

Доклады Российской академии наук. Химия, науки о материалах , 2022, T. 506, № 1, стр. 14-19
Синтетические подходы к (3-тиенил)-содержащим производным 2,2'-бипиридинов как потенциальным мономерам для электрополимеризации
А. П. Криночкин 1, 2, *, М. И. Валиева 1, 2, Е. С. Старновская 1, 2, Я. К. Штайц 1, С. С. Рыбакова 1, Э. Р. Шарафиева 1, 3, Д. С. Копчук 1, 2, Г. В. Зырянов 1, 2, член-корреспондент РАН В. Л. Русинов 1, 2
1 Уральский федеральный университет
620002 Екатеринбург, Россия
2 Институт органического синтеза,
Уральское отделение Российской академии наук
620219 Екатеринбург, Россия
3 Уральский Медицинский университет
Министерства здравоохранения Российской Федерации
620028 Екатеринбург, Россия
* E-mail: a.p.krinochkin@urfu.ru
Поступила в редакцию 31.05.2022
После доработки 18.07.2022
Принята к публикации 15.08.2022
- EDN: EAXHKS
- DOI: 10.31857/S2686953522700133
Аннотация
Разработаны эффективные синтетические подходы к новым 2,2'-бипиридиновым лигандам, функционализированным 3-тиенильным фрагментом, которые представляют интерес в качестве мономерных звеньев для электрополимеризации. Синтез выполнен с использованием “1,2,4-триазиновой” методологии.
ВВЕДЕНИЕ
Олиго- и политиофены, а также их производные являются перспективными материалами для применения в органической электронике и молекулярной сенсорике [1–4] благодаря своим подходящим электрохимическим и механическим свойствам, в том числе высокой устойчивости к воздействию окружающей среды. Отдельный интерес представляют поли/олигомеры, в составе которых содержатся фрагменты олигопиридина [5]. В ряде случаев эти фрагменты могут быть одновременно введены в состав одного полимера [6], например, с целью получения материалов для электрокатализа [7]. Также следует отметить, что рутениевые комплексы лигандов, включающих остатки тиофена и пиридина, перспективны благодаря возможностям связывания теломерных G‑квадруплексов ДНК человека [8], а также в качестве материалов для создания покрытий для формирования нанокомпозитов золота и серебра в различных средах [9].
Однако синтез структур, одновременно включающих фрагменты 2,2′-бипиридина и 3-тиенила, представлен к настоящему времени лишь в ограниченном количестве публикаций [10–13]. В данной статье мы предлагаем удобные синтетические подходы к потенциальным мономерам 2,2′-бипиридинового ряда с 3-тиенильным заместителем.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
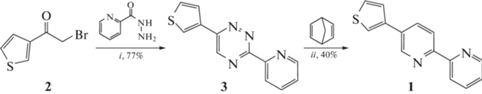
Для синтеза целевых структур мы использовали “1,2,4-триазиновую” методологию [14, 15]. Так, первой стадией синтеза 2,2′-бипиридина 1 с непосредственно связанным с ним 3-тиенильным фрагментом является гетероциклизация 2-бром-1-(тиофен-3-ил)этанона 2 с двумя эквивалентами гидразида пиколиновой кислоты (схема 1). Такой подход к 3,6-дизамещенным 1,2,4-триазинам известен достаточно давно [16]. В данном случае была использована процедура, предполагающая нагрев исходных реагентов в диметилформамиде (ДМФА) в отсутствие дополнительных реагентов [17], что позволяет избежать образования побочных продуктов – дигидразонов арилглиоксаля [18, 19]. Последующая реакция аза-Дильса–Альдера промежуточного триазина 3 с 2,5-норборнадиеном позволила получить С5-тиофензамещенный целевой 2,2′-бипиридин 1. Относительно низкий выход (40%) продукта 1 в данном случае обусловлен необходимостью использования достаточно высококипящего растворителя – 1,2-дихлорбензола вместо о‑ксилола, часто используемого в подобных случаях [20], ввиду заметного электронодонорного характера фрагмента 3‑тиенила.
Также разработан еще один подход, который включает применение недавно обнаруженной нами реакции 1,2,4-триазин-5-карбонитрилов с производными 2-амино-4-арилоксазолов как диенофилами, одностадийно приводящей к 4-арил-3-гидроксипиридинам [21]. В данном случае путем реакции 5-циано-1,2,4-триазина 4 [22] с ранее не описанным 4-(3-тиенил)оксазолом 5 был получен 2,2′-бипиридин 6, имеющий в положении С4 фрагмент 3-тиенила (схема 2).

Выход продукта 6 составил 51%, что соответствует средним значениям выходов соединений в аналогичных ранее описанных превращениях, когда 2‑амино-4-арилоксазолы используются в качестве диенофилов [21].
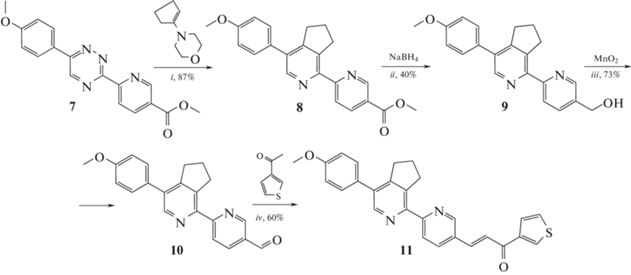
Наконец, в данной работе предложен третий метод получения тиофен-замещенных 2,2′-бипиридинов, который основан на использовании реакции Кневенагеля [23, 24] между натриевой солью 3‑ацетилтиофена, получаемой in situ, и производным 2,2′-бипиридин-5-карбальдегида. Для этого в качестве исходного соединения использовали ранее описанный 3-(5-метоксикарбонилпиридин-2-ил)-1,2,4-триазин 7 [25] (схема 3). Так, реакцией аза-Дильса–Альдера между соединением 7 и 1‑морфолиноциклопентеном получен 2,2′-бипиридин 8. Следует отметить необходимость модификации синтетической процедуры в данном случае по сравнению с соединениями, имеющими сложноэфирную группу в положении С6 остатка 2-пиридила [26]. А именно, для ароматизации нового пиридинового кольца требуется кратковременное нагревание в ледяной уксусной кислоте. Ранее такая процедура применялась, например, при использовании 1,2,4-триазин-5-карбонитрилов в качестве исходных соединений в данной реакции [22]. Последующее восстановление сложноэфирной группы, согласно описанной ранее процедуре [27], привело к образованию гидроксиметилбипиридина 9. Выход продукта, равный 40%, в данном случае обусловлен конкурентной реакцией щелочного гидролиза сложноэфирной группы. Дальнейшее окисление соединения 9 активированным MnO2 [28] привело к формированию альдегида 10. На последней стадии по реакции Кневенагеля между альдегидом 10 и 3-ацетилтиофеном был получен целевой 2,2′-бипиридин 11.
Схема 3.
Реагенты и условия: i – о-ксилол, 143°C, 10 ч, затем AcOH, 118°C, 5–10 мин; ii – EtOH + CHCl3 (8 : 1 (об.)), кипячение, 10 ч, затем EtOH, кипячение, 10 ч; iii – 1,2-дихлорэтан, 50°C, 12 ч; iv – NaOH/EtOH + H2O (9 : 1 (об.)), комнатная температура, 24 ч.
Структура всех полученных соединений была подтверждена на основании данных спектроскопии 1Н, 13С ЯМР, масс-спектрометрии и элементного анализа. Так, для бипиридина 1 в спектре 1Н ЯМР могут быть отмечены сигналы протонов остатка 3-тиенила, а также 5-замещенного 2,2′-бипиридина. В случае соединения 6 в спектре 1Н ЯМР присутствуют сигналы протонов остатков 3-тиенила и 2-пиридила, а также фенильного заместителя. Соединение 11 характеризуется наличием в спектре 1Н ЯМР сигналов протонов алкенового фрагмента в области 7.82 и 8.07 м. д., КССВ (константа спин-спинового взаимодействия) между ними составляет 15.6 Гц, что соответствует транс-конфигурации данного фрагмента. Кроме этого, в спектре 1Н ЯМР имеются сигналы протонов алифатического карбоцикла в области 2.00–3.47 м. д., остатка 4‑метоксифенила, АВХ-системы пиридинового кольца, остатка 3-тиенила, а также синглет протона фрагмента 6,7-дигидро-5Н-циклопента[c]пиридина.
ЗАКЛЮЧЕНИЕ
Таким образом, продемонстрирована возможность использования “1,2,4-триазиновой” методологии для получения новых 2,2′-бипиридиновых лигандов, содержащих в своей структуре фрагмент 3-тиенила. Синтез осуществлен или в результате использования синтонов, заранее включающих остаток 3-тиенила, или за счет его последующего введения на различных стадиях (реакция аза-Дильса–Альдера или модификация альдегидной группы в составе готового бипиридина). Полученные структуры являются перспективными в качестве мономерных звеньев для электрополимеризации.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Спектры 1H и 13С ЯМР записаны на спектрометрах Bruker Avance-400 (400 МГц) и Bruker Avance-500 (500 МГц), внутренний стандарт – SiMe4. Масс-спектры (тип ионизации – электроспрей) записаны на приборе MicrOTOF-Q II фирмы “Bruker Daltonics” (Бремен, Германия). Элементный анализ выполнен на CHN-анализаторе РЕ 2400 II фирмы Perkin Elmer (Уолтем, США). Исходные соединения 2 [29], 4 [22] и 7 [25] были синтезированы по описанным методикам. Все остальные реагенты коммерчески доступны. Для очистки соединений колоночной хроматографией была использована смесь хлористый метилен : этилацетат (9 : 1 (об.)).
5-(3-Тиенил)-2,2′-бипиридин 1. 1,2,4-Триазин 3 (303 мг, 1.26 ммоль) суспендировали в 1,2-дихлорбензоле (25 мл). К суспензии добавили 2,5-норборнадиен (6.30 ммоль, 0.64 мл) и полученную смесь кипятили 8 ч. Затем добавили вторую порцию 2,5-норборнадиена (6.30 ммоль, 0.64 мл) и полученную смесь кипятили еще 8 ч. Затем добавили третью порцию 2,5-норборнадиена (6.30 ммоль, 0.64 мл) и полученную смесь кипятили еще 8 ч. Растворитель удаляли при пониженном давлении, продукт очищали методом колоночной хроматографии, Rf = 0.7). Аналитический образец был получен перекристаллизацией из ацетонитрила. Выход 120 мг (0.50 ммоль, 40%). 1Н ЯМР (ДМСО-d6, δ, м. д.): 7.38–7.40 (м, 1H, H-5′), 7.61–7.64 (м, 2H, H-5тиофен, H-3), 7.88–7.91 (ддд, 1Н, Н-4', 3J 8.0 Гц, 8.0 Гц, 4J 1.8 Гц), 7.97–7.98 (м, 1H, H-2тиофен), 8.18–8.20 (м, (1H, H-4тиофен), 8.42–8.44 (м, 2H, H-4, H-3′), 8.64–8.65 (м, (H, H‑6'), 9.01 (д, 1H, 4J 1.8 Гц, H-6). Масс-спектр (m/z, Iотн., %): 239.07 [М + Н]+. Найдено, %: С, 70.58; Н, 4.21; N, 11.74. Вычислено для С14Н10N2S, %: С, 70.56; Н, 4.23; N 11.76.
3-(Пиридин-2-ил)-6-(3-тиенил)-1,2,4-триазин 3. Смесь соединения 2 (335 мг, 1.63 ммоль) и гидразида пиридин-2-карбоновой кислоты (448 мг, 3.27 ммоль) в ДМФА перемешивали при 120°С в течение 10 ч в атмосфере аргона. Затем растворитель упарили при пониженном давлении. Продукт был очищен колоночной хроматографией, Rf = 0.4. Выход 303 мг (1.26 ммоль, 77%). 1Н ЯМР (ДМСО-d6, δ, м. д.): 7.61–7.64 (м, 1Н, Н-5Py), 7.84–7.87 (м, 1H, H-5тиофен), 8.01–8.05 (м, 1Н, Н-4тиофен), 8.07 (ддд, 1Н, 3J 8.0 Гц, 8.0 Гц, 4J 2.0 Гц, Н-4Py), 8.48 м (1Н, Н-3Py), 8.65–8.68 (м, 1Н, Н-2тиофен), 8.82–8.85 (м, 1Н, Н-6Py), 9.51 (с, 1H, H-5). Масс-спектр, (m/z, Iотн., %): 241.06 [М + Н]+.
2-Амино-4-(3-тиенил)оксазол 5 получен по ранее предложенной методике [30] для аналогичных соединений. Тпл. = 176–178°C. Выход 185 мг (1.11 ммоль, 41%). 1Н ЯМР (CDCl3, δ, м. д.): 4.67–5.00 (уш. с, 2H, NH2), 7.20–7.25 (м, 1H, H-4тиофен), 7.30–7.36 (м, 2H, H-5тиофен, H-5), 7.49–7.53 (м, 1H, H-2тиофен). 13C ЯМР (DMSO-d6, δ, м. д.): 120.5, 125.7, 127.2, 127.3, 134.1, 136.1, 161.9. Масс-спектр, (m/z, Iотн., %): 167.01 [М + Н]+. Найдено, %: С, 50.47; Н, 3.51; N, 16.71. Вычислено для С7Н6N2OS, %: С, 50.59; Н, 3.64; N, 16.86.
3-Гидрокси-4-(3-тиенил)-5-фенил-2,2ʹ-бипиридин-6-карбонитрил 6. Смесь 5‑циано-1,2,4-триазина 4 (100.0 мг, 0.39 ммоль) и 2-аминооксазола 5 (70.5 мг, 0.42 ммоль) перемешивали в атмосфере аргона при 155°С в течение 8 ч в отсутствие растворителя. Продукт был очищен колоночной хроматографией, Rf = 0.7. Аналитический образец был получен перекристаллизацией из этанола. Тпл. = 240–242°C. Выход 70 мг (0.20 ммоль, 51%). 1Н ЯМР (CDCl3, δ, м. д.): 6.89–6.90 (дд, 1H, H-5тиофен), 7.15–7.16 (м, 1H, H-2тиофен), 7.18–7.20 (м, 1H, H-4тиофен), 7.23–7.25 (м, 2H, Ph), 7.34–7.36 (м, 3H, Ph), 7.45–7.48 (м, 1H, H-5'), 8.01–8.04 (м, 1H, H-4'), 8.51–8.52 (м, 1H, H-3'), 8.72–8.74 (д, 1H, 3J 8.5 Гц, H-6'). 13C ЯМР (CDCl3, δ, м. д.): 117.6, 121.7, 123.4, 124.2, 124.3, 127.1, 128.5, 128.8, 129.3, 129.9, 132.2, 133.1, 135.0, 137.3, 138.7, 142.9, 144.9, 156.6, 157.1. Масс-спектр, (m/z, Iотн., %): 356.07 [М + Н]+. Найдено, %: С, 70.98; Н, 3.71; N, 11.84. Вычислено для С21Н14N3OS, %: С, 70.97; Н, 3.69; N, 11.82.
Метил 6-{4-(4-метоксифенил)-6,7-дигидро-5H-циклопента[c]пиридин-1-ил}никотинат 8. 1,2,4-Триазин 7 (663 мг, 2.06 ммоль) суспендировали в о-ксилоле (25 мл). К суспензии добавили 1-морфолиноциклопентен (10.3 ммоль, 1.65 мл) и полученную смесь кипятили 10 ч. Растворитель удалили при пониженном давлении, к остатку добавили ледяную уксусную кислоту и полученную смесь кипятили в течение 5–10 мин. Затем растворитель удаляли при пониженном давлении и продукт очищали колоночной хроматографией, Rf = 0.8. Выход 645 мг (1.79 ммоль, 87%). 1Н ЯМР (ДМСО-d6, δ, м. д.): 2.05–2.14 (м, 2H, CH2-6), 3.04 (т, 2H, 3J 7.6 Гц, CH2-7), 3.51 (т, 2H, 3J 7.6 Гц, CH2-5), 3.85 (с, 3H, OСН3), 3.95 (с, 3H, OСН3), 6.98–7.04 (м, 2H, C6H4OСН3), 7.41–7.48 (м, 2H, C6H4OСН3), 8.36 (дд, 1H, 3J 8.4 Гц, 4J 2.0 Гц, H-4'), 8.45 (c, 1H, Н-6), 8.47 (д, 1H, 3J 8.4 Гц, H-3'), 9.19 (д, 1H, 4J 2.0 Гц, H-6'). Масс-спектр, (m/z, Iотн., %): 361.16 [М + Н]+.
{6-[4-(4-Метоксифенил)-6,7-дигидро-5H-циклопента[c]пиридин-1-ил]пиридин-3-ил}метанол 9. К раствору бипиридина 8 (308 мг, 0.85 ммоль) в 50 мл смеси этилового спирта и хлороформа (8 : 1) добавляли NaBH4 (162 мг, 4.27 ммоль) и полученную смесь перемешивали при кипячении в течение 8 ч. Растворитель удаляли при пониженном давлении. Остаток суспендировали в 40 мл этилового спирта, к смеси добавили NaBH4 (81 мг, 2.13 ммоль) и полученную смесь перемешивали при кипячении в течение 8 ч. Растворитель удаляли при пониженном давлении, к осадку добавляли воду и продукт экстрагировали хлористым метиленом. Полученный продукт использовали на следующей стадии без дополнительной очистки. Выход 110 мг (0.33 ммоль, 40%). 1Н ЯМР (CDCl3, δ, м. д.): 2.02–2.12 (м, 2H, CH2-6), 3.03 (т, 2H, 3J 7.6 Hz, CH2-7), 3.38 (м, 2H, 3J 7.6 Гц, CH2-5), 3.86 (с, 3H, OСН3), 4.73 (с, 2H, CH2OH), 6.98–7.03 (м, 2H, C6H4OСН3), 7.40–7.46 (м, 2H, C6H4OСН3), 7.75 (дд, 1H, 3J 8.0 Гц, 4J 1.6 Гц, H-4'), 8.07 (д, 1H, 3J 8.0 Гц, H-3'), 8.51 (с, 1H, H-6), 8.62 (д, 1H, 4J 1.6 Гц, H-6'). Масс-спектр, (m/z, Iотн., %): 333.15 [М + Н]+.
6-{4-(4-Метоксифенил)-6,7-дигидро-5H-циклопента[c]пиридин-1-ил}никотинальдегид 1. К раствору гидроксиметилбипиридина 9 (110 мг, 0.33 ммоль) в 40 мл 1,2-дихлорэтана добавили MnO2 (288 мг, 3.31 ммоль), полученную смесь перемешивали при 50°С в течение 12 ч. Прохождение реакции контролировалось по ТСХ (элюент – смесь хлористый метилен : этилацетат (9 : 1)). Затем реакционную массу отфильтровывали, фильтрат упаривали при пониженном давлении. Полученный продукт использовали на следующей стадии без дополнительной очистки. Тпл. = 108–110°C. Выход 80 мг (0.24 ммоль, 73%. 1Н ЯМР (ДМСО-d6, δ, м. д.): 2.05–2.15 (м, 2H, CH2-6), 3.06 (т, 2H, 3J 7.6 Гц, CH2-7), 3.53 (т, 2H, 3J 7.6 Гц, CH2-5), 3.86 (с, 3H, OСН3), 7.00–7.06 (м, 2H, C6H4OСН3), 7.45–7.51 (м, 2H, C6H4OСН3), 8.33 (дд, 1H, 3J 8.0 Гц, 4J 1.2 Гц, H-4'), 8.49 (с, 1H, H-6), 8.55 (д, 1H, 3J 8.0 Гц, H-3'), 9.15 (д, 1H, 4J 1.2 Гц, H-6'), 10.18 (с, 1H, CHO). 13C ЯМР (CDCl3, δ, м. д.): 25.4, 32.7, 33.8, 55.4, 114.2, 123.1, 129.8, 129.8, 130.0, 134.3, 136.1, 140.8, 146.8, 148.8, 151.3, 153.3, 159.5, 163.1, 190.8. Масс-спектр, (m/z, Iотн., %): 331.13 [М + Н]+. Найдено, %: С, 76.24; Н, 5.36; N, 8.35. Вычислено для С21Н18N2O2, %: С, 76.34; Н, 5.49; N, 8.48.
(E)-3-{5-[4-(4-метоксифенил)-6,7-дигидро-5H-циклопента[c]пиридин-1-ил]пиридин-2-ил}-1-(3-тиенил)проп-2-ен-1-он 11. К раствору 3-ацетилтиофена (45 мг, 0.36 ммоль) и альдегида 10 (118 мг, 0.36 ммоль) в этаноле (9 мл) добавляли раствор гидроксида натрия (21 мг, 0.54 ммоль) в воде (1 мл). Полученную смесь перемешивали при комнатной температуре в течение 24 ч. Выпавший осадок отфильтровывали, промывали этиловым спиртом и высушивали при пониженном давлении. Аналитический образец получен перекристаллизацией из этанола. Тпл. > 250°C. Выход 94 мг (0.21 ммоль, 60%). 1Н ЯМР (ДМСО-d6, δ, м. д.): 2.00–2.08 (м, 2H, CH2-6), 3.03 (т, 2H, 3J 7.6 Гц, CH2-7), 3.47 (т, 2H, 3J 7.6 Гц, CH2-5), 3.83 (с, 3H, OСН3), 7.06–7.11 (м, 2H, C6H4OСН3), 7.53–7.58 (м, 2H, C6H4OСН3), 7.69–7.74 (м, 2H, H-4,5тиофен), 7.82 и 8.07 (оба д, 1H, CH=CH, 3J 15.6 Гц), 8.41 (д, 1H, H-3', 3J 7.2 Гц), 8.49 (дд, 1H, 3J 8.4 Гц, 4J 2.0 Гц, H-4'), 8.54 (с, 1H, H-6), 8.89–8.91 (м, 1H, H-2тиофен), 9.13 (д, 1H, 4J 2.0 Гц, H-6'). 13C ЯМР (CDCl3, δ, м. д.): 25.5, 32.7, 33.5, 55.4, 114.2, 122.9, 124.1, 126.7, 127.5, 129.3, 129.7, 130.0, 132.4, 133.8, 134.6, 140.0, 140.4, 142.9, 146.7, 149.3, 149.4, 153.1, 159.4, 159.7, 183.3. Масс-спектр, (m/z, Iотн., %): 439.15 [М + Н]+. Найдено, %: С, 73.94; Н, 5.07; N, 6.37. Вычислено для С14Н10N2S, %: С, 73.95; Н, 5.06; N, 6.39.
Список литературы
Sirringhaus H., Tessler N., Friend R.H. // Science. 1998. V. 280. P. 1471–1474. https://doi.org/10.1126/science.280.5370.1741
Somani P.R., Radhakrishnan S. // Mater. Chem. Phys. 2003. V. 77. P. 117–133. https://doi.org/10.1016/S0254-0584(01)00575-2
McQuade D.T., Pullen A.E., Swager T.M. // Chem. Rev. 2000. V. 100. № 7. P. 2537–2574. https://doi.org/10.1021/cr9801014
Pathiranage T.M.S.K., Dissanayake D.S., Niermann C.N., Ren Y., Biewer M.C., Stefan M.C. // J. Polym. Sci., A. 2017. V. 55. P. 3327–3346. https://doi.org/10.1002/pola.28726
Yamamoto T., Maruyama T., Zhou Z., Ito T., Fukuda T., Yoneda Y., Begum F., Ikeda T., Sasaki S., Takezoe H., Fukuda A., Kubotal K. // J. Am. Chem. Soc. 1994. V. 116. P. 4832–4845. https://doi.org/10.1021/ja00090a031
Abd-El-Aziz A.S., Dalgakiran S., Vandel M., Owen E.M., Wagner B.D. // J. Electrochem. Soc. 2013. V. 160. № 3. G61–G67. https://doi.org/10.1149/2.004304jes
Wang J., Keene F.R. // Electrochim. Acta. 1996. V. 41. № 16. P. 2563–2569. https://doi.org/10.1016/0013-4686(96)00070-9
Wang X., Pei L., Fan X., Shi S. // Inorg. Chem. Commun. 2016. V. 72. P. 7–12. https://doi.org/10.1016/j.inoche.2016.07.010
Mayer C.R., Dumas E., Miomandre F., Méallet-Renault R., Warmont F., Vigneron J., Pansu R., Etcheberrya A., Sécheresse F. // New J. Chem. 2006. V. 30. P. 1628–1637. https://doi.org/10.1039/B607889C
Wang J., Pappalardo M., Keene F.R. // Aust. J. Chem. 1995. V. 48. № 8. P. 1425–1436. https://doi.org/10.1071/CH9951425
Prasad K.S., Pillai R.R., Shivamallu C., Prasad S.K., Jain A.S., Pradeep S., Armaković S., Armaković S.J., Srinivasa C., Kallimani S., Amachawadi R.G., Ankegowda V.M., Marraiki N., Elgorban A.M., Syed A. // Molecules. 2020. V. 25. № 12. P. 2865. https://doi.org/10.3390/molecules25122865
Zhao K., Xu X.-P., Zhu S.-L., Shi D.-Q., Zhang Y., Ji S.-J. // Synthesis. 2009. V. 16. P. 2697–2708. https://doi.org/10.1055/s-0029-1216873
O’Sullivan T.J., Djukic B., Dube P.A., Lemaire M.T. // Can. J. Chem. 2009. V. 87. P. 533–538. https://doi.org/10.1139/V09-012
Prokhorov A.M., Kozhevnikov D.N. // Chem. Heterocycl. Compd. 2012. V. 48. P. 1153–1176. https://doi.org/10.1007/s10593-012-1117-9
Anderson E.D., Boger D.L. // J. Am. Chem. Soc. 2011. V. 133. № 31. P. 12285–12292. https://doi.org/10.1021/ja204856a
Saraswathi T.V., Srinivasan V.R. // Tetrahedron. 1977. V. 33. P. 1043–1051. https://doi.org/10.1016/0040-4020(77)80223-8
Shtaitz Y.K., Savchuk M.I., Kopchuk D.S., Taniya O.S., Santra S., Zyryanov G.V., Suvorova A.I., Rusinov V.L., Chupakhin O.N. // Russ. J. Org. Chem. 2020. V. 56. № 3. P. 548–551. https://doi.org/10.1134/S1070428020030306
Kopchuk D.S., Kovalev I.S., Zyryanov G.V., Khasanov A.F., Medvedevskikh A.S., Rusinov V.L., Chupakhin O.N. // Chem. Heterocycl. Compd. 2013. V. 49. P. 500–502. https://doi.org/0.1007/s10593-013-1275-4
Korotkikh N.I., Chervinskiy A.Y., Baranov S.N., Kap-kan L.M., Shvayka O.P. // J. Org. Chem. USSR. 1979. V. 15. P. 860.
Kozhevnikov V.N., Shabunina O.V., Kopchuk D.S., Ustinova M.M., König B., Kozhevnikov D.N. // Tetrahedron. 2008. V. 64. № 37. P. 8963–8973. https://doi.org/10.1016/j.tet.2008.06.040
Krinochkin A.P., Reddy G.M., Kopchuk D.S., Slepukhin P.A., Shtaitz Y.K., Khalymbadzha I.A., Kovalev I.S., Kim G.A., Ganebnykh I.N., Zyryanov G.V., Chupakhin O.N., Charushin V.N. // Mendeleev Commun. 2021. V. 31. P. 542–544. https://doi.org/10.1016/j.mencom.2021.07.035
Kozhevnikov V.N., Kozhevnikov D.N., Nikitina T.V., Rusinov V.L., Chupakhin O.N., Zabel M., König B. // J. Org. Chem. 2003. V. 68. № 7. P. 2882–2888. https://doi.org/10.1021/jo0267955
Heravi M.M., Janati F., Zadsirjan V. // Monatsh. Chem. 2020. V. 151. P. 439–482. https://doi.org/10.1007/s00706-020-02586-6
van Beurden K., de Koning S., Molendijk D., van Schijndel J. // Green Chem. Lett. Rev. 2020. V. 13. № 4. P. 349–364. https://doi.org/10.1080/17518253.2020.1851398
Kozhevnikov D.N., Shabunina O.V., Kopchuk D.S., Slepukhin P.A., Kozhevnikov V.N. // Tetrahedron Lett. 2006. V. 47. № 39. P. 7025–7029. https://doi.org/10.1016/j.tetlet.2006.07.111
Kopchuk D.S., Krinochkin A.P., Kozhevnikov D.N., Slepukhin P.A. // Polyhedron. 2016. V. 118. P. 30–36. https://doi.org/10.1016/j.poly.2016.07.025
Kopchuk D.S., Pavlyuk D.E., Kovalev I.S., Zyryanov G.V., Rusinov V.L., Chupakhin O.N. // Can. J. Chem. 2016. V. 94. № 7. P. 599–603. https://doi.org/10.1139/cjc-2015-0576
Kopchuk D.S., Kim G.A., Kovalev I.S., Santra S., Zyryanov G.V., Majee A., Rusinov V.L., Chupakhin O.N. // Can. J. Chem. 2018. V. 96. P. 419–424. https://doi.org/10.1139/cjc-2017-0485
Deng L., Kleij A.W., Yang W. // Chem. Eur. J. 2018. V. 24. Is. 72. P. 19156–19161. https://doi.org/10.1002/chem.201805295
Turner W.W., Arnold L.D., Maag H., Zlotnick A. Patent WO 2015138895A. 2015.
Дополнительные материалы отсутствуют.
Инструменты
Доклады Российской академии наук. Химия, науки о материалах