

Доклады Российской академии наук. Химия, науки о материалах , 2023, T. 508, № 1, стр. 39-49
Синтез гидрированных мезо-тетрапиридилпорфиринов с регулируемым составом
А. В. Любимцев 1, *, А. С. Семейкин 1, М. О. Койфман 1, академик РАН О. И. Койфман 1
1 Федеральное государственное бюджетное образовательное учреждение высшего образования “Ивановский государственный химико-технологический университет”
153000 Иваново, Россия
* E-mail: lyubimtsev_av@isuct.ru
Поступила в редакцию 01.07.2022
После доработки 24.10.2022
Принята к публикации 26.10.2022
- EDN: EVXGPT
- DOI: 10.31857/S2686953522600465
Аннотация
Работа посвящена разработке метода получения фоточувствительной композиции контролируемого состава, содержащей катионные водорастворимые 5,10,15,20-тетракис(N-Me-пиридин-3-ил)хлорин и 5,10,15,20-тетракис(N-Me-пиридин-3-ил)бактериохлорин. Изучен процесс диимидного восстановления 5,10,15,20-тетракис(пиридин-3-ил)порфина в пиридине и в отсутствие растворителя. Показано, что проведение восстановления в плаве значительно снижает продолжительность реакции (с 12 до 1.5 ч) и позволяет получать композиции с выходом до 90%.
ВВЕДЕНИЕ
Тетрапиррольные макрогетероциклические соединения, благодаря их уникальному строению и широкому спектру физико-химических и фотохимических свойств, находят применение в качестве катализаторов различных процессов, в фото- и электрокатализе, оптических хемосенсоров для катионов металлов, селективных рецепторов органических соединений, индукторов и селекторов, в нелинейной оптике, органической электронике, в качестве магнитов, в медицине. Медицинское направление применения этих соединений связано в первую очередь с использованием их в качестве фотосенсибилизаторов для фотодинамической терапии (ФДТ) ряда онкологических заболеваний и для антимикробной ФДТ [1]. В медицинской практике успешно применяются препараты на основе природных и синтетических соединений хлоринового ряда. Подавляющее большинство фотосенсибилизаторов являются производными хлорина е6: Фотодитазин, (Деко, Россия), Фоторан е6 (Ранфарма, Россия), Фотолон (Медфармхим, Республика Беларусь), Радахлорин (РадаФарма, Россия) и др., – эффективность которых подтверждена клиническим применением. Одним из недостатков природных производных является их сложный состав. Этого недостатка лишены синтетические фотосенсибилизаторы (например, Фотосенс (ФГУП ГНЦ “НИОПИК”, Россия), Фоскан (Biolitec AG, Германия), состав которых контролируется условиями их синтеза. Возможность контролируемого синтеза и выполнения основных требований к фотосенсибилизаторам позволяет отнести водорастворимые восстановленные формы мезо-пиридилпорфиринов к потенциальным конкурентам их природных и синтетических аналогов. Начиная с 2002 г. ряд исследовательских групп опубликовали результаты по получению водорастворимого фотосенсибилизатора – “тетрагидропорфирина”, представляющего собой тетратозилат 5,10,15,20-тетракис(N-Me-пиридин-3-ил)бактериохлорина (TNMe3PyBCh) [2–7], а также его предшественника – 5,10,15,20-тетракис(пиридин-3-ил)бактериохлорина (T3PyBCh) и его тетра- и октакатионных производных с различной длиной алкильной цепи [8–15]. В настоящей работе мы приводим исследование процесса диимидного восстановления 5,10,15,20-тетракис(пиридин-3-ил)порфина (T3PyP) в пиридине и в отсутствие растворителя. Впервые выделены в индивидуальном виде 5,10,15,20-тетракис(пиридин-3-ил)хлорин (T3PyCh), водорастворимые 5,10,15,20-тетракис(N-Me-пиридин-3-ил)хлорин (TNMe3PyCh) и 5,10,15,20-тетракис(N-Me-пиридин-3-ил)бактериохлорин (TNMe3PyBCh), а также предложен эффективный метод получения фоточувствительной композиции контролируемого состава, содержащей TNMe3PyCh и TNMe3PyBCh.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Исходный 5,10,15,20-тетракис(пиридин-3-ил)порфин (T3PyP) получали конденсацией пиррола с 3-пиридилкарбальдегидом (схема 1).

Нами установлено, что наибольший выход порфирина (26%) наблюдается при проведении реакции конденсации в среде уксусная кислота–нитробензол с добавкой уксусного ангидрида для связывания выделяющейся воды. Проведение реакции в уксусной кислоте значительно снижало выход порфирина 12%, который повышается в пропионовой кислоте до 22%. Использование для проведения реакции малополярного ксилола, аналогично [16], также значительно снижало выход (4.5%).
На первом этапе для получения 5,10,15,20-тетракис(пиридин-3-ил)хлорина (T3PyCh) и 5,10,15,20-тетракис(пиридин-3-ил)бактериохлорина (T3PyBCh) мы использовали классическую процедуру Уитлока [17], нагревая T3PyP в пиридине при 100–105°С в присутствии поташа с п-толуолсульфонилгидразидом (pTSH) при постепенном прибавлении последнего (схема 2).

Реакцию проводили до максимального соотношения оптических плотностей при максимумах поглощения 745 и 653 нм, отвечающих поглощению бактериохлорина и хлорина соответственно. Показано, что данное соотношение непостоянно и достигает максимальных значений (8–10) в течение периода времени от 12 до 16 ч.
При этом не всегда увеличение продолжительности нагревания приводит к увеличению содержания T3PyBCh в реакционной смеси. Кроме того, установлено, что больший избыток восстановителя и увеличение времени реакции приводят к суммарному снижению содержания T3PyBCh и T3PyCh, что связано с образованием продуктов более глубокого восстановления.
Разделение смеси продуктов в случае пиридилзамещенных хлорина и бактериохлорина методом “кислотной отмывки” компонентов не пригодно по причине протонирования периферийных атомов азота. Поэтому для их разделения мы использовали метод колоночной хроматографии, предложенный в работе [18]. Хроматографическое разделение проводили на окиси алюминия, как описано в [18], и на силикагеле с использованием хлороформа или дихлорметана и их смесей (до 5%) с метанолом. В целом хроматография на обоих сорбентах приводит практически к одинаковым результатам, однако, разделение на силикагеле более эффективно, считая на выход продуктов.
Таким образом, в результате хроматографического эксперимента были выделены индивидуальные T3PyCh и T3PyBCh, которые были полностью охарактеризованы методами электронной (рис. 1), 1Н ЯМР-спектроскопии (рис. 2) и масс-спектрометрии (рис. 3). Следует отметить тот факт, что в литературе [3–5, 18] представлены спектральные характеристики бактериохлорина T3PyBCh. При этом в некоторых работах авторы оговаривают наличие хлорина T3PyCh [3–5]. Поскольку целевым продуктом в этих исследованиях являлся именно T3PyBCh, спектральных характеристик T3PyCh в литературе не приводится.


Как следует из рис. 1, при переходе от порфирина T3PyP к хлорину T3PyCh происходят заметные изменения в электронных спектрах их растворов. В частности, значительно снижается интенсивность полосы Соре, наблюдается ее уширение с образованием “плеча” с одновременным ростом интенсивности и небольшим батохромным сдвигом последней полосы в Q-области. Еще более заметны изменения в электронных спектрах поглощения (ЭСП) раствора бактериохлорина T3PyBCh, который характеризуется появлением трех интенсивных полос поглощения – двух в коротковолновой области при 355 и 378 нм и одной при 745 нм.
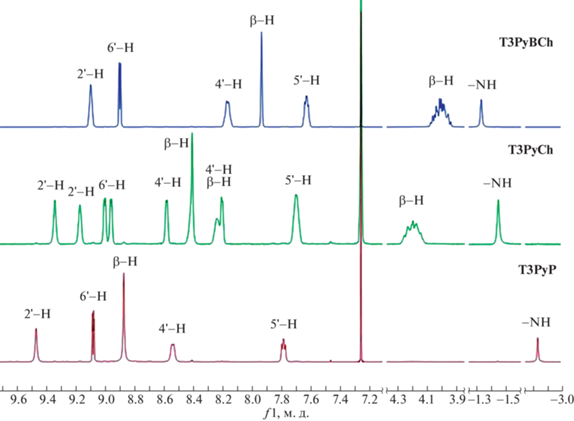
Положение сигналов протонов и анализ констант спин-спинового взаимодействия в спектрах 1Н ЯМР синтезированных соединений полностью подтверждают их индивидуальность и строение. Так, в 1Н ЯМР спектрах этих макроциклов можно выделить три характерные области (рис. 2). В слабом поле обнаруживаются сигналы ароматических протонов пиридильных фрагментов и β-протонов. При этом характер спектра усложняется при переходе от порфирина к хлорину и становится более “простым” при дальнейшем переходе к бактериохлорину, что связано с изменением симметрии молекул этих соединений. В области 4.3–3.8 м. д. для восстановленных форм порфирина присутствуют сигналы насыщенных β-протонов, причем сигналы четырех β-протонов хлорина наблюдаются в более слабом поле по сравнению с сигналами восьми β-протонов бактериохлорина. В противоположность этому сигналы протонов внутрициклических имино-групп хлорина обнаруживаются в более сильном поле. Синглет имино-протонов T3PyP проявляется при –2.83 м. д.
Незначительная разница молекулярных масс T3PyP и его восстановленных форм T3PyCh и T3PyBCh фиксируется и в масс-спектрах этих соединений (рис. 3). Так, хлорин и бактериохлорин характеризуются пиком [M + H]+, в то время как сигналу в спектре порфирина соответствует пик молекулярного иона [M]+. Следует отметить, что метод масс-спектрометрии будет менее информативным при анализе смесей T3PyP, T3PyCh и T3PyBCh.
В результате хроматографического разделения, наряду с зонами чистых T3PyCh и T3PyBCh, выделены зоны с их переменным составом. Нами показано, что медленное увеличение полярности элюента приводит к уменьшению выхода T3PyBCh и практически не сказывается на выходе T3PyCh. Кроме того, в результате хроматографического разделения выделено несколько малоподвижных зон, в электронных спектрах которых отмечается присутствие нескольких полос поглощения, гипсохромно сдвинутых по отношению к длинноволновой полосе поглощения чистого бактериохлорина T3PyBCh (рис. 4a).
Рис. 4.
Электронные спектры поглощения растворов (дихлорметан) (а) и фрагмент масс-спектра (MALDI TOF) хроматографических зон (б).

Проведенный масс-спектрометрический анализ этих зон (рис. 4б) показывает присутствие сигнала, соответствующего увеличенной на 16, 32 и 48 единиц массы молекулярного иона T3PyBCh. Данный факт можно объяснить присоединением гидроксильных групп по восстановленным связям пиррольного фрагмента. Это подтверждается литературными данными [19], в которых приводятся экспериментальные результаты по получению гидрокси- и оксохлоринов в результате взаимодействия с окисью алюминия. Факт подобных превращений на силикагеле нами установлен впервые. При этом процесс гидроксилирования быстрее протекает на окиси алюминия, чем на силикагеле, что объясняет наши данные по выходам продуктов при хроматографии на этих сорбентах.
Чтобы избежать нежелательных процессов гидроксилирования, происходящих на сорбентах, нами предложен эффективный метод разделения смеси хлорин-бактериохлорин с получением желаемого конечного продукта. Данный метод основан на различной способности хлорина и бактериохлорина к окислению и различной способности этих соединений к комплексообразованию.
Так, осторожное окисление смеси хлорина и бактериохлорина о-хлоранилом в органическом растворителе, контролируемое по исчезновению полосы в области 750 нм, приводит к получению хлорина с минимальным содержанием бактериохлорина [20–25]. Таким образом, T3PyCh получен окислением T3PyBCh в дихлорметане о-хлоранилом или DDQ. Процесс проводили до полного исчезновения полосы поглощения бактериохлорина при 745 нм. После этого реакционную смесь хроматографировали на силикагеле, элюируя смесью дихлорметан : метанол = 95 : 5 (об.).
T3PyBCh не образует цинковый комплекс в отличие от порфирина и хлорина [18], а способность к комплексообразованию между порфирином и хлорином выражена значительно меньше, чем между хлорином и бактериохлорином. Для получения T3PyBCh, не содержащего примесь хлорина (T3PyCh), раствор смеси этих соединений нагревали с ацетатом цинка в дихлорметане с добавлением метанола до тех пор, пока полоса поглощения хлорина при 653 нм не исчезнет (при этом появляется полоса поглощения цинкового комплекса хлорина в области 630 нм). После чего реакционную смесь хроматографировали на окиси алюминия смесью дихлорметан : метанол (95 : 5) под небольшим избыточным давлением. Минимальный контакт с сорбентом не приводил к продуктам гидроксилирования. Используя эти подходы, нами выделены индивидуальные T3PyCh и T3PyBCh с приемлемыми выходами, по сравнению с методом колоночной хроматографии.
Несмотря на популярность, процедура Уитлока не лишена недостатков, основными из которых являются продолжительность процесса и постепенное добавление восстановителя в ходе всего процесса. Технологичность процесса также снижает использование токсичных растворителей, которыми в различных модификациях метода выступают пиридин, пиколины, ксилол, ДМФА и др. В этой связи метод получения хлоринов (бактериохлоринов) “в плаве”, предложенный в работе [24, 25], представляется более перспективным. Этот метод апробирован на нескольких мезо-арилпорфиринах, содержащих на периферии атомы галогена, сульфамоильные и сульфогруппы. К восстановлению других замещенных мезо-арилпорфиринов данный метод не применялся.
Восстановление 5,10,15,20-тетра(пиридин-3-ил)порфина проводили в плаве pTSH в мольном соотношении 1 : 30, как и в случае восстановления в пиридине при нагревании до 115°С.
Экспериментальный мониторинг взаимодействия показал, что для достижения максимального содержания бактериохлорина в плаве требуется не более 1.5 ч. Таким образом, проведение реакции в плаве значительно сокращает время процесса и не требует применения токсичных растворителей и дополнительных источников генерации диимида (поташа). Кроме того, содержание T3PyBCh в реакционной смеси выше при проведении реакции в плаве, чем при восстановлении в пиридине (рис. 5).
Рис. 5.
Электронные спектры поглощения растворов (дихлорметан) реакционных масс восстановления T3PyP в плаве и в пиридине. Интенсивности приведены к оптической плотности T3PyBCh.

N-Метилирование T3PyCh и T3PyBCh проводили метиловым эфиром п-толуолсульфокислоты (метилтозилатом) и йодистым метилом в ДМФА при температуре 95°С в течение 1.5 ч. Продукты осаждали добавлением бензола, отфильтровывали и сушили под вакуумом при комнатной температуре. Строение полученных тетратозилатов и тетрайодидов 5,10,15,20-тетракис(N-Me-пиридин-3-ил)хлорина (TNMe3PyCh) и 5,10,15,20-тетракис(N-Me-пиридин-3-ил)бактериохлорина (TNMe3PyBCh) подтверждено с применением электронной, 1Н ЯМР-спектроскопии и масс-спектрометрии (MALDI-TOF).
В электронных спектрах поглощения N-метилированных производных TNMe3PyCh и TNMe3PyBCh наблюдается батохромный сдвиг полос поглощения, соответствующих поглощению хлорина и бактериохлорина до 660 и 761 нм соответственно, по сравнению с исходными соединениями T3PyCh и T3PyBCh (рис. 6).

Характер спектров 1Н ЯМР (рис. 7) практически не изменяется при переходе от хлорина T3PyCh и бактериохлорина T3PyBCh к их N-метилированным производным. Общим является смещение сигналов ароматических протонов в слабое поле и более выраженное различие положения сигналов внутрициклических протонов, которые фиксируются при –1.53 и –1.25 м. д. для метилированных хлорина TNMe3PyCh и бактериохлорина TNMe3PyBCh соответственно. Сигналы протонов N-метильных групп проявляются в спектре в виде двух синглетов для TNMe3PyCh и в виде синглета для TNMe3PyBCh в области 4.6 м. д. Для тетратозилатов TNMe3PyCh и TNMe3PyBCh дополнительно появляются сигналы ароматических протонов тозильного остатка при 7.38 м. д. и около 7.00 м. д. в виде двух дублетов и сигналы метильных групп в виде синглетов при 2.16 м. д.

Ранее [5] нами показано, что смесь мети-лированных тетратозилатов TNMe3PyCh и TNMe3PyBCh можно получать с выходом до 90% без промежуточной хроматографической очистки продуктов восстановления T3PyP. Мы применили этот подход в совокупности с предложенным в данной работе методом восстановления в плаве. Для этого полученный плав разбавляли метанолом и раствор приливали к водному раствору ацетата натрия с добавлением концентрированного аммиака. Выпавший осадок отфильтровывали и сушили на воздухе. Полученный технический продукт метилировали как описано выше. Для определения пороговой концентрации pTSH, при которой происходит полная конверсия порфирина в продукты восстановления, мы провели несколько опытов с мольным соотношением порфирин : pTSH, равным 1 : 15, 1 : 20 и 1 : 30. Для определения отсутствия в реакционной смеси исходного порфирина мы применили анализ продуктов методом 1Н ЯМР-спектроскопии (рис. 8). Присутствие в конечных смесях нескольких соединений значительно усложняет спектр, особенно в области ароматических протонов. В свою очередь в области сильного поля сигналы протонов внутрициклических имино-групп четко разделены между собой, что позволяет определить не только присутствие того или иного продукта в смеси, но и позволяет оценить их процентное содержание.
Рис. 8.
Положение (м. д.) и соотношение интегральных интенсивностей сигналов иминных NH-протонов T3PyBCh / T3PyCh / T3PyP при восстановлении T3PyP в плаве в избытке pTSH 15 экв. (а), 20 экв. (б) и 30 экв. (в).
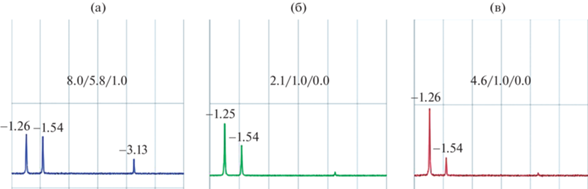
На рис. 8 приведены фрагменты 1Н ЯМР-спектров в области сигналов внутрициклических NH-протонов продуктов реакции при различных соотношениях порфирин : pTSH.
Как следует из представленных на рис. 8 данных, полная конверсия порфирина в продукты гидрирования достигается при 20-кратном избытке восстановителя, а максимальное количество бактериохлорина в смеси может быть достигнуто при соотношении 1 : 30. Воспроизводимость предложенного метода показана на примере пяти повторений (рис. 9) для соотношения порфирин : pTSH = 1 : 20. Представленные на диаграмме данные показывают высокую воспроизводимость метода как по выходу хлорина (бактериохлорина), так и по их содержанию в смеси.
Содержание основного вещества в полученных композициях оценивали методом ВЭЖХ (рис. 10). ВЭЖХ-анализ композиции на длине волны детектора 254 нм содержит не менее 96% смеси TNMe3PyCh и TNMe3PyBCh.

В заключение мы оценили стабильность растворов полученных композиций. Для этого регистрировали количественные электронные спектры поглощения раствора в воде в течение двух месяцев в темноте (рис. 11). В качестве отклика выбрано отношение коэффициентов экстинкции ε, определенных на полосах поглощения бактериохлорина и хлорина.

Результаты диаграммы показывают высокую стабильность растворов композиции N-Me-хлорина и N-Me-бактериохлорина в течение, как минимум, двух месяцев.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все растворители, если специально не указано, были марок ч., х. ч., ч. д. а. Осушку растворителей проводили по стандартным методикам. Пиррол и 3-пиридилкарбоксальдегид были коммерчески доступными (Sigma Aldrich). ЭСП регистрировались на спектрофотометре Shimadzu UV-1800, 1Н ЯМР спектры – на ЯМР-спектрометре “Bruker” Avance 500 MГц, масс-спектры (MALDI-TOF) – на времяпролетном масс-спектрометре Shimadzu Axima Confidence.
Синтез 5,10,15,20-тетракис(пиридин-3-ил)порфина (T3PyP). При кипении к смеси 350 мл уксусной кислоты, 150 мл нитробензола и 35 мл уксусного ангидрида постепенно добавляли смесь 10 мл (0.144 моль) пиррола и 13.5 мл (0.144 моль) 3-пиридилкарбоксальдегида. Смесь кипятили 1.5 ч при барботировании воздухом, затем отгоняли нитробензол с водяным паром. Реакционную массу охлаждали, разбавляли водой, отфильтровывали, промывали водой и высушивали на воздухе. Сухой осадок экстрагировали хлороформом в аппарате Сокслета до бесцветного вытека и экстракт упаривали до половины объема. Раствор хроматографировали на оксиде алюминия II степени активности по Брокману. Элюат упаривали до минимального объема, осаждали метанолом, отфильтровывали порфирин, промывали метанолом и высушивали при 70°C. Выход 5.8 г (26%).
Масс-спектр (MALDI-TOF), m/z: найдено 618.4 [M]+; вычислено для C40H26N8: 618.23. ЭСП (CHCl3, λmax, нм, (ε)): 647 (3500), 590 (6100), 550 (7600), 516 (18300), 419 (439300). 1H ЯМР (CDCl3, δ, м. д.): 9.47 (с, 4H, 2'-H), 9.08 (дд, 4H, 6'-H, J 5.0 Гц, J 1.5 Гц), 8.87 (с, 8H, β-H), 8.54 (д, 4H, 4'-H, J 6.0 Гц), 7.79 (т, 4Н, 5'-H, J 6.2 Гц), –2.83 (с, 2Н, NH).
Диимидное восстановление 5,10,15,20-тетракис(пиридин-3-ил)порфина (T3PyP) в пиридине. В двугорлой колбе с дефлегматором нагревали при перемешивании 100 мг (0.16 ммоль) 5,10,15,20-тетракис(пиридин-3-ил)порфина, 60 мг (0.32 ммоль) п-толуолсульфонилгидразида и 0.2 г карбоната калия в 10 мл пиридина при 100°С в течение 12 ч. Каждые 1.5 ч в реакционную массу добавляли по 60 мг (0.32 ммоль) п-толуолсульфонилгидразида. После охлаждения реакционную массу постепенно выливали в раствор 0.5 г ацетата натрия и 1 мл 25%-го водного аммиака в 10 мл воды. Полученную суспензию перемешивали 15 мин, отфильтровывали, осадок промывали водой до нейтральной реакции, высушивали при комнатной температуре до постоянной массы. Полученный остаток растворяли в дихлорметане (ДХМ) и хроматографировали на силикагеле или оксиде алюминия ДХМ, затем смесью ДХМ–метанол (до 5%). Собирали первую темно-малиновую зону 5,10,15,20-тетракис(пиридин-3-ил)бактериохлорина (T3PyBCh) и интенсивно окрашенную следующую зону 5,10,15,20-тетракис(пиридин-3-ил)хлорина (T3PyCh).
5,10,15,20-Тетракис(пиридин-3-ил)хлорин (T3PyCh). Выход: 12 мг (силикагель и оксид алюминия). Масс-спектр (MALDI-TOF), m/z: найдено 621.6 [M + H]+; вычислено для C40H28N8: 620.24. ЭСП (CHCl3, λmax, нм, (ε)): 653 (37 700), 599 (6200), 544 (10 600), 518 (16 000), 419 (185 000). 1H ЯМР (CDCl3, δ, м. д.): 9.34 (с, 2H, 2'-H), 9.17 (с, 2H, 2'-H) 9.00 (д, 2H, 6'-H, J 4.7 Гц), 8.96 (д, 2H, 6'-H, J 4.7 Гц), 8.58 (д, 2H, 4'-H, J 4.8 Гц), 8.41 (с, 4H, β-H), 8.31–8.13 (м, 4Н, β-H, 4'-H), 7.70 (уш.с, 4Н, 5'-H), 4.31–4.07 (м, 4H, β-H), –1.44 (с, 2Н, NH).
5,10,15,20-тетракис(пиридин-3-ил)бактериохлорин (T3PyBCh). Выход 52 мг (силикагель), 40 мг (оксид алюминия). Масс-спектр (MALDI-TOF), m/z: найдено 623.2 [M + H]+; вычислено для C40H30N8: 622.26. ЭСП (CHCl3, λmax, нм, (ε)): 746 (140 000), 520 (66 000), 378 (160 000), 355 (132 000). 1H ЯМР (CDCl3, δ, м. д.): 9.10 (уш.с, 4H, 2'-H), 8.90 (дд, 4H, 6'-H, J 5.0 Гц, J 1.6 Гц), 8.17 (д, 4H, 4'-H, J 7.6 Гц), 7.94 (уш.с, H, β-H), 7.63 (т, 4Н, 5'-H, J 6.0 Гц), 4.10–3.92 (м, 8H, β-H), –1.33 (с, 2Н, NH).
N-метилирование 5,10,15,20-тетракис(пиридин-3-ил)хлорина T3PyCh и 5,10,15,20-тетракис(пиридин-3-ил)бактериохлорина T3PyBCh. 0.1 ммоль хлорина T3PyCh или бактериохлорина T3PyBCh и 1 ммоль метилтозилата или йодистого метила в 5 мл осушенного ДМФА нагревали при перемешивании и температуре 95°С в течение 1.5 ч. По окончании выдержки реакционную массу охлаждали до комнатной температуры и разбавляли 5 мл осушенного бензола. Смесь перемешивали еще 2 ч, осадок отфильтровывали, промывали осушенным бензолом, высушивали под вакуумом при комнатной температуре.
Тетратозилат 5,10,15,20-тетракис(N-Me-пиридин-3-ил)хлорина (TNMe3PyCh). Выход 126 мг (92%). Масс-спектр (MALDI TOF), m/z: 633.5 [M-4Ts-3Me + 5H]+, 647.6 [M-4Ts-2Me + 5H]+, 661.6 [M-4Ts-Me + 5H]+. ЭСП (этанол, λmax, нм, (ε)): 660 (43 000), 606 (3800), 541 (4100), 514 (14 000), 424 (143 000). 1H ЯМР (DMSO-d6, δ, м. д.): 9.87 (уш.с, 2H, 2'-H), 9.75 (уш.с, 2H, 2'-H), 9.49 (д, 2H, 6'-H, J 5.5 Гц); 9.43 (д, 2H, 6'-H, J 5.5 Гц), 9.17 (уш.с, 4H, β-H), 9.01 (уш.с, 2H, 4'-H), 8.65 (уш.с, 2H, 4'-H), 8,59 (с, 2H, β-H), 8,55 (т, 4Н, 5'-H, J 6.0 Гц), 7,39 (д, 8H, J 7.7 Гц, 3,5-H-Ts), 6.97 (д, 8H, J 7.7 Гц, 2,6-H-Ts), 4.64 (с, 6H, –NCH3), 4.61 (с, 6H, –NCH3), 4.40–4.15 (м, 4H, β-H), 2.16 (с, 12H, –CH3), –1.53 (с, 2Н, NH).
Тетратозилат 5,10,15,20-тетракис(N-Me-пиридин-3-ил)бактериохлорина (TNMe3PyBCh). Выход 124 мг (91%). Масс-спектр (MALDI-TOF), m/z: 621.7 [M-4Ts-4Me + H]+, 633.7 [M-4Ts-3Me + 4H]+, 647.8 [M-4Ts-2Me + 5H]+, 661.8 [M-4Ts-Me + 6H]+, 685.8 [M-4Ts + 3H]+. ЭСП (этанол, λmax, нм, (ε)): 761 (104 500), 515 (48 700), 375 (90 000), 355 (86 000). 1H ЯМР (DMSO-d6, δ, м. д.): 9.68–9.57 (м, 4H, 2'-H), 9.36 (д, 4Н, 6'-H, J 6.2 Гц), 9.13–9.03 (м, 4H, 4'-H), 8.53–8.43 (м, 4H, 5'-H), 8.27 (с, 4H, β-H), 7.39 (д, 8H, J 7.9 Гц, 3,5-H-Ts), 6.95 (д, 8H, J 7.8 Гц, 2,6-H-Ts), 4.58 (с, 12H, –NCH3), 4.24–4.15 (м, 8H, β-H), 2.15 (с, 12H, –CH3), –1.25 (с, 2Н, NH).
Cмесь 5,10,15,20-тетракис(N-Me-пиридин-3-ил)хлорина (TNMe3PyCh) и 5,10,15,20-тетракис(N-Me-пиридин-3-ил)бактериохлорина (TNMe3PyBCh) контролируемого состава. Тонко перетертую смесь 0.5 г (0.81 ммоль) 5,10,15,20-тетра(пиридин-3-ил)порфина и 12.15 ммоль (15 экв.), 16.12 (20 экв.) или 24.30 ммоль (30 экв.) п-толуолсульфонилгидразида нагревали в круглодонной колбе объемом 25 мл при перемешивании на магнитной мешалке при температуре 115°С в течение 1.5 ч. По окончании выдержки реакционную массу охлаждали до 50°С и добавляли 10 мл метанола. Полученный раствор охлаждали до комнатной температуры и постепенно выливали в раствор 2.5 г ацетата натрия и 1 мл 25%-го водного аммиака в 50 мл воды. Полученную суспензию выдерживали 15 мин, отфильтровывали, осадок промывали водой до нейтральной реакции, высушивали при комнатной температуре до постоянной массы. Полученный сухой остаток растворяли в 20 мл осушенного диметилформамида и добавляли 8.10 ммоль метилтозилата или йодистого метила. Полученный раствор нагревали при перемешивании на магнитной мешалке 1.5 ч при температуре 95°С. По окончании выдержки реакционную массу охлаждали до комнатной температуры и добавляли 20 мл осушенного бензола, выдерживали при перемешивании 2 ч, осадок отфильтровывали, промывали осушенным бензолом и высушивали при комнатной температуре под вакуумом. Продукты реакции анализировали методом 1Н ЯМР- спектроскопии. Выходы и соотношение продуктов реакции приведены в тексте (рис. 9).
ЗАКЛЮЧЕНИЕ
Впервые предложен метод получения фоточувствительной композиции контролируемого состава, содержащей хлорин и бактериохлорин, заключающийся в восстановлении T3PyP в плаве с последующим метилированием полученной смеси хлорина и бактериохлорина без стадии хроматографической очистки. Предложенный метод значительно сокращает продолжительность процесса (на стадии восстановления с 12 до 1.5 ч) и является высоко воспроизводимым. Определена минимальная концентрация восстановителя, при которой происходит полная конверсия порфирина в продукты реакции.
Список литературы
Koifman O.I., Ageeva T.A., Beletskaya I.P., Averin A.D., Yakushev A.A., Tomilova L.G., Dubinina T.V., Tsiva-dze A.Yu., Gorbunova Yu.G., Martynov A.G., Konarev D.V., Khasanov S.S., Lyubovskaya R.N., Lomova T.N., Korolev V.V., Zenkevich E.I., Blaudeck T., von Borczyskowski Ch., Zahn D.R.T., Mironov A.F., Bragina N.A., Ezhov A.V., Zhdanova K.A., Stuzhin P.A., Pakhomov G.L., Rusakova N.V., Semenishyn N.N., Smola S.S., Parfenyuk V.I., Vashurin A.S., Makarov S.V., Dereven’kov I.A., Mamardashvili N.Zh., Kurtikyan T.S., Martirosyan G.G., Burmistrov V.A., Aleksandriiskii V.V., Novikov I.V., Pritmov D.A., Grin M.A., Suvorov N.V., Tsigankov A.A., Fedorov A.Yu., Kuzmina N.S., Nyu-chev A.V., Otvagin V.F., Kustov A.V., Belykh D.V., Bere-zin D.B., Solovieva A.B., Timashev P.S., Milaeva E.R., Gracheva Yu.A., Dodokhova M.A., Safronenko A.V., Shpakovsky D.B., Syrbu S.A., Gubarev Yu.A., Kiselev A.N., Koifman M.O., Lebedeva N.Sh., Yurina E.S. // Macroheterocycles. 2020. № 13. P. 311–467. https://doi.org/10.6060/mhc200814k
Shastak S., Shulga A., Berr F., Wiedemann P. Porphyrins and their use as photosensitizer. Patent US 6410568 B1. 2002.
Oertel M., Shastak S., Tannapfel A., Hermann R., Sack U., Moessner J., Berr F. // J. Photochem. Photobiol., B. 2003. V. 71. P. 1–10. https://doi.org/10.1016/S1011-1344(03)00091-5
Shastak S., Jean B., Handzel R., Kostenich G., Hermann R., Sack U., Orenstein A., Wang Yu., Wiedemann P. // J. Photochem. Photobiol., B. 2005. V. 78. P. 203–213. https://doi.org/10.1016/j.jphotobiol.2004.11.006
Койфман О.И., Пономарев Г.В., Сырбу С.А., Жаров Е.В., Сергеева Т.В., Луковкин А.В. Фотосенсибилизатор и способ его получения. Патент РФ № 2535097 C1. 2014.
Berndt-Paetz M., Weimann A., Sieger N., Shastak S., Riyad Y., Griebel J., Arthanareeswaran V., Stolzenburg J-U., Neuhaus J. // Photodiagnostics Photodyn. Ther. 2017. V. 18. P. 244–251. https://doi.org/10.1016/j.pdpdt.2017.02.017
Hambsch P., Istomin Yu., Tzerkovsky D., Patties I., Neuhaus J., Kortmann R.-D., Shastack S., Glasow A. // Oncotarget. 2017. V. 8. № 42. P. 72411–72423. https://doi.org/10.18632/oncotarget.20403
Тиганова И.Г., Макарова Е.А., Меерович Г.А., Алексеева Н.В., Толордава Э.Р., Жижимова Ю.С., Лукьянец Е.А., Романова Ю.М. // Biomed. Photonics. 2017. Т. 6. № 4. С. 27–36. https://doi.org/10.24931/2413-9432-2017-6-4-27-36
Морозова Н.Б., Плотникова Е.А., Плютинская А.Д., Страмова В.О., Воронцова М.С., Панкратов А.А., Якубовская Р.И., Макарова Е.А., Лукъянец Е.А., Каприн А.Д. // Российский Биотерапевтический Журнал. 2018. Т. 17. № 3. С. 55–64. https://doi.org/10.17650/1726-9784-2018-17-3-55-64
Макарова Е.А., Меерович Г.А., Лукьянец Е.А., Тиганова И.Г., Романова Ю.М., Лощенов В.Б., Алексее-ва Н.В., Ахлюстина Е.В. Фотосенсибилизаторы для фотодинамической инактивации бактерий, в том числе в биопленках. Патент РФ № 2670201. 2018.
Меерович Г.А., Ахлюстина Е.В., Тиганова И.Г., Макарова Е.А., Филипова Н.И., Романишкин И.Д., Алексеева Н.В., Лукьянец Е.А., Романова Ю.М., Лощенов В.Б. // Вестник РГМУ. 2018. № 6. С. 80–85. https://doi.org/10.24075/brsmu.2018.087
Meerovich G.A., Akhlyustina E.V., Tiganova I.G., Lukyanets E.A., Makarova E.A., Tolordava E.R., Yuzhako-va O.A., Romanishkin I.D., Philipova N.I., Zhizhimo-va Yu.S., Romanova Yu.M., Loschenov V.B., Gintsburg A.L. // Adv. Exp. Med. Biol. 2019. P. 1–19. https://doi.org/10.1007/5584_2019_431
Meerovich G.A., Akhlyustina E.V., Tiganova I.G., Lukyanets E.A., Makarova E.A., Tolordava E.R., Yuzhako-va O.A., Romanishkin I.D., Philipova N.V., Zhizhimo-va Yu.S., Gonchukov S.A., Romanova Yu.M., Losche-nov V.B. // Laser Phys. Lett. 2019. V. 16. P. 115603. https://doi.org/10.1088/1612-202X/ab4806
Tiganova I.G., Zhizhimova Yu.S., Philipova N.I., Tolordava E.R., Alekseeva N.V., Makarova E.A., Lukyanets E.A., Meerovich G.A., Romanova Yu.M., Gintsburg A.L. // Mol. Gen. Microbiol. Virol. 2020. V. 35. № 4. P. 248–256. https://doi.org/10.3103/S0891416820040096
Kogan E.A., Meerovich G.A., Karshieva S.Sh., Akhlyustina E.V., Makarova E.A., Dalina A.A., Romanishkin I.D., Zharkov N.V., Gonchukov S.A., Angelov I.P., Losche-nov V.B. // Laser Phys. Lett. 2022. V. 19. № 3. P. 035601. https://doi.org/10.1088/1612-202X/ac4e96
Семейкин А.С., Койфман О.И., Березин Б.Д. // ХГС. 1986. № 6. С. 798–801.
Whitlock H., Hanauer J., Oester M., Bower B. // J. Am. Chem. Soc. 1969. P. 7485–7489. https://doi.org/10.1021/ja01054a044
Dudkin S., Makarova E., Slivka L., Lukyanets E. // J. Porphyrins Phthalocyanines. 2014. V. 18. P. 107–114. https://doi.org/10.1142/S1088424613501162
Burns D., Li Yu., Shi D., Delaney M. // Chem. Commun. 1998. P. 1677–1678. https://doi.org/10.1039/A802165A
Laville I., Pigaglio S., Blais J.-C., Loock B., Maillard Ph., Grierson D., Blais J. // Bioorg. Med. Chem. 2004. V. 12. P. 3673–3682. https://doi.org/10.1016/j.bmc.2004.04.022
Hirohara S., Obata M., Ogata S., Ohtsuki S., Higashida S., Ogura S., Okura I., Takenaka M., Ono H., Sugai Y., Mikata Y., Tanihara M., Yano M. // J. Photochem. Photobiol., B. 2005. V. 78. P 7–15. https://doi.org/10.1016/j.jphotobiol.2004.09.003
Bonnett R., White R., Winfield U-J., Berenbaum M. // Biochem. J. 1989. V. 261. P. 277–280. https://doi.org/10.1042/bj2610277
Laville I., Figueiredo T., Loock B., Pigaglio S., Mail-lard Ph., Grierson D., Carrez A., Blais J. // Bioorg. Med. Chem. 2003. V. 11. P. 1643–1652. https://doi.org/doi:10.1016/S0968-0896(03)00050-6
Pereira M., Abreu A., Goncalves N., Calvete M., Simões A., Monteiro C., Arnaut L., Eusébio M., Canotilho J. // Green Chem. 2012. V. 14. P. 1666–1672. https://doi.org/10.1039/C2GC35126A
Dabrowski J., Arnaut L., Pereira M., Monteiro C., Urbanska K., Sims S., Stochel G. // Chem. Med. Chem. 2010. V. 5. P. 1770–1780. https://doi.org/10.1002/cmdc.201000223
Дополнительные материалы отсутствуют.
Инструменты
Доклады Российской академии наук. Химия, науки о материалах