трифторацетатными группами PIFA [44, 45] (схема 15).

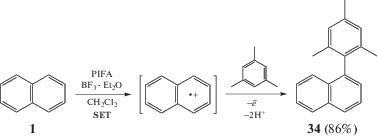
Данная методика применима и для межмолекулярных сочетаний двух аренов. Так, например, реакция нафталина 1 с мезитиленом в присутствии PIFA позволяет получать продукт С–Н/С–Н-сочетания аренов с высокими выходами. Важно отметить, что при этом не образуются продукты С–С-сочетания между двумя одинаковыми аренами, т.е. бинафтилы или бис-мезитилы, так как, по-видимому, комплекс переноса заряда легче образуется с нафталином [46] (схема 16).
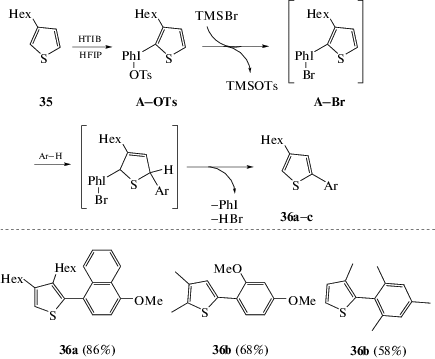
К сожалению, метод С–Н/С–Н-сочетания, основанный на одноэлектронном переносе, не работает в случае реакций аренов с π-избыточными гетероароматическими соединениями (тиофены, пирролы, индолы), поскольку последние подвержены димеризации. Альтернативой этому методу являются синтезы с использованием окислительного потенциала арилиодониевых солей, получаемых конденсацией гетероаренов с соединениями гипервалентного иода. Так, реакция 3-гексилтиофена 35 с гидрокси(тозилокси)иодбензолом (HTIB) проходит по положению С-2 гетероароматического кольца с образованием продукта конденсации А–OTs, далее А–OTs взаимодействует с бромтриметилсиланом (TMSBr) с образованием более реакционноспособной арилиодониевой соли A–Br. Дальнейшая реакция A–Br приводит к присоединению арена в положение С-5 с последующим элиминированием фенилиодида и бромоводородной кислоты [47] (схема 17).
2.2.2. Арилирование под действием дихлордициано-пара-бензохинона. Описан также метод окислительного биарильного внутри- и межмолекулярного С–Н/С–Н-сочетания с использованием дихлордициано-пара-бензохинона (2,3-dichloro-5,6-dicyano-1,4-benzoquinone, DDQ) в качестве окислителя. Так, орто-терфенил 37 гладко трансформируется в трифенилен 38 в растворе дихлорметана при температуре 0°С в присутствии метансульфоновой кислоты и 1 эквивалента DDQ в атмосфере аргона [48]. Аналогично протекает межмолекулярное С–Н/С–Н-сочетание двух аренов 39, содержащих электронодонорные заместители (схема 18).

2.2.3. Электрохимическое арилирование. Электрохимический синтез открывает широкие возможности для развития новых синтетических методов, в которых окислители, способствующие формированию новых С–С-связей, а также отщеплению водорода от С–Н-связей, можно заменить электрохимической активацией этих процессов. При этом отпадает необходимость вводить в ароматические субстраты вспомогательные, легко уходящие группы, что снижает количество отходов, а также стадий синтеза. Таким образом, использование электрохимических методов вписывается в концепцию “зеленой химии”.
Один из электрохимических путей синтеза биарильных производных – С–Н-активация исходных аренов на аноде. Основной проблемой кросс-сочетания двух аренов является селективность реакции, ведущей к несимметричным би-арилам, а нежелательными являются процессы, ведущие к образованию смеси продуктов из-за неселективного окисления промежуточных продуктов. В этих реакциях, помимо возможного гомо-сочетания исходных ароматических субстратов, может наблюдаться избирательное окисление одного из исходных аренов. В большинстве случаев арен, содержащий электронодонорные заместители, легче окисляется, а также является более нуклеофильным, что и приводит к получению продуктов гомо-сочетания. В противном случае, в результате неселективного окисления образуются продукты гомо- и кросс-сочетания, что в лучшем случае приводит к умеренным выходам продукта кросс-сочетания (схема 19) [49].

Контролировать реакцию можно по значениям потенциалов окисления реагентов, которые в идеале должны быть ниже, чем у конечных продуктов.
Среди реагентов, которые катализируют окислительные сочетания аренов, особое место принадлежит соединениям Mo(V) [50]. При использовании молибденового анода в среде гексафторизопропанола на поверхности электрода формируется слой высоковалентного молибдена, за счет чего эта активная анодная система может заменить молибденовые реагенты в реакциях окислительного сочетания (схемы 20, 21).
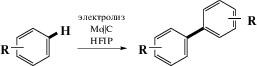

Первые примеры электрохимических гомо-сочетаний производных ди- и триметоксибензолов были получены еще в начале 1930-х годов. Показано, что при анодном окислении этих веществ образуются соответствующие продукты С–Н/С–Н-гомо-сочетаний с выходами до 85% [51].
На основе результатов данных исследований разработан метод анодного окисления замещенных анизолов, который использовали для получения соответствующих бифенилов. При этом наилучшие результаты были достигнуты при использовании в качестве электролита смеси дихлорметана и трифторуксусной кислоты (2 : 1 по объему). Предполагается, что трифторуксусная кислота стабилизирует образующиеся катион-радикалы. Этой же группой исследователей осуществлен первый синтез трифениленов путем анодного окисления вератролов [52] (схема 22).

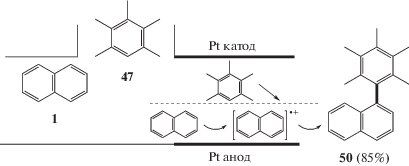
Первые примеры электрохимических кросс-сочетаний были описаны в начале 1970-х годов. Сочетание нафталина с пентаметилбензолом было проведено в неразделенной электрохимической ячейке с выходом 64%. Селективность этой реакции можно объяснить сравнительной легкостью образования катион-радикала нафталина [53] (схема 23).

Кросс-сочетание нафталина с пентаметилбензолом позднее было усовершенствовано. Если проводить анодное окисление нафталина при низких температурах, то полученный катион-радикал остается стабильным достаточно долго. Второй арен, а именно пентаметилбензол, добавляется к полученному интермедиату на второй стадии, и смесь нагревают до комнатной температуры. Благодаря разделению этих двух стадий электрохимического окисления во времени и пространстве достигается селективное образование продуктов кросс-сочетания и исключается избыточное окисление. Так называемый метод “cation pool” позволяет получать несимметричные биарилы электрохимическим окислением и обеспечивает высокую селективность образующихся продуктов с выходом около 90% [54] (схема 24).
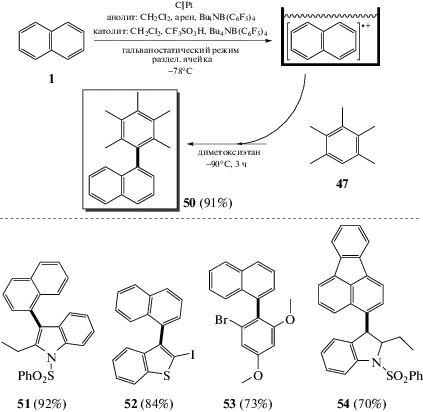
Сообщалось об анодном кросс-сочетании двух аренов с использованием параллельного ламинарного потока в микрореакторе, а также о реакции нафталинов и алкилбензолов, в частности пентаметилбензола и нафталина. При этом применялась аналогичная концепция отделения стадии окисления арена от стадии образования связи С–С, которая уже была исследована в методе “cation pool”. Через первое входное отверстие раствор, содержащий нафталин, подается в микропоточный реактор, тогда как во второй вход подается раствор другого арена – партнера по реакции кросс-сочетания. Из-за образования ламинарного потока “жидкость–жидкость” второй арен будет защищен от окисления, что делает возможным селективное окисление нафталина на аноде с образованием реакционноспособных катион-радикалов. Данный прием позволяет увеличить выход продукта с 49 до 85%, по сравнению с обычной электрохимической ячейкой [55], (схема 25).
Изучение С–Н/С–Н-сочетаний в ряду ароматических соединений фенольного ряда показало, что помимо гомо-сочетаний могут осуществляться реакции кросс-сочетания, что наблюдалось, к примеру, при попытках синтезировать бифенилы из производных гваякола. Вместо ожидаемого С–С-сочетания в орто-положение к гидроксильной группе в результате реакции были получены преимущественно орто-/мета-связанные биарилы [56] (схема 26).

Для этих превращений был предложен следующий механизм реакции (схема 27). Реакция начинается с образования на аноде феноксильных радикалов I, которые далее подвергаются атаке вторым ареном. Промежуточные соединения II и III окисляются на аноде с элиминированием атома водорода в виде катионной частицы и получением биарильного продукта. Среди электродов наиболее эффективным для данного процесса оказался допированный бором алмазный электрод (boron doped diamond electrode, BDD), а селективность данного процесса обеспечивается использованием 1,1,1,3,3,3-гексафторпропан-2-ола в качестве растворителя. Последний способен стабилизировать радикалы и, в то же время, остается устойчивым в условиях электрохимического окисления.
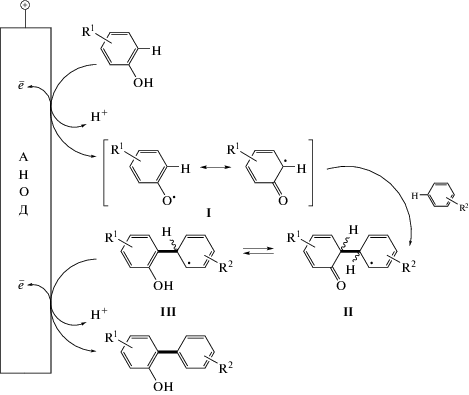
Этот факт можно объяснить, если принять во внимание, что в полярных растворителях фенолы сольватируются лучше, чем арены, из-за способности фенолов образовывать водородные связи. Следовательно, фенолы сильно экранированы чистым HFIP, что предотвращает атаку на генерируемые радикалы. Ареновый субстрат при этом селективно окисляется и подвергается нежелательным реакциям гомо-сочетания. Метанол действует как основание при добавлении в раствор HFIP: он не только ослабляет сольватацию фенолов, но также облегчает их депротонирование за счет взаимодействия посредством водородных связей. Это приводит в некоторых случаях к сдвигу потенциалов окисления, создавая подходящие пары для селективных анодных кросс-сочетаний.
Помимо несимметричных производных гваякола, было достигнуто кросс-сочетание фенолов с замещенными аренами с выходами до 69% в неразделенной ячейке [57, 58].
2.3. Другие С–С-сочетания, ведущие к образованию связи С(sp2)–C(sp2)
Одним из синтетических путей образования связи С(sp2)–C(sp2) являются реакции кросс-сочетания аренов или гетероаренов с алкинами. В данной главе приведены примеры превращений, отличных от классических кросс-сочетаний Хека, Судзуки, Соногаширы и других именных реакций, позволяющих построить С–С-связь между углеродом С(sp2) и неактивированным ареном. Сообщалось, к примеру, о катализируемой палладием реакции алкинов 56 с бензолом 32, приводящей к С–Н-функционализации бензола путем его алкенилирования в присутствии CF3COOH. В данную реакцию легко вступают не только бензол, но и его производные, содержащие электронодонорные группировки (схема 28).

Механизм превращений включает в себя генерацию электрофильной частицы IV, а именно трифторацетатного палладий-катиона, который способен реагировать по двум возможным путям (схема 29). Путь А – взаимодействие IV с ареном I – приводит к арилпалладиевому комплексу V, который далее вступает в реакцию с алкином II с получением интермедиата VII. Альтернативный путь формирования VII заключается в реакции IV с тройной связью алкина II, ведущей к образованию соединения VI, с последующим электрофильным замещением в арене I. На последнем этапе превращений группа Pd(O2CCF3)+ в структуре VII замещается протоном с образованием конечного продукта алкенилирования III.
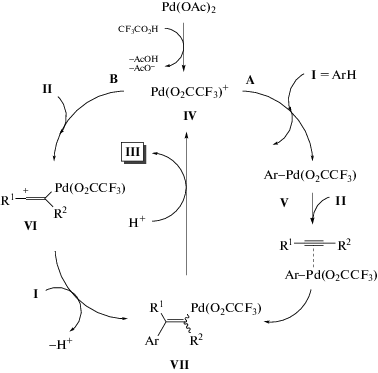
Полагают, что арены, содержащие электронодонорные заместители, реагируют по пути B в мягких условиях (при комнатной температуре) с образованием продуктов анти-присоединения к тройной связи. В то же время бензол или арены с функциональными группами, способные координировать атомы палладия в орто-положение, реагируют при повышенных температурах по пути А [59–62].
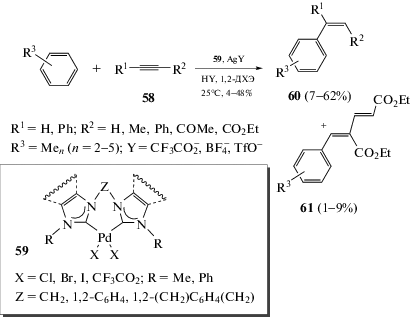
Интересным направлением С–Н-функционализации аренов являются превращения с участием карбеновых комплексов Pd. Имеются данные об успешном алкенилировании полиметилбензолов хелатирующими дикарбенпалладиевыми комплексными катализаторами 59, что приводит к получению соединений 60 и 61 (схема 30) [63, 64].
Сложный палладиевый комплекс 63 был использован в качестве катализатора стереоселективного алкенилирования полиалкиларенов (схема 31) [65].

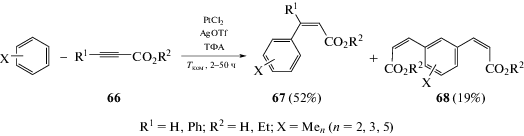
Гидроарилирование пропиновых кислот и их производных аренами, содержащими донорные заместители, протекает при катализе соединениями Pt(II) в присутствии трифторуксусной кислоты и приводит к образованию соединений 67 с выходами до 95%, наряду с побочными бис-замещенными продуктами 68 (схема 32). Увеличение температуры реакции приводит к повышению конверсии алкилпропиноатов и сопровождается гидролизом сложных эфиров 67 и 68 до соответствующих кислот [66].
2.4. Ацилирование
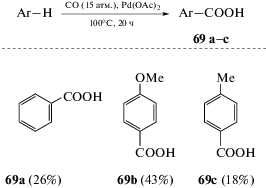
Окислительное карбоксилирование неактивированных аренов впервые было проведено под действием CO в присутствии ацетата палладия [67]. Реакция проводилась при температуре 100°С в течение 15 ч, давление CO составляло 15 атм. В качестве ароматических субстратов были использованы бензол, толуол и анизол. В реакции с бензолом выход бензойной кислоты составил 26%. В случае замещенных бензолов карбоксилирование протекало в пара-положения [68] (схема 33).
Роль окислителя в данных превращениях до конца не выяснена. На первый взгляд, для проведения реакции присутствие кислорода не является обязательным, однако в поисках оптимальных условий использовались окислители, такие как t-BuOOH. Также был расширен и круг ароматических субстратов. К примеру, проведено карбоксилирование нафталина с получением нафтойной кислоты с выходом 25%.
Наилучшие результаты были достигнуты при проведении карбоксилирования в мягких условиях (давление 1 атм, комнатная температура) в присутствии трифторуксусной кислоты и использовании в качестве окислителя K2S2O8 [69] (схема 34).

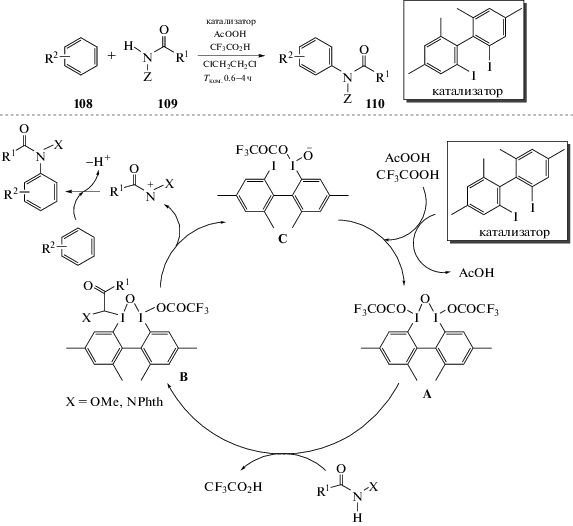
Предполагаемый механизм реакции приведен на схеме 35. В начале каталитического цикла формируется электрофильная частица Pd+(OCOCF3)2II за счет взаимодействия Pd(OAc)2I и трифторуксусной кислоты. Затем электрофильная атака Pd+(OCOCF3)2 на бензольное кольцо приводит к формированию арилпалладиевого интермедиата, который претерпевает внедрение группы CO с образованием арилпалладия (II) III. Последующее восстановительное элиминирование приводит к получению Pd(0) и ангидрида IV, который реагирует с трифторуксусной кислотой с образованием Ar–COOH V и (CF3CO)2O. Pd(0) повторно окисляется K2S2O8 до Pd(II), возобновляя каталитический цикл. Данная реакция протекает в более мягких условиях, чем в обычной системе Pd(OAc)2 : AcOH, что можно объяснить образованием более активных электрофильных частиц Pd+(OCOCF3)2 при использовании трифторуксусной кислоты, по сравнению с Pd+(OCOCH3).

Другим способом введения карбоксильной группы в ароматическое ядро являются реакции аренов с углекислым газом, протекающие в присутствии катализаторов на основе Rh. Одним из примеров подобных реакций является карбоксилирование бензола и толуола с использованием комбинации реагентов, включающей в свой состав соединение родия [RhCl(dcype)]2, где (dcype) – 1,2-бис(дициклогексилфосфино)этан, и метильное производное алюминия в качестве метилирующего агента (схема 36) [70].

Реакция предположительно начинается с генерации 14-электронного метилродиевого (I) комплекса А путем трансметилирования [RhCl(dcype)]2 соединениями алюминия. Внедрение комплекса А в C–H-связь арена приводит к получению фенил(гидридо)(метил)родия (III) B. Элиминирование метана из структуры B позволяет получить реакционноспособный 14-электронный комплекс С, вступающий во взаимодействие с углекислым газом и приводящий в результате этой реакции к образованию комплекса D, который трансметилируется метилалюминием с регенерацией комплекса А.
Электрохимическое карбоксилирование полициклических ароматических углеводородов может быть также осуществлено под действием углекислого газа под высоким давлением, однако этот процесс ведет не к замещению атома водорода С–Н-связи в аренах, а к введению двух карбоксильных остатков и к потере ароматичности. Простой и эффективный электросинтез позволяет получать исключительно транс-дикарбоновые кислоты с выходом до 90% [71] (схема 37).

2.5. Цианирование
Разработка методов введения цианогруппы в производные аренов представляет собой актуальную задачу с учетом того, что ароматические нитрилы широко используются в синтезе кислот, альдегидов, аминов, амидов и других производных аренов. Классическими примерами подобных превращений являются реакции Зандмайера и Розенмунда–фон Брауна, в которых используются токсичные цианистые соединения Cu(I); кроме того, эти процессы требуют достаточно жестких условий. Альтернативой является окислительное цианирование, катализируемое переходными металлами, позволяющее осуществлять прямую функционализацию C–H-связи в аренах [72].
Большое число исследований по цианированию ароматических соединений касается прямого замещения атома водорода С–Н-связи в аренах. К примеру, окислительное цианирование неактивированных аренов гладко протекает под действием трифлатов арил(циано)иодония 72 в качестве источников цианогруппы и ацетата Fe(II) в качестве окислителя (схема 38) [73].

Оптимизация условий реакции выявила, что именно использование иодониевой соли 72 и Fe(OAc)2 является удачным сочетанием реагентов. В данных условиях ряд арилцианидов был получен с выходами от 48 до 87%.
Показана важная роль катион-радикалов в реакциях прямого C–H-цианирования замещенных бензолов. Отметим, что катион-радикалы аренов генерируются путем фотоиндуцированного переноса электрона к акридиниевому катализатору 74 (схема 39). Это превращение не требует наличия направляющей группы в аренах и протекает селективно в пара-положение монозамещенных бензолов [74].
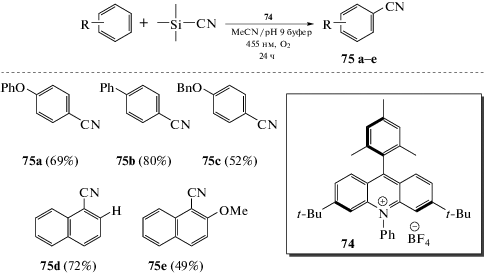
Реакции цианирования ароматических соединений сильно выигрывают при проведении окислительного процесса в электрохимическом варианте, поскольку нуклеофильные цианиды доступны и многочисленны, тогда как источники цианогруппы электрофильной природы требуют особых условий обращения. Электрохимическое сочетание NaCN с аренами приводит к образованию бензонитрилов, и эту реакцию можно проводить при постоянном токе, используя простую двухэлектродную установку с неразделенной ячейкой. Первые работы в этом направлении, посвященные цианированию метиланизолов и алкилнафталинов, были опубликованы в начале 70-х годов прошлого века (схема 40).
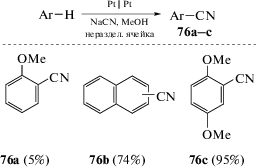
Помимо цианида калия в метаноле в этих реакциях применяли цианид тетраметиламмония в ацетонитриле, а в качестве ароматических субстратов использовали бензол, нафталин, антрацен и другие обогащенные электронами арены. К сожалению, в данных условиях реакции цианирования протекают с низким или умеренным выходом [75] (максимально 45%).
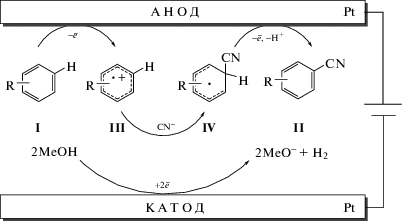
Работы по цианированию аренов, инициированные в прошлом веке, сегодня дополняются новыми данными по механизму превращений, который включает ступенчатое анодное окисление арена до катион-радикала III, нуклеофильную атаку последнего цианидом, что приводит к циклогексадиенильному радикалу IV, который далее окисляется в соответствующий бензонитрил [76] (схема 41).
3. ОКИСЛИТЕЛЬНАЯ С–Н-ФУНКЦИОНАЛИЗАЦИЯ НЕАКТИВИРОВАННЫХ АРЕНОВ: ПОСТРОЕНИЕ СВЯЗЕЙ УГЛЕРОД–ГЕТЕРОАТОМ
В данной главе рассматриваются реакции построения связей углерод–гетероатом с ароматическим кольцом в ряду неактивированных аренов. Поскольку обзор посвящен реакциям окислительного кросс-сочетания, авторы посчитали уместным привести несколько наиболее интересных примеров подобных превращений, катализируемых комплексами с участием переходных металлов. В большинстве из них комплексы выступают в качестве окислителей, что дает право относить их к окислительным дегидрогенизационным кросс-сочетаниям (Oxidative Cross Dehydrogenative Coupling), рассмотрение которых является одной из целей данного обзора.
3.1. Построение связи С–О
Прямое гидроксилирование ароматических соединений может быть осуществлено путем свободнорадикального процесса с использованием ионов железа и перекиси водорода, т.е. реактива Фентона. В этой реакции гидроксильный радикал образуется в результате взаимодействия перекиси водорода с железом (II). Активные гидроксильные радикалы реагируют с ароматическим соединением с образованием гидроксициклогексадиенильного радикала, который далее окисляется под действием ионов железа (III) и отщепляет протон с образованием соответствующего фенола 77 (схема 42).
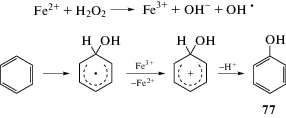
К сожалению, низкие выходы фенола (21%) и значительные объемы образования бифенила (24%) в качестве побочного продукта ограничивают практическое применение реакции Фентона.
Как селективность по фенолу, так и общий выход могут быть улучшены при использовании других катализаторов. Действительно, в присутствии меди (II) выход фенола увеличивается до 57%, а соотношение фенола к бифенилу – до 140 : 1. Аналогичные результаты были получены с окислительной системой пероксид–сульфат железа (II) (схема 43). Дальнейшая оптимизация условий реакции Фентона с применением в качестве лиганда 1-оксида пиразин-3-карбоновой кислоты приводит к увеличению выхода фенола до 78% [77]. Несмотря на значительное улучшение, выходы фенола все еще недостаточно высоки, чтобы иметь широкое практическое применение. Катализаторы на основе переходных металлов, такие как комплексы пероксованадия (V) и никель–ванадиевые (V) оксидные соединения, также оказались эффективными в качестве катализаторов гидроксилирования бензола [78].
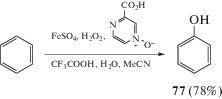
Прямое превращение ароматических соединений в фенолы путем гидроксилирования в присутствии кислорода/воздуха является привлекательным методом синтеза фенолов, однако, несмотря на многие усилия, в ходе которых в качестве катализаторов использовались соединения различных переходных металлов (железо, палладий, медь, рений, никель или платина) [79–85], его практическое применение сдерживается умеренными выходами. Примером является жидкофазное каталитическое окисление бензола в мягких условиях на медьсодержащих цеолитных катализаторах с использованием молекулярного кислорода и аскорбиновой кислоты в качестве окислителя и восстановителя соответственно (схема 44).
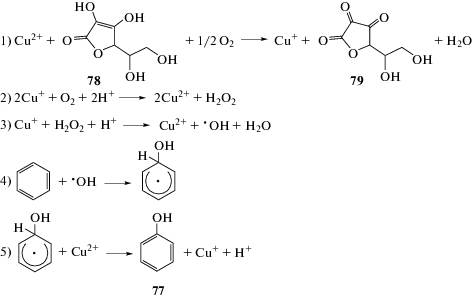
Установлено, что фенол образуется как единственный продукт окисления, причем его выход увеличивается при одновременном увеличении количества Cu и аскорбиновой кислоты [86–88].
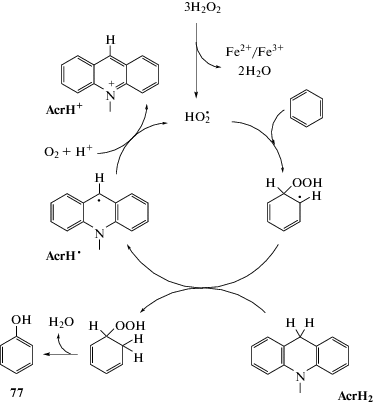
Окислительное гидроксилирование бензола, ведущее к образованию фенола, наблюдалось также под действием молекулярного кислорода (O2) при участии 10-метил-9,10-дигидроакридина (AcrH2) в качестве аналога NADH (nicotinamide adenine dinucleotide, никотинамидадениндинуклеотид) и каталитических количеств Fe(ClO4)3 (схема 45). Каталитическое окисление бензола начинается с образования H2O2 из AcrH2, O2 и H+. Гидропероксильный радикал (HO$_{2}^{\centerdot }$) получают из H2O2 в присутствии окислительно-восстановительной пары Fe3+/Fe2+. HO$_{2}^{\centerdot }$ реагирует с бензолом, что приводит к сигма-аддукту радикальной природы, который отрывает атом водорода от AcrH2 с образованием соответствующего гидропероксида и акридинильного радикала (AcrH•) [89].
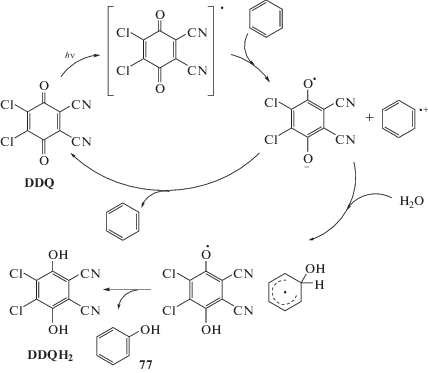
Окислительное гидроксилирование бензола кислородом обычно требует применения гетерогенных катализаторов, работающих при высоких температурах. Вместе с тем описано фотохимическое гидроксилирование бензола в присутствии 2,3-дихлор-5,6-дициано-п-бензохинона (DDQ) в качестве окислителя и воды, которое реализуется при облучении видимым светом и ведет к получению фенола с выходом 99% и DDQH2 (cхема 46). Реакция протекает через промежуточное образование катион-радикала бензола за счет переноса электрона от бензола к DDQ [90, 91].
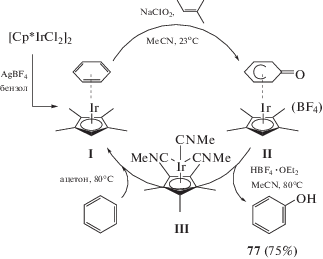
Возможно также гидроксилирование C−H-связи в ароматических соединениях за счет активации последних через η6-координацию с комплексом иридия (III). Механизм реакции представлен на схеме 47. Комплекс I легко может быть получен из коммерчески доступного [Cp*IrCl2]2 под действием AgBF4. При добавлении NaClO2 и 2-метил-2-бутена в качестве поглотителя HOCl, соединение I превращается в η5-феноксокомплекс II. Превращение I в II − это трансформация связи C−H в аренах в связь C−O. После протонирования соединения II и нагревания при 80°C в ацетонитриле фенол 77 выделяют с выходом 75%, а трис-ацетонитрильный комплекс III − с выходом 85%. Иридиевый комплекс I можно регенерировать и использовать в новом каталитическом цикле путем превращения соединения III в комплекс I при нагревании в смеси бензола и ацетона 1 : 1 по объему (выход 81%) [92].
Еще одним примером окислительного гидроксилирования ароматической связи С−Н является комбинированное применение фотокатализа и кобальтового катализатора. Механизм реакции представлен на схеме 48. В качестве фотокатализатора использованы соли N-метилхинолиния (PC+). Генерируемое облучением возбужденное состояние фотокатализатора (PC•+) вызывает одноэлектронный перенос от молекулы бензола с образованием соответствующего катион-радикала и радикала фотокатализатора (PC•). Образовавшиеся соединения могут участвовать в переносах электрона для получения Co(II) и фотокатализатора в основном состоянии (PC+), завершая, таким образом, цикл фотокатализа. Катион-радикал бензола реагирует с анионным нуклеофилом (HO–) с образованием циклогексадиенильного радикала. Этот радикальный аддукт может переносить электрон на Co(II), образуя Co(I) и циклогексадиенильный катион, который далее трансформируется в фенол. Два электрона, освобождающиеся в каждом цикле катализа, используются для восстановления двух протонов с образованием молекулы H2 и, отщепляясь, они снова генерируют Co(III), завершая каталитический цикл [93].

Анодное окисление мезитилена в электролите на основе СН3СN, Н2О и Н2SО4 протекает более сложно, сопровождается перегруппировкой и ведет к образованию гидрохинона 81 с выходом 57% (схема 49) [94].

Моногидроксилирование аренов также возможно, если анодное окисление вести в присутствии трифторуксусной кислоты с последующим гидролизом трифторацетата, в результате которого освобождается фенол 77. С помощью этого метода удалось получить фенол из бензола с выходом 67% (схема 50), а также гидроксилировать различные производные бензола [95].

3.2. Построение связи C–N
Окислительное кросс-сочетание ароматических соединений с аминами (или амидами) представляет собой один из наиболее проработанных вариантов прямой C–H-функционализации аренов. Впервые каталитическое построение связи С–N с ароматическим кольцом в реакциях аминирования (или амидирования) аренов было осуществлено Бухвальдом (Buchwald) с использованием ацетата палладия и комбинированного окислителя, включающего O2 и Cu(OAc)2 (схема 51). Примером внутримолекулярного построения связи С–N является метод получения N-ацилкарбазолов, в котором использование в качестве окислителя PhI(OAc)2 позволило осуществить эту реакцию при комнатной температуре. Кроме палладиевых катализаторов могут использоваться комплексные соединения меди или железа [96].
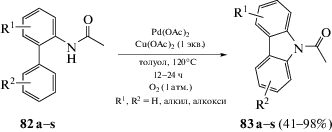
Использование направляющих групп, связанных с атомом азота, служит эффективным приемом при проведении реакций внутримолекулярного C–H/N–H-кросс-сочетания. К примеру, описаны палладий-катализируемые C–H/N–H-кросс-сочетания с использованием пиколинамидов в качестве направляющей группы (схема 52) [97, 98]. Реакция проводится в мягких условиях и применима для широкого ряда функциональных заместителей.

Пиколинамидная направляющая группа была использована также в медь-катализируемом внутримолекулярном С–Н-амидировании при получении карбазолов 87 (схема 53) [99]. Эти реакции обладают большой практической ценностью и затрагивают широкий круг аренов.

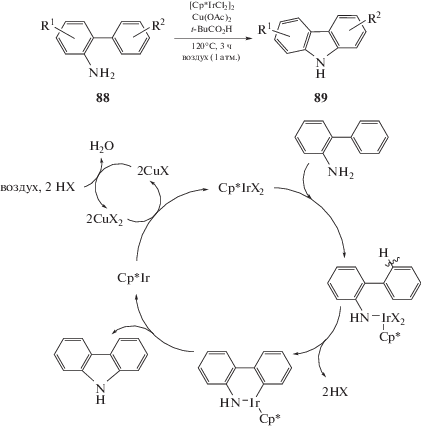
Свободные аминогруппы редко вступают в подобные металл-катализируемые реакции из-за координации самой аминогруппы с переходными металлами. Тем не менее в литературе имеются примеры, когда в реакции окислительного каталитического аминирования удается ввести свободную аминогруппу в присутствии иридиевого катализатора [100]. Данная реакция позволяет получить широкий ряд карбазолов из 2-аминобифенилов в одну стадию. Аналогичное превращение было осуществлено с использованием Pt/C в качестве катализатора, но в более жестких условиях – при температуре выше 250°С. Указанный на схеме механизм предполагает включение иридия по связи С–Н, участие аминогруппы в качестве направляющей и последующее формирование С–N-связи (схема 54) [101].
Осуществлен синтез фенантридинов с использованием медных катализаторов (схема 55) [102, 103]. Показано, что в результате медь-катализируемой реакции 2-азидо-2-(бифенил-2-ил)замещенного этилацетата фенантридин 92 образуется через стадию формирования медь-иминиевого интермедиата. Альтернативный путь получения фенантридинов 93 включает в себя реакцию 2-цианобифенила с реактивом Гриньяра с последующим образованием медь-иминиевого комплекса.

Сообщалось также о медь-катализируемом С–Н-амидировании аренов под действием N-тозилоксикарбаматов (схема 56) [104].

Разработан метод фотокатализируемого С–Н-аминирования простых аренов при комнатной температуре [105]. Ключевой стадией является формирование фталимидного радикала из N-ацилоксифталимида, инициируемое иридиевым фотокатализатором (схема 57). Наличие в аренах электронодонорных заместителей обеспечивает орто-/пара-ориентацию, а электроноакцепторных – направленность реакции в мета-положение.
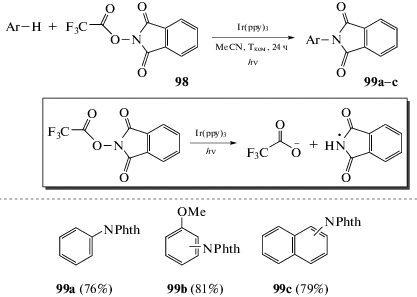
Разработан метод C–N-амидирования толуола в присутствии PhI(OAc)2 в качестве окислителя [106], позволяющий контролировать хемоселективность реакции, направляя ее по sp2-атому углерода, либо по sp3-атому углерода боковой цепи (схема 58). Так, фталимид реагирует с толуолом по ароматическому кольцу, а N-(фенилсульфонил)-бензосульфамид – с атомом углерода метильной группы.
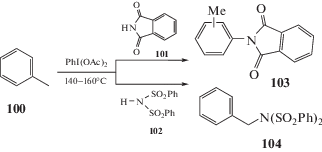
Обнаружено, что добавление Pd(OAc)2 в данную систему позволяет провести амидирование толуола в мета- или пара-положение, а катализ соединениями золота способствует селективному введению аминогруппы исключительно в пара-положение (схема 59) [107–110].

Вышеописанный метод был усовершенствован путем добавления каталитических количеств арилиодидов в присутствии стехиометрических количеств надуксусной кислоты. В ряду монозамещенных бензолов реакция протекает в орто-/пара-положения. Механизм реакции, показанный на схеме, включает следующие стадии: (1) соединение гипервалентного иода (III) A генерируется in situ, (2) A претерпевает замещение лиганда с получением соединения B, (3) формируется нитрениевый ион, который через интермедиат С регенерирует А. Электронодефицитный нитрениевый ион атакует арен с получением продукта амидирования (схема 60) [111].
Показана возможность проведения ферроцен-катализируемого имидирования аренов под действием N-сукцинимидильного пероксиэфира (succinimidyl perester, NSP) в качестве источника имидильных радикалов (схема 61) [112]. Ферроцен в данной реакции играет роль переносчика электрона, участвуя в генерации имидильных радикалов.

Проведено С–Н-амидирование аренов с использованием N-фтор-N-(фенилсульфонил)бензосульфонамида (N-fluorobis(benzenesulfon)-imide, NFSI) в присутствии бромида меди в качестве катализатора и 6,6'-диметилбипиридина в качестве лиганда (схема 62) [113]. Диметильные группы в 6,6'-положениях лиганда играют ключевую роль в катализе данной реакции. Хотя механизм полностью не изучен, в качестве реакционноспособного интермедиата предполагается имидильный радикал. Эта реакция применима к широкому кругу субстратов, включая полициклические арены.

Поскольку реализация прямого анодного аминирования аренов под действием аммиака или первичных аминов сталкивается с большими трудностями, в том числе связанными с низкими потенциалами окисления ожидаемых продуктов аминирования, широкое развитие получили непрямые методы электрохимического аминирования. Примером использования подобных методов является электрохимический вариант реакции Зинке, протекающей через раскрытие цикла [114] (схема 63). При этом положительный заряд пиридиниевого интермедиата эффективно защищает атом азота от окисления. Также данный метод обладает высокой селективностью, позволяя получать исключительно продукты моноаминирования. Данный метод применим к аренам, содержащим различные функциональные группы [115, 116].

Позднее метод был адаптирован к ряду других неактивированных аренов за счет использования в качестве анода допированного бором электрода, что, в частности, позволило провести электрохимическое аминирование нафталина (схема 64) [117, 118].

Показана также возможность проводить непрямой электролиз с использованием катализаторов на основе переходных металлов. Так, аминирование бензола и анизола протекает в присутствии редокс-пары Ti(IV)/Ti(III), которая служит медиатором катодного процесса (схема 65) [119].

3.3. Построение связей С–Р и С–S с ароматическим кольцом
Осуществлен синтез метиларилтиоэфиров из аренов и соединений серы, в основе которого лежит электрохимическое окисление диметилдисульфида при постоянном потенциале в разделенной ячейке (схема 66) [120, 121]. Реакция генерируемого таким образом электрофильного агента с аренами протекает региоселективно и приводит к продуктам монозамещения.

Аналогичным образом синтезированы диарилтиоэфиры с использованием “cation pool”, генерируемых электрохимически катионов Ar–S+, которые вводят в реакции с аренами (схема 67) [122].
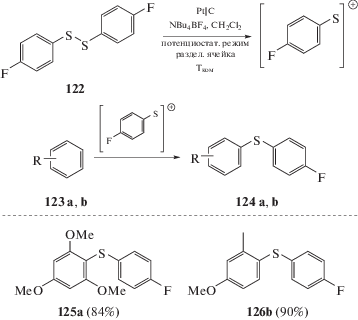
Разработан также метод получения диарилтиоэфиров из ароматических тиолов и катехолов, в котором использован проточный электрохимический микрореактор, причем выходы по току были намного выше, чем в ячейке периодического действия (схема 68) [123, 124].

Ключевой стадией прямого электрохимического тиоцианирования анизола является образование на аноде тиоцианогена (схема 69) [125, 126]. В условиях постоянного потенциала пара-тиоцианоанизол был получен с выходом 77%. Разработанный метод также применим для тиоцианирования замещенных анизолов, толуола и производных анилина.

Электрохимическое арилирование соединений фосфора – тема, редко освещаемая в литературе. Недавно описано успешное электрохимическое фосфорилирование 2-фенилпиридина (схема 70) [127, 128] с применением Pd(OAc)2 в качестве медиатора, в результате которого был получен продукт фосфорилирования 132 с выходом 78%.

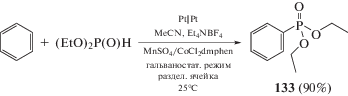
Осуществлено электрохимическое фосфорилирование бензола с использованием электрохимической установки в присутствии катализаторов на основе переходных металлов. Были использованы комплексы и соли со степенью окисления металла (II), окисляемые электрохимическим путем до степени окисления (III). Так, использование комбинации MnSO4 и комплекса CoCl2 c 2,9-диметил-10-фенантролином (CoCl2dmphen), в системе MeCN/AcOH (2 : 1), позволяет получить продукт фосфорилирования 133 с выходом 90% (схема 71) [129].
4. ЗАКЛЮЧЕНИЕ
В рамках краткого обзора, касающегося весьма обширной темы прямой С–Н-функционализации ароматических соединений, мы попытались привлечь внимание исследователей к сравнительно малоизученному разделу, связанному с введением реагентов формально нуклеофильной природы (карбанионов, аминов, ариламинов, гидроксидов, фенолов, цианидов и др.) во взаимодействие с неактивированными аренами путем замещения атома водорода С–Н-связи.
Анализ приведенных в обзоре данных показывает, что в реакциях кросс-сочетания с участием С–Н-связи аренов важнейшее значение имеет окислитель, всегда принимающий участие либо в прямой, либо в опосредованной формах. В самом деле, окислитель участвует в разнообразных процессах, сопровождаемых С–Н-функционализацию аренов: химических и электрохимических, каталитических и некаталитических. Окислитель может менять электронное состояние участвующих в реакции аренов, содержащих электронодонорные группы, или π-избыточных гетаренов, трансформируя их в катион-радикалы или другие электрофильные частицы (umpolung). Окислитель может воздействовать на формально анионные или анионоидные реагенты и генерировать из них электрофильные или радикальные частицы. Наконец, в реакциях аренов с нуклеофильными реагентами, окислитель просто необходим для переноса электрона от промежуточных σH-аддуктов, образуемых в результате присоединения нуклеофилов к атому углерода С–Н-связи арена, и последующего элиминирования атома водорода С–Н-связи в привычной для него катионной форме.
Важно отметить, что ряд упомянутых выше функций окислителя, в частности таких, как активация аренов к нуклеофильной атаке путем генерации из них катион-радикалов, может успешно выполнять анод в электрохимических процессах. В других случаях – электрохимический анод трансформирует нуклеофильные частицы в электрофильные, которые становятся способными реагировать с неактивированным кольцом. Кроме того, окислитель или электрохимический анод исключительно важны для поддержания каталитического цикла и в тех случаях, когда С–Н-функционализация аренов протекает с участием катализаторов.
Приведенные в статье примеры указывают на многогранность функций окислителя как третьего компонента С–Н-функционализации аренов, что порой затрудняет классификацию реакций из-за наложения процессов каталитического цикла, в которых окислитель должен поддерживать регенерацию активной формы катализатора, и исключительно окислительной (без участия катализирующих металлов) прямой С–Н-функционализации аренов.
В заключение необходимо отметить, что окислительная С–Н-функционализация аренов занимает все более заметное место в арсенале методов “зеленой химии” XXI века в качестве эффективного инструмента прямой модификации С–Н-связи.
Список литературы
Grimmer G., Jacob J., Naujack K.W. // Fresenius. J. Anal. Chem. 1983. V. 314. № 1. P. 29–36. https://doi.org/10.1007/BF00476507
Niu M., Song Z., Pan L., Yan Y., Liu N., Li D., Li W. // Energy and Fuels. 2020. V. 34. № 11. P. 13614–13624. https://doi.org/10.1021/acs.energyfuels.0c02096
Aumaitre C., Morin J.F. // Chem. Rec. 2019. V. 19. № 6. P. 1142–1154. https://doi.org/10.1002/tcr.201900016
Murakami K., Yamada S., Kaneda T., Itami K. // Chem. Rev. 2017. V. 117. № 13. P. 9302–9332. https://doi.org/10.1021/acs.chemrev.7b00021
Smith M.B., March J. March’s Advanced Organic Chemistry: Reactions, Mechanism and Structure, 6th ed., John Wiley and Sons Inc., Hoboken, 2007. 2354 pp.
Anastas P.T., Warner J.C. Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998. 135 pp.
De Meijere A., Diederich F. (Eds.). Metal-catalyzed Cross-coupling Reactions, 2nd ed., Wiley-VCN, Weinheim, 2004. 938 pp. https://doi.org/10.1002/9783527619535
Chupakhin O.N., Charushin V.N., van der Plas H.C. Nucleophilic Aromatic Substitution of Hydrogen. Academic Press, San Diego; New York, 1994. 367 pp. https://doi.org/10.1016/C2009-0-21214-1
Charushin V.N., Chupakhin O.N. (Eds). Metal Free C–H Functionalization of Aromatics. Nucleophilic Displacement of Hydrogen. In: Topics in Heterocyclic Chemistry. Maes B.U.W., Cossy J., Poland S. (Series Eds.) V. 37. Springer, Heidelberg; New York; Dordrecht; London, 2014.
Yi H., Zhang G., Wang H., Huang Z., Wang J., Singh A.K., Lei A. // Chem. Rev. 2017. V. 117. № 13. P. 9016–9085. https://doi.org/10.1021/acs.chemrev.6b00620
Yeung C.S., Dong V.M. // Chem. Rev. 2011. V. 111. № 3. P. 1215–1292. https://doi.org/10.1021/cr100280d
Seebach D. // Angew. Chemie Int. Ed. English. 1979. V. 18. № 4. P. 239–258. https://doi.org/10.1002/anie.197902393
Yoshida J.I., Suga S., Suzuki S., Kinomura N., Yamamoto A., Fujiwara K. // J. Am. Chem. Soc. 1999. V. 121. № 41. P. 9546–9549. https://doi.org/10.1021/ja9920112
Lund H. // J. Solid State Electrochem. 2011. V. 15. № 7–8. P. 1733–1751. https://doi.org/10.1007/s10008-010-1265-8
Prier C.K., Rankic D.A., MacMillan D.W.C. // Chem. Rev. 2013. V. 113. № 7. P. 5322–5363. https://doi.org/10.1021/cr300503r
Shi L., Xia W. // Chem. Soc. Rev. 2012. V. 41. № 23. P. 7687–7697. https://doi.org/10.1039/c2cs35203f
Meng Q.Y., Gao X., Lei T., Liu Z., Zhan F., Li Z.-J., Wu L.-Z., Zhong J.-J., Xiao H., Feng K., Chen B., Teo Y., Tung C.-H. // Sci. Adv. 2017. V. 3. № 8. Article no. e1700666. https://doi.org/10.1126/sciadv.1700666
Charushin V.N., Chupakhin O.N. // Russ. Chem. Bull. 2019. V. 68. № 3. P. 453–471. https://doi.org/10.1007/s11172-019-2441-3
Charushin V.N., Chupakhin O.N. // Mendeleev Commun. 2007. V. 17. № 5. P. 249–254. https://doi.org/10.1016/j.mencom.2007.09.001
Chupakhin O.N., Charushin V.N. // Pure Appl. Chem. 2017. V. 89. № 8. P. 1195–1208. https://doi.org/10.1515/pac-2017-0108
Chupakhin O.N., Charushin V.N. // Tetrahedron Lett. Elsevier Ltd, 2016. V. 57. № 25. P. 2665–2672. https://doi.org/10.1016/j.tetlet.2016.04.084
Sharma N., Jung J., Ohkubo K., Lee Y.M., El-Khouly M., Nam W., Fukuzumi S. // J. Am. Chem. Soc. 2018. V. 140. № 27. P. 8405–8409. https://doi.org/10.1021/jacs.8b04904
Dixon J.A., Fishman D.H., Dudinyak R.S. // Tetrahedron Lett. 1964. V. 5. № 12. P. 613–616. https://doi.org/10.1016/0040-4039(64)83014-8
Eppley R.L., Dixon J.A. // J. Am. Chem. Soc. 1968. V. 90. № 6. P. 1606–1612. https://doi.org/10.1021/ja01008a034
Bryce-Smith D., Wakefield B.J. // Tetrahedron Lett. 1964. V. 5. № 45. P. 3295−3298. https://doi.org/10.1016/0040-4039(64)83086-0
Hixson S.S. // J. Chem. Soc., Chem. Commun. 1974. V. 15. P. 574−575. https://doi.org/10.1039/C39740000574
Winkler H.J.S., Winkler H. // J. Org. Chem. 1967. V. 32. № 6. P. 1695–1699. https://doi.org/10.1021/jo01281a002
Brinkmann A.W., Gordon M., Harvey R.G., Rabideau P.W., Stothers J.B., Ternay A.L. // J. Am. Chem. Soc. 1970. V. 92. № 20. P. 5912–5916. https://doi.org/10.1021/ja00723a016
Fox M.A., Ranade A.C., Madany I. // J. Organomet. Chem. 1982. V. 23. № 2. P. 269–277. https://doi.org/10.1016/S0022-328X(00)95252-5
Russell G.A., Weiner S.A. // J. Org. Chem. 1966. V. 31. № 1. P. 248–251. https://doi.org/10.1021/jo01339a056
Yamamoto Y., Nisimura T., Nozaki H. Condensation reactions of sylfonyl carbanion // Bull. Chem. Soc. Jpn. 1971. V. 44. P. 541–545. https://doi.org/10.1246/bcsj.44.541
Al Mamari H.H., Štefane B., Žugelj H.B., Brodnik H. // Tetrahedron. 2020. V. 76. № 9. P. 1–22. https://doi.org/10.1016/j.tet.2020.130925
Kulkarni A.A., Daugulis O. // Synthesis. 2009. № 24. P. 4087–4109. https://doi.org/10.1055/s-0029-1217131
Kramer S. // Synthesis. 2020. V. 52. № 14. P. 2017–2030. https://doi.org/10.1055/s-0039-1690882
Ackermann L., Vicente R., Kapdi A.R. // Angew. Chemie. Int. Ed. 2009. V. 48. № 52. P. 9792–9826. https://doi.org/10.1002/anie.200902996
Sinha S.K., Bhattacharya T., Maiti D. // React. Chem. Eng. 2018. V. 4. № 2. P. 244–253. https://doi.org/10.1039/c8re00225h
De Sarkar S., Liu W., Kozhushkov S., Ackermann L. // Adv. Synth. Catal. 2014. V. 356. № 7. P. 1461–1479. https://doi.org/10.1002/adsc.201400110
Kozhushkov S.I., Ackermann L. // Chem. Sci. 2013. V. 4. № 3. P. 886–896. https://doi.org/10.1039/c2sc21524a
Kuhl N., Hopkinson M., Wencle-Delord J., Glorius F. // Angew. Chemie. Int. Ed. 2012. V. 51. № 41. P. 10236–10254. https://doi.org/10.1002/anie.201203269
Fricke C., Reid W.B., Schoenebeck F. // Eur. J. Org. Chem. 2020. V. 2020. № 46. P. 7119–7130. https://doi.org/10.1002/ejoc.202000856
Fañanás-Mastral M. // Synth. 2017. V. 49. № 9. P. 1905–1930. https://doi.org/10.1055/s-0036-1589483
Kita Y., Tohma H., Hatanaka K., Takada T., Fujita S., Mitoh S., Sakurai H., Oka S. // J. Am. Chem. Soc. 1994. V. 116. № 9. P. 3684–3691. https://doi.org/10.1021/ja00088a003
Kita Y., Egi M., Ohtsubo M., Saiki T., Takada T., Tohma H. // Chem. Commun. 1996. P. 2225–2226. https://doi.org/10.1039/CC9960002225
Takada T., Arisawa M., Gyoten M., Hamada R., Tohma H., Kita Y. // J. Org. Chem. 1998. V. 63. № 22. P. 7698–7706. https://doi.org/10.1021/jo980704f
Hamamoto H., Anikumar G., Tohma H., Kita Y. // Chem. Commun. 2002. V. 2. № 5. P. 450–451. https://doi.org/10.1039/b111178g
Kita Y., Dohi T., Morimoto K. // Journal Synth. Org. Chem. Jpn. 2011. V. 69. № 11. P. 1241–1250. https://doi.org/10.5059/yukigoseikyokaishi.69.1241
Dohi T., Ito M., Yamaoka N., Morimoto K., Fujioka H., Kita Y. // Angew. Chem. 2010. V. 122. № 19. P. 3406–3409. https://doi.org/10.1002/ange.200907281
Zhai L., Shukla R., Wadumethrige S., Rathore R. // J. Org. Chem. 2010. V. 75. № 14. P. 4748–4760. https://doi.org/10.1021/jo100611k
Waldvogel S.R., Lips S., Selt M., Riehl B., Kampf C.J. // Chem. Rev. 2018. V. 118, № 14. P. 6706–6765. https://doi.org/10.1021/acs.chemrev.8b00233
Beil S.B., Müller T., Sillart S., Franzmann P., Bomm A., Holkamp M., Karst U., Schade W., Waldvogel S.R. // Angew. Chemie. Int. Ed. 2018. V. 57. № 9. P. 2450–2454. https://doi.org/10.1002/anie.201712718
Erdtman H.G.H. // Proc. R. Soc. London, Ser. A. 1933. V. 143. № 848. P. 191−222. https://doi.org/10.1098/rspa.1933.0215
Bechgaard K., Parker V.D. // J. Am. Chem. Soc. 1972. V. 94. № 13. P. 4749–4750. https://doi.org/10.1021/ja00768a063
Nyberg K., Ekström B., Sjöberg B., Husebye S., Klæboe P., Swahn C.-G. // Acta Chem. Scand. 1973. V. 27. № 2. P. 503−509.
Morofuji T., Shimizu A., Yoshida J. // J. Am. Chem. Soc. 2013. V. 135. № 13. P. 5000–5003. https://doi.org/10.1021/ja402083e
Arai T., Taneto K., Nakabayashi K., Kashiwagi T., Atobe M. // Chem. Commun. 2015. V. 51. № 23. P. 4891–4894. https://doi.org/10.1039/c5cc01253h
Kirste A., Schnakenburg G., Stecker F., Fischer A., Waldvogel S.R. // Angew. Chem. Int. Ed. 2010. V. 49. № 5. P. 971–975. https://doi.org/10.1002/anie.200904763
Elsler B., Wiebe A., Schollmeyer D., Dyballa K.M., Franke R., Waldvogel S.R. // Chem. Eur. J. 2015. V. 21. № 35. P. 12321–12325. https://doi.org/10.1002/chem.201501604
Elsler B., Schollmeyer D., Dyballa K., Franke R., Waldvogel S.R. // Angew. Chem. Int. Ed. 2014. V. 53. № 20. P. 5210–5213. https://doi.org/10.1002/anie.201400627
Jia C., Lu W., Oyamada J., Kitamura T., Matsuda K., Irie M., Fujiwara Y. // J. Am. Chem. Soc. 2000. V. 4. № 122. P. 7252–7263. https://doi.org/10.1021/ja0005845
Jia C., Piao D., Oyamada J., Lu W., Kitamura T., Fuji-wara Y. // Science. 2000. V. 287. № 5460. P. 1992−1995. https://doi.org/10.1126/science.287.5460.1992
Jia C., Kitamura T., Fujiwara Y. // Acc. Chem. Res. 2001. V. 34. № 8. P. 633−639. https://doi.org/10.1021/ar000209h
Nevado C., Echavarren A.M. // Synthesis. 2005. № 2. P. 167–182. https://doi.org/10.1055/s-2005-861781
Biffis A., Tubaro C., Buscemi G., Basato M. // Adv. Synth. Catal. 2008. V. 350. № 1. P. 189–196. https://doi.org/10.1002/adsc.200700271
Biffis A., Gazzola L., Tubaro C., Basato M. // ChemSusChem. 2010. V. 3. № 7. P. 834–839. https://doi.org/10.1002/cssc.201000039
Gazzola L., Tubaro C., Biffis A., Basato M. // New J. Chem. 2010. V. 34. № 3. P. 482–486. https://doi.org/10.1039/b9nj00506d
Saravanakumar R., Ramkumar V., Sankararaman S. // Organometallics. 2011. V. 30. № 6. P. 1689–1694. https://doi.org/10.1021/om1011984
Oyamada J., Kitamura T. // Tetrahedron. 2007. V. 63. № 51. P. 12754–12762. https://doi.org/10.1016/j.tet.2007.09.076
Jintoku T., Fujiwara Y., Kawata I., Kawauchi T., Taniguchi H. // J. Organomet. Chem. 1990. V. 385. № 2. P. 297–306. https://doi.org/10.1016/0022-328X(90)87292-L
Lu W., Yamaoka Y., Taniguchi Y., Kitamura T., Takaki K., Fujiwara Y. // J. Organomet. Chem. 1999. V. 580. № 2. P. 290–294.https://doi.org/10.1016/S0022-328X(98)01160-7
Suga T., Saitou T., Takaya J., Iwasawa N. // Chem. Sci. 2017. V. 8. № 2. P. 1454–1462. https://doi.org/10.1039/C6SC03838G
Yuan G., Li L., Jiang H., Qi C., Xie F. // Chinese J. Chem. 2010. V. 28. № 10. P. 1983–1988. https://doi.org/10.1002/cjoc.201090331
Shu Z., Ji W., Wang X., Zhou Y., Zhang Y., Wang J. // Angew. Chem. 2014. V. 126. № 8. P. 2218–2221. https://doi.org/10.1002/ange.201309791
Mcmanus J.B., Nicewicz D. // J. Am. Chem. Soc. 2017. V. 139. № 8. P. 2880–2883. https://doi.org/10.1021/jacs.6b12708
Yoshida K., Nagase S. // J. Am. Chem. Soc. 1979. V. 101. № 15. P. 4268–4272. https://doi.org/10.1021/ja00509a037
Andreades S., Zahnow E.W. // J. Org. Chem. 1969. V. 91. № 15. P. 4181–4190. https://doi.org/10.1021/ja01043a028
Hayrapetyan D., Rit R.K., Kratz M., Tschulik K., Gooßen L.J. // Chem. Eur. J. 2018. V. 24. № 44. P. 11288-11291. https://doi.org/10.1002/chem.201802247
Bianchi D., Bertoli M., Tassinari R., Ricci M., Vignola R. // J. Mol. Catal. A: Chem. 2003. V. 204–205. P. 419–424. https://doi.org/10.1016/S1381-1169(03)00323-6
Bonchio M., Conte V., DiFuria F., Modena G., Moro S. // J. Org. Chem. 1994. V. 59. № 21. P. 6262–6267. https://doi.org/10.1021/jo00100a029
Udenfriend S., Clark C.T., Axelrod J., Brodie B.B. // J. Biol. Chem. 1954. V. 208. № 2. P. 731–739. https://doi.org/10.1016/S0021-9258(18)65598-X
Clark C.T., Dowing R.D., Martin Jr. J.B. // J. Org. Chem. 1962. V. 27. № 3. P. 4698–4701. https://doi.org/10.1021/jo01059a538
Seo Y.-J., Mukai Y., Tagawa T., Goto S. // J. Mol. Catal. A: Chem. 1998. V. 120. № 1–3. P. 149–154. https://doi.org/10.1016/S1381-1169(96)00426-8
Kitajima N., Ito M., Fukui H., Moro-oka Y. // J. Chem. Soc. Chem. Commun. 1991. V. 1991. № 2. P. 102–104. https://doi.org/10.1039/C39910000102
Passoni L.C., Cruz A.T., Buffon R., Schuchardt U. // J. Mol. Catal. A: Chem. 1997. V. 120. № 1–3. P. 117–123. https://doi.org/10.1016/S1381-1169(96)00440-2
Kimura E., Machida R. // J. Chem. Soc., Chem. Commun. 1984. № 8. P. 499–500. https://doi.org/10.1039/C39840000499
Bal R., Tada M., Sasaki T., Iwasawa Y. // Angew. Chem. 2006. V. 118. № 3. P. 462–466. https://doi.org/10.1002/ange.200502940
Kitamura T., Kanzaki H., Hamada R., Nishiyama S., Tsuruya S. // Can. J. Chem. 2004. V. 82. № 11. P. 1597–1605. https://doi.org/10.1139/v04-123
Shibata Y., Hamada R., Ueda T., Ichihashi Y., Nishiyama S., Tsuruga S. // Ind. Eng. Chem. Res. 2005. V. 44. № 23. P. 8765–8772. https://doi.org/10.1021/ie050821i
Yamanaka H., Hamada R., Nibuta H., Nishiyama S., Tsuruga S. // J. Mol. Catal. A: Chem. 2002. V. 178. № 1–2. P. 89–95. https://doi.org/10.1016/S1381-1169(01)00279-5
Yamada M., Karlin K.D., Fukuzumi S. // Chem. Sci. 2016. V. 7. P. 2856–2863. https://doi.org/10.1039/C5SC04312C
Ohkubo K., Fujimoto A., Fukuzumi S. // J. Am. Chem. Soc. 2013. V. 135. № 14. P. 5368–5371. https://doi.org/10.1021/ja402303k
Chang X-P., Cui G., Fang W.-H., Thiel W. // ChemPhysChem. 2015. V. 16. № 5. P. 933–937. https://doi.org/10.1002/cphc.201402897
D’Amato E.M., Neumann C.N., Ritter T. // Organometallics. 2015. V. 34. № 18. P. 4626–4631. https://doi.org/10.1021/acs.organomet.5b00731
Zheng Y.W., Chen B., Ye P., Feng K., Wang W., Meng Q.-Y., Wu Li-Z., Tung C.-H. // J. Am. Chem. Soc. 2016. V. 138. № 32. P. 10080–10083. https://doi.org/10.1021/jacs.6b05498
Oberrauch E., Eberson L. // J. Appl. Electrochem. 1986. V. 16. № 4. P. 575–582. https://doi.org/10.1007/BF01006852
Fujimoto K., Maekawa H., Tokuda Y., Matsubara Y., Mizuno T., Nishiguchi I. // Synlett. 1995. V. 6. P. 661–662. https://doi.org/10.1055/s-1995-5036
Tsang W.C.P., Zheng N., Buchwald S.L. // J. Am. Chem. Soc. 2005. V. 127. № 42. P. 14560–14561. https://doi.org/10.1021/ja055353i
He G., Zhao Y., Zhang S., Lu C., Chen G. // J. Am. Chem. Soc. 2012. V. 134. № 1. P. 3–6. https://doi.org/10.1021/ja210660g
Nadres E.T., Daugulis O. // J. Am. Chem. Soc. 2012. V. 134. № 1. P. 7–10. https://doi.org/10.1021/ja210959p
Takamatsu K., Hirano K., Satoh T., Miura M. // Org. Lett. 2014. V. 16. № 11. P. 2892–2895. https://doi.org/10.1021/ol501037j
Suzuki C., Hirano K., Satoh T., Miura M. // Org. Lett. 2015. V. 17. № 6. P. 1597–1600. https://doi.org/10.1021/acs.orglett.5b00502
Chiba S. // Bull. Chem. Soc. Jpn. 2013. V. 86. № 12. P. 1400–1411. https://doi.org/10.1246/bcsj.20130220
Chiba S., Zhang L., Anget G., Hui B.W.-Q. // Org. Lett. 2010. V. 12. № 9. P. 2052–2055. https://doi.org/10.1021/ol100522z
John A., Byun J., Nicholas K.M. // Chem. Commun. 2013. V. 49. № 93. P. 10965–10967. https://doi.org/10.1039/c3cc46412a
Allen L.J., Cabrera P., Lee M., Sanford M.S. // J. Am. Chem. Soc. 2014. V. 136. № 15. P. 5607–5610. https://doi.org/10.1021/ja501906x
Kim H.J., Kim J., Cho S.H., Chang S. // J. Am. Chem. Soc. 2011. V. 133. № 41. P. 16382–16385. https://doi.org/10.1021/ja207296y
Kantak A.A., Potavathri S., Barham R., Romano K.M., DeBoef B. // J. Am. Chem. Soc. 2011. V. 133. № 49. P. 19960–19965. https://doi.org/10.1021/ja2087085
Shrestha R., Mukherjee P., Tan Y., Litman Z.C., Hartwig J.F. // J. Am. Chem. Soc. 2013. V. 135. № 23. P. 8480–8483.https://doi.org/10.1021/ja4032677
Marchetti L., Kantak A., Davis R., DeBoef B. // Org. Lett. 2015. V. 17. № 2. P. 358–361. https://doi.org/10.1021/ol5034805
Samanta R., Bauer J., Strohmann C., Antonchick A.P. // Org. Lett. 2012. V. 14. № 21. P. 5518–5521. https://doi.org/10.1021/ol302607y
Antonchick A.P., Samanta R., Kulikov K., Lategahn J. // Angew. Chem. Int. Ed. 2011. V. 50. № 37. P. 8605–8608.https://doi.org/10.1002/anie.201102984
Foo K., Sella E., Thome I., Eastgate M.D., Baran P.S. // J. Am. Chem. Soc. 2014. V. 136. № 14. P. 5279–5282. https://doi.org/10.1021/ja501879c
Kawakami T., Murakami K., Itami K. // J. Am. Chem. Soc. 2015. V. 137. № 7. P. 2460–2463. https://doi.org/10.1021/ja5130012
Morofuji T., Shimizu A., Yoshida J. // J. Am. Chem. Soc. 2013. V. 135. № 13. P. 5000–5003. https://doi.org/10.1021/ja402083e
Morofuji T., Shimizu A., Yoshida J. // J. Am. Chem. Soc. 2013. V. 135. № 13. P. 5000–5003.https://doi.org/10.1021/ja402083e
Yoshida J.I., Shimizu A., Ashikari Y., Morofuji T., Hayashi R., Nokami T., Nagaki A. // Bull. Chem. Soc. Jpn. 2015. V. 88. № 6. P. 763–775. https://doi.org/10.1246/bcsj.20150100
Herold S., Möhle S., Zirbes M., Richter F., Nefzger H., Waldvogel S.R. // European J. Org. Chem. 2016. V. 2016. № 7. P. 1274–1278. https://doi.org/10.1002/ejoc.201600048
Waldvogel S.R., Möhle S., Herold S., Richter F., Nefz-ger H. // ChemElectroChem. 2017. V. 4. № 9. P. 2196–2210. https://doi.org/10.1002/celc.201700476
Lisitsyn Y.A., Kargin Y.M. // Rus. J. Electrochem. 2000. V. 36. P. 89–99. https://doi.org/10.1007/BF02756893
Lisitsyn Y.A., Sukhov A.V. // Russ. J. Gen. Chem. 2017. V. 87. P. 929–933. https://doi.org/10.1134/S1070363217010042
Do Q.T., Elothmani D., Le Guillanton G. // Tetrahedron Lett. 1998. V. 39. P. 4657–4658. https://doi.org/10.1016/S0040-4039(98)00871-5
Do Q.T., Elothmani D., Simonet J., Le Guillanton G. // Electrochim. Acta. 2005. V. 50. № 24. P. 4792–4799. https://doi.org/10.1016/j.electacta.2005.02.033
Matsumoto K., Kozuki Y., Ashikari Y., Suga S., Kashimura S., Yoshida J.I. // Tetrahedron Lett. 2012. V. 53. № 15. P. 1916–1919. https://doi.org/10.1016/j.tetlet.2012.01.131
Kashiwagi T., Amemiya F., Fuchigami T., Atobe M. // Chem. Commun. 2012. V. 48. № 22. P. 2806–2808. https://doi.org/10.1039/c2cc17979b
Kashiwagi T., Amemiya F., Fuchigami T., Atobe M. // J. Flow Chem. 2013. V. 3. № 1. P. 17–22. https://doi.org/10.1556/JFC-D-12-00019
Krishnan P., Gurjar V.G. // J. Appl. Electrochem. 1993. V. 23. № 3. P. 268–270. https://doi.org/10.1007/BF00241920
Krishnan P., Gurjar V.G. // Synth. Commun. 1992. V. 22. № 19. P. 2741–2744. https://doi.org/10.1080/00397919208021538
Yakhvarov D.G., Budnikova Y.H., Tazeev D.I., Sinya-shin O.G. // Russ. Chem. Bull. 2002. V. 51. № 11. P. 2059–2064. https://doi.org/10.1023/A:1021611926712
Grayaznova T.V., Dudkina Y.B., Islamov D.R., Katae-va O.N., Sinyashin O.G., Vicic D.A., Budnikova Y.H. // J. Organomet. Chem. 2015. V. 785. P. 68–71. https://doi.org/10.1016/j.jorganchem.2015.03.001
Khrizanforov M.N., Strekalova S.O., Gryaznova T.V., Khrizanforova V.V., Budnikova Yu.H. // Russ. Chem. Bull. 2015. V. 64. № 8. P. 1926–1932. https://doi.org/10.1007/s11172-015-1095-z
Дополнительные материалы отсутствуют.
Инструменты
Доклады Российской академии наук. Химия, науки о материалах