

Электрохимия, 2019, T. 55, № 1, стр. 95-105
Эволюция состава анoлита при окислительном электролизе сернокислого раствора бромида натрияМ. М. Петров, П. А. Локтионов, Д. В. Конев, А. Е. Антипов, Е. А. Астафьев, М. А. Воротынцев
М. М. Петров a, b, П. А. Локтионов a, Д. В. Конев a, c, *, А. Е. Антипов a, b, Е. А. Астафьев c, М. А. Воротынцев a, b, c, d, **
a Российский химико-технологический университет им. Д.И. Менделеева
125047 Москва, А-47, Миусская пл., 9, Россия
b Московский государственный университет им. М.В. Ломоносова
119992 Москва, ГСП-1, Ленинские горы, 1, Россия
c Институт проблем химической физики РАН
142432 Московская обл., Черноголовка, просп. академика Семенова, 1, Россия
d Институт молекулярной химии университета Бургундии
Дижон, Франция
* E-mail: dkfrvzh@gmail.com
** E-mail: mivo2010@yandex.com
Поступила в редакцию 30.08.2018
После доработки 30.08.2018
Принята к публикации 13.09.2018
Аннотация
Исследован процесс окисления 10 мМ водного раствора бромида натрия в сернокислой среде на поверхности платинового электрода в ячейке с разделенными простанствами, который важен ввиду использования бром-бромидной редокс-пары в проточных редокс-батареях. В работе применялись методы циклической вольтамперометрии, а также окислительного электролиза в режиме хроноамперометрии, в ходе которого проводилась снятие серии оптических спектров поглощения в УФ-видимом диапазоне, а также измерения потенциала редокс-электрода сравнения. Разработана численная процедура обработки экспериментально измеренных спектров раствора для их разделения на вклад молекулярного брома Br2 и остаточный сигнал, отнесенный на основе сопоставления с литературными данными к поглощению трибромид-аниона ${\text{Br}}_{3}^{ - }.$ Установленные таким образом экспериментальные зависимости концентраций Br2 и ${\text{Br}}_{3}^{ - }$ для окислительного электролиза раствора NaBr в сернокислой среде хорошо согласуются с теоретическими предсказаниями. Оценен выход по току для процесса образования брома.
ВВЕДЕНИЕ
Бром и его соединения широко применяются в различных областях. Они используются для производства антипиренов [1], пестицидов [2], в системах охлаждения воды и воздуха [3]. Кроме того, ввиду возросшего интереса к электрохимическими источникам тока в последние годы появились работы, посвященные водород-бромным проточным редокс-батареям (ПРБ) с бром-бромидной редокс-парой [4, 5].
Лучшие образцы водород-бромных ПРБ показывают удельные мощности вплоть до 1.5 Вт/см2 [5] и в перспективе хорошо подходят для различных стационарных энергетических приложений – например, в качестве источников резервного питания или для хранения электроэнергии, производимой в непостоянном режиме солнечными и ветряными электростанциями. Однако применение водородно-бромных проточных батарей ограничено коррозионной активностью брома, деградацией платинового катализатора на поверхности водородного электрода вследствие кроссовера брома, а также сравнительно высокой стоимостью брома.
Предложенные недавно водородно-броматные ПРБ [6], в которых бромат-анион ${\text{BrO}}_{3}^{ - }$ используется в качестве высокоэнергетического многоэлектронного окислителя, лишены этих недостатков. Показано, что 6-электронное восстановление бромат-анионов до бромид-анионов [7], протекающее в автокаталическом режиме, соответствует огромным теоретическими значениям плотностей тока разряда, а также максимальной среди всех проточных батарей энергоемкости. Однако на данный момент продемонстрирована только работа водородно-броматных проточных батарей в режиме генератора тока [8], в то время, как для реализации полноценного энергетического цикла необходимо обеспечить еще эффективный режим регенерации электроэнергии, то есть зарядки батареи.
В работе [8] показано, что работающая в режиме разряда водородно-броматная проточная батарея дает в качестве основного продукта кислые бромид-содержащие растворы, которые во время регенерации необходимо окислить обратно до бромат-аниона. Известно, что бромид-анион самопроизвольно переходит в бромат-анион при высоких pH [9]. Также существуют некоторые другие способы превращения бромид-иона в бромат-ионы, например, с помощью хлорирования и обработки ультрафиолетом [10, 11] или озонирования в сочетании с обработкой перекисью водорода H2O2 [12]. Однако с точки зрения замкнутого энергетического цикла проточной батареи наиболее выгодным и удобным было бы превращать бромид-ион в бромат-ион с помощью электроокисления – по возможности без какой-либо коррекции pH.
Во многих работах описывается электроокисление бромид-анионов как до бромат-анионов, так и до соединений брома даже более высоких степеней окисления [13–18]. Однако они не подходят для режима регенерации топлива водородно-броматных батарей, поскольку в них проводится электролиз щелочных или нейтральных бромидсодержащих растворов [14–18] с использованием таких дорогостоящих электродных материалов как допированный бором алмаз (ДБА) [13, 14, 16], в то время как электроокисление бромид-иона в реакторе регенерации для наибольшей эффективности замкнутого энергетического цикла выгодней всего проводить в его кислых растворах, т.е. без коррекции рН. Кроме того, во многих из приведенных работ проводятся исследования только с низкими концентрациями бромид-ионов [13, 14, 16, 17], а электролиз бромид-содержащих растворов ведется при потенциалах, выходящих за пределы электрохимического окна стабильности водных электролитов [14, 16–18], что приводит к сопутствующим реакциям электролиза воды и, как следствие, неизбежным энергопотерям.
В литературе имеются работы, описывающие электроокисление бромид-иона до брома, проведенное на платиновых электродах в кислых водных средах [19–22]. Некоторые из них говорят о возможности дальнейшего электроокисления до соединений более высокой степени окисления – например, гипобромит-анионов BrO–. Так, в исследованиях [19, 20] на циклических вольтамперограммах в анодной области наблюдается две пары редок-пиков: первая в области 0.94 В относительно каломельного электрода (ее ассоциируют с редокс-парой Br2/Br–) и вторая в области 1.29 В относительно каломельного электрода (ее сопоставляют с переходом Br–/BrO–). Эти данные представляют большой интерес с точки зрения электрохимической регенерации отработанного топлива водородно-броматной проточной батареи, поскольку гипобромит-анион – это один из обязательных промежуточных продуктов в серии реакций восстановления бромат-ионов [23]. Однако гипотеза об электрохимическом получении гипобромита BrO– не подвергается в работах [20, 21] никакой дополнительной проверке, а другие авторы интерпретируют аналогичные данные, полученные при электроокислении бромид-ионов в нитробензоле и ацетонитриле [24, 25], как пики редокc-пар Br–/${\text{Br}}_{3}^{ - }$ и ${\text{Br}}_{3}^{ - }$/Br2.
Цель данного исследования – это разработка методики оперативного определения состава бромсодержащей среды, подходящей для исследования в будущем реакций электролизного окисления бромид-анионов при различных условиях проведений процесса. Для достижения этой цели в настоящей работе была проведена серия экспериментов в проточной ячейке специальной конструкции, способной одновременно регистрировать оптические спектры в диапазоне видимого света и ближнего УФ, оценивать общую редокс-емкость системы, а также определять равновесный потенциал индикаторного платинового электрода в бромсодержащей среде, находящейся в анодной камере трехэлектродной электрохимической ячейки с разделенными пространствами.
Применимость новой методики проверена на процессе электролизного окисления 10 мМ NaBr в кислой среде (0.7 M H2SO4) при потенциале 1.1 В относительно Ag/AgCl значения концентраций Br2 и ${\text{Br}}_{3}^{ - }$ в зависимости от количества прошедшего заряда, которые были сопоставлены с теоретическими предсказаниями на основе термодинамических данных из литературы.
МЕТОДИКА ЭКСПЕРИМЕНТА
Подготовка образцов
Для приготовления раствора электролита использовались следующие вещества: серная кислота (H2SO4, ХЧ, производство “Химмед”, массовая концентрация – 95%), бромид натрия (NaBr, ≥99%, Sigma Aldrich), сульфат натрия (Na2SO4, 99%, Sigma Aldrich) и бромат натрия (NaBrO3, ≥99%, Sigma Aldrich). pH системы был измерен на стационарном pH-метре АНИОН 4100 с использованием электрода ЭСК-10601/7 и составлял 0.41.
Для приготовления эталонного водного раствора брома (5 мМ Br2) смешивались точные объемы растворов 0.3 М NaBr и 0.15 М NaBrO3 и 2 М H2SO4, обеспечивающие стехиометрическое соотношение бромида и бромата натрия (5 : 1) в реакции (1) на фоне избытка серной кислоты:
(1)
$\begin{gathered} {\text{5NaBr}} + {\text{NaBr}}{{{\text{O}}}_{{\text{3}}}} + {\text{3}}{{{\text{H}}}_{{\text{2}}}}{\text{S}}{{{\text{O}}}_{{\text{4}}}} = \\ = {\text{3B}}{{{\text{r}}}_{{\text{2}}}} + {\text{3}}{{{\text{H}}}_{{\text{2}}}}{\text{O}} + {\text{3N}}{{{\text{a}}}_{{\text{2}}}}{\text{S}}{{{\text{O}}}_{{\text{4}}}}. \\ \end{gathered} $Циклическая вольтамперометрия
Для измерений методом циклической вольтамперометрии (ЦВА) использовалась трехэлектродная схема с хлоридсеребряным электродом сравнения (XCЭ, Ag/AgCl в 3.5 M KCl), платиновой проволокой в качестве вспомогательного электрода и дисковым платиновым электродом диаметра 1 мм в качестве рабочего электрода. Величины потенциалов здесь и далее приводятся относительно Ag/AgCl-электрода в 3.5 M KCl. Измерения проводились на потенциостате ELINS-50-Pro в экранированной от внешних электромагнитных полей клетке Фарадея. Используемый раствор был обескислорожен до проведения опыта, а измерения проводились в инертной среде аргона.
ЦВА измерялись в пределах от –220 до 1500 мВ, скорость сканирования составляла 100 мВ/c.
Окислительный электролиз бромида натрия с регистрацией эволюции оптического спектра поглощения анолита
Электролиз 10 мМ бромидсодержащих растворов проводился в атмосфере аргона в герметизированной кварцевой оптической кювете специальной конструкции с погруженной на дно мешалкой. В тефлоновой крышке кюветы были предусмотрены отверстия для четырех электродов: платинового противоэлектрода, хлоридсеребряного электрода сравнения (Ag/AgCl-электрод в 3.5 M KCl), платинового рабочего электрода в виде квадратной сеточки со стороной 1.5 и шагом в 1.5 мм, а также индикаторного электрода в виде платиновой проволоки длиной 3 см и диаметром 1 мм. Объем исследуемого раствора V внутри кюветы составлял 2.3 мл.
Использовался потенциостатический режим с наложением на 90 мин потенциала 1.1 В относительно Ag/AgCl с помощью потенциостата Autolab PGSTAT302N, регистрировавшего экспериментальные зависимости тока от пропущенного заряда.
Оптические спектры поглощения раствора регистрировались периодически в ходе электролиза с помощью оптоволоконного спектрофотометра Avantes StarLine AvaSpec2048. Интервал между последовательными записями оптических спектров составлял 10 с. Длина оптического пути – 10 мм. В качестве спектра сравнения регистрировался спектр кварцевой кюветы с тридистиллированной водой. Все измерения проводились при включенной мешалке для равномерного распределения всех компонентов раствора по его объему, работа потенциостата и спектрофотометра синхронизировалась при помощи ПК.
Индикаторный электрод (платиновая проволока) и хлоридсеребряный электрод сравнения были подключены к входному каналу АЦП потенциостата Autolab 302N через дифференциальный вычитающий повторитель, обеспечивающий высокое входное сопротивление и отсутствие влияния измерительного тока на регистрируемую разницу потенциалов.
Математическая обработка оптических спектров поглощения
Алгоритм программы (предложенный и подробно описанный в [26]) реализуется в программной среде Mathcad. Он позволяет разложить экспериментальный оптический спектр, снятый в определенный момент в ходе электролиза, на вклад поглощения растворенным молекулярным Br2 и остаточный спектр.
Спектр Aexp(λ) (поглощение в зависимости от длины волны) может быть представлен в виде суммы двух компонентов
где λ – длина волны, Aet(λ) – эталонный оптический спектр поглощения 5 мМ Br2 (см. выше о его синтезе), предварительно измеренный в таких же условиях, Ares(λ) – остаточный оптический спектр, а X – коэффициент от 0 до 1, при котором достигается минимальная сумма выражения при суммирования вкладов для длин волн λ1 в пределах от 380 до 430 нм, т.е. в узкой окрестности максимума поглощения растворенного брома. Таким образом, найденное значение X позволяет вычислить степень конверсии бромид-аниона Br– в молекулярный бром Br2 в момент измерения спектра. При X = 0 в системе отсутствует Br2, при X = 1 весь Br– переведен окислением в Br2, а XAet(l) показывает спектр брома, который накопился в растворе к данному моменту времени. Для каждого момента времени измерения спектра Aexp(1) после нахождения соответствующего значения Х можно использовать соотношение (2), чтобы вычислить остаточный оптический спектр Ares(1).Теоретические зависимости концентраций компонентов раствора от пропущенного заряда
Теоретические виды зависимостей концентраций соединений брома от равновесного потенциала системы рассчитаны по аналогии с работой [27]. Для заданных значений начальной концентрации бромид-аниона ctot, а также равновесного потенциала E решается система следующих уравнений:
(4)
$\frac{{\left[ {{\text{B}}{{{\text{r}}}_{2}}} \right]}}{{{{{\left[ {{\text{B}}{{{\text{r}}}^{ - }}} \right]}}^{2}}}} = {{10}^{{2A\left( {E - E_{1}^{^\circ }} \right)}}},$(5)
$\frac{{\left[ {{\text{Br}}_{3}^{ - }} \right]}}{{{{{\left[ {{\text{B}}{{{\text{r}}}^{ - }}} \right]}}^{3}}}} = {{10}^{{2A\left( {E - E_{2}^{^\circ }} \right)}}},$(6)
$\left[ {{\text{B}}{{{\text{r}}}^{ - }}} \right] + {{\left[ {{\text{Br}}_{3}^{ - }} \right]} \mathord{\left/ {\vphantom {{\left[ {{\text{Br}}_{3}^{ - }} \right]} 3}} \right. \kern-0em} 3} + {{\left[ {{\text{B}}{{{\text{r}}}_{2}}} \right]} \mathord{\left/ {\vphantom {{\left[ {{\text{B}}{{{\text{r}}}_{2}}} \right]} 2}} \right. \kern-0em} 2} = {{c}_{{{\text{tot}}}}},$РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Анализ ЦВА
На рис. 1 представлена циклическая вольтамперограмма (ЦВА) для процесса электроокисления кислого раствора NaBr на платиновом электроде. В области отрицательных потенциалов видны характерные для сернокислой среды пики адсорбции/десорбции водорода на поверхности платины [28]. В области положительных потенциалов видны три группы волн: (1) пара пиков окисления/восстановления с потенциалом полуволны E1/2 = Ера + Ерс = 0.952 В, соответствующих редокс-паре Br2/Br–; (2) слабая катодная волна в области –0.55 В, соответствующая обратному восстановлению поверхностного оксида платины Pt–OH, образующегося в конкурирующем с окислением бромид-иона процессе [20]; (3) пик окисления в области 1.40 В с плохо разрешенным обратным пиком восстановления в области 1.33 В.
Рис. 1.
Циклическая вольтамперограмма Pt-электрода в 10 мМ NaBr + 0.3 М Na2SO4 + 0.7 М H2SO4. pH 0.41. Скорость сканирования: 100 мВ/c.

Интерпретация последней пары редокс-пиков в литературе различается. В ряде работ ее ассоциируют с электроокислением Br2 до соединений брома в более высоких степенях окисления [21] – например, гипобромит-аниона BrO–. При этом отсутствие ярко выраженного обратного пика восстановления BrO– в данном случае может объясняться высокой скоростью гомогенной реакции конпропорционирования (8) в кислой среде [23]:
(8)
${\text{HOBr}} + {\text{B}}{{{\text{r}}}^{--}} + {{{\text{H}}}^{ + }} = {\text{B}}{{{\text{r}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}.$Однако в других исследованиях аналогичные данные интерпретируются иным образом. Так, в работе [29] похожий по форме и положению пик связывали электроокислением до HOBr адсорбированного на поверхности бромид-аниона Br–, а в работах [24, 25], где изучали электроокисление бромид-ионов в неводных средах, две волны схожей формы в области положительных потенциалов связывали с редокс-парами Br–/${\text{Br}}_{3}^{ - }$ и ${\text{Br}}_{3}^{ - }$/Br2.
Окислительный электролиз бромид-аниона
В дальнейших экспериментах электролиз бромид-аниона проводился в потециостатическом режиме при потенциале E = 1.1 В. Такое значение было выбрано в соответствии с данными ЦВА из следующих соображений: (1) оно находится внутри окна электрохимической стабильности водных электролитов при использовании платины в качестве материала рабочего электрода; (2) потенциал E = 1.1 В расположен между двумя анодными волнами на ЦВА-кривой (рис. 1), так что в таких условиях не должен проходить процесс, обуславливающий вторую анодную волну около потенциала 1.4 В, поэтому анализ состава раствора в ходе электролиза может предоставить информацию о процессах в ходе обеих анодных волн.
Заметим, что из термодинамических и кинетических данных для реакций (1) и (8) следует отсутствие в растворе бромат- и гипобромит-ионов по крайней мере до истощения бромид-ионов в растворе из-за прохождения быстрых реакций конпропорционирования. В то же время термодинамическими данными не исключалось накопление в значительных количествах трибромид-аниона ввиду реакции:
На рис. 2а приведены экспериментальные зависимости I(t) и Q(t), полученные при проведении окислительного электролиза сернокислого 10 мM NaBr в течении 90 мин. Видно, что за время эксперимента через систему проходит Q = 2.20 Кл.
Рис. 2.
(а) Хроноамперограмма (сплошная линия) и хронокулонограмма (пунктирная линия), зарегистрированные в ходе потенциостатического электролиза (E = 1.1 В) раствора состава 10 мМ NaBr + 0.3 М Na2SO4 + 0.7 М H2SO4 на платиновом электроде. Стрелками показаны соответствующие кривым оси ординат. (б) Спектры поглощения анолита в ходе электролиза (рис. 1) в различные моменты времени, t, c: 1 – 1, 2 – 10, 3 – 100, 4 – 250, 5 – 500, 6 – 1000, 7 – 2500 и 8 – 5400.

Этот заряд следует сопоставить с зарядом, необходимым для полного одноэлектронного окисления Br– до молекулярного брома Br2:
После подстановки F = 96485 Кл/моль, ctot = = 10 мМ и V = 2.3 мл получается, что Qэкв = 2.219 Кл (будем называть эту величину эквивалентным зарядом), а для величины Q/Qэкв введем обозначение Y (относительный заряд).
В ходе электролиза регистрировались спектры поглощения системы (рис. 2б). Видно, что с началом электролиза (см. кривую для t = 30 с) на спектрах начинает быстро расти пик в области 265 нм, который можно приписать трибромид-аниону ${\text{Br}}_{3}^{ - }$ [30]. Молярный коэффициент экстинкции ${\text{Br}}_{3}^{ - }$ на длине волны 260 нм ε(${\text{Br}}_{3}^{ - }$) является очень большой величиной: 37 200 M–1 cм–1 [31], т.е. более чем на два порядка превышает коэффициент экстинкции Br2 в области его максимума поглощения: ε(Br2) = 167 М–1 cм–1 при 400 нм [32, 33]. Рост концентрации бромид-аниона в ходе электролиза быстро приводит к превышению порога чувствительности прибора (см. кривые для t = 100, 250, 500 с). Одновременно с этим на оптических спектрах становится хорошо различим широкий пик поглощения в области 400 нм, соответствующий молекулярному брому Br2 [30]. При дальнейшем электролизе на оптических спектрах начинает уменьшаться вклад трибромид-аниона (можно сравнить кривую t = 1000 с, где вклад ${\text{Br}}_{3}^{ - }$ еще явно выражен, и кривые t = 2500 и 5400 с, вид которых уже близок к оптическим спектрам чистого Br2 c широким пиком в области 400 нм и резким подъемом в коротковолновой области, начиная с 290–300 нм), а также стабилизируется интенсивность полосы поглощения Br2 (см. кривые для t = = 1000, 2500 и 5400 с; небольшое уменьшение высоты пика для t = 5400 по сравнению с t = 2500 с будет рассмотрено ниже).
Результаты процедуры разложения экспериментальных оптических спектров
Рисунок 2б подходит для качественного анализа поведения системы, однако количественный анализ на основе таких данных затруднен, потому что оптические спектры различных соединений брома накладываются друг на друга в определенных интервалах длин волн. Поэтому нами была применена процедура разделения экспериментальных спектров Aexp(λ) на две составляющие: вклад за счет поглощения молекулярным бромом XAet(λ) и остаточный спектр Ares(λ). Вклад молекулярного брома определялся как произведение эталонного спектра на коэффициент Х, подбор которого осуществлялся по описанному в разделе “математическая обработка спектров поглощения” алгоритму и пересчитывался в текущее содержание брома в анолите. Типичный пример обработки графических результатов на основе предложенного алгоритма приведен на рис. 3. Видно, что после проведения процедуры разделения в качестве остаточного спектра остается спектр трибромид-аниона [30], что служит подтверждением корректности выбранной процедуры.
Рис. 3.
Графическая иллюстрация результатов работы алгоритма разложения экспериментального спектра поглощения анолита (Aexp(λ), кривая 1) на спектр брома (XAet(λ), кривая 2) и остаточный спектр (Ares(λ), кривая 3). Для разложения использовался эталонный спектр раствора брома (5 мМ Br2, кривая 4). Экспериментальный спектр соответствует t = 1070 с (относительный заряд Y = 0.9), X = 0.837.
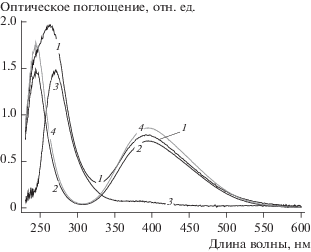
Из рис. 2б видно, что на оптических спектрах поглощения отсутствуют характерные признаки каких-либо других соединений брома, кроме молекулярного брома Br2 и трибромид-аниона ${\text{Br}}_{3}^{ - }$ – например бромат-аниона ${\text{BrO}}_{3}^{ - }$ с характерным подъемом в коротковолновой области, начиная с длины волны 270 нм [33].
Отсутствие других соединений брома в значимых концентрациях относительно общей аналитической концентрации брома ожидаемо в системе с выбранными нами экспериментальными условиями. В кислой среде равновесие реакций конпропорционирования (8) и (1) с участием HOBr и ${\text{BrO}}_{3}^{ - }$ сильно сдвинуто в сторону образования Br2 [34], что исключает получение путем гомогенных химических реакций значительного количества соединений брома в более высоких степенях окисления, чем нулевая (Br2) [23].
Поэтому в пределах прошедшего заряда вплоть до Qэкв в системе присутствуют значимые количества только трех соединений брома: Br–, Br2 и ${\text{Br}}_{3}^{ - }.$ При этом концентрация Br2 может быть количественно оценена по спектру XAet(λ), полученному после разложения экспериментального оптического спектра системы по оптическому поглощению на длине волны 396 нм (соответствует максимуму оптического поглощения Br2), а концентрация ${\text{Br}}_{3}^{ - }$ может быть оценена из остаточного спектра Ares(λ) по оптическому поглощению на длине волны 310 нм (такая длина волны выбрана для того, чтобы избежать попадания в область превышения чувствительности прибора).
Зависимость концентраций различных соединений брома от количества прошедшего заряда. Сопоставление с термодинамическими предсказаниями
Для расчета экспериментальных значений концентраций Br2 и ${\text{Br}}_{3}^{ - }$ использовали молярные коэффициенты экстинкции $\varepsilon _{{{\text{B}}{{{\text{r}}}_{{\text{2}}}}}}^{{396}}$ = 167 М–1 см–1 и $\varepsilon _{{{\text{Br}}_{3}^{ - }}}^{{310}}$ = 7400 М–1 см–1 [23]. Результаты расчетов приводятся на рис. 4 и сопоставляются с термодинамическими предсказаниями, сделанными по аналогии с работой [28]. В качестве свободных параметров модели, описанной выше в пункте “теоретические зависимости” используется две величины: $E_{1}^{^\circ }$ – стандартный потенциал редокс-пары Br–/Br2 и $E_{2}^{^\circ }$ – стандартный потенциал редокс-пары Br–/${\text{Br}}_{3}^{ - }.$ $E_{1}^{^\circ }$ = 1.0874 [34], а $E_{2}^{^\circ }$ подбирается из соображений наилучшего совпадения термодинамических предсказаний для вида зависимости [${\text{Br}}_{3}^{ - }$] от относительного заряда Y и результатов эксперимента (см. рис. 4б).
Рис. 4.
(а) Зависимость концентрации ${\text{Br}}_{3}^{ - }$ от относительного заряда. Точки – результат обработки спектрофотометрических данных, сплошные линии – расчет согласно системе уравнений (4)–(6) для различных значений $E_{2}^{^\circ },$ B: 1 – 1.053, 2 – 1.052, 3 – 1.051. (б) Зависимость концентрации Br2 от относительного заряда: точки – результат обработки спектрофотометрических данных, сплошная линия – расчет согласно системе уравнений (4)–(6) при $E_{2}^{^\circ }$ = 1.052 В, пунктирная линия – гипотетическая ситуация, когда молекулярный бром является единственным продуктом электроокисления бромида со 100% выходом по току.

На рис. 4а видно, что наилучшее совпадение результатов эксперимента наблюдается для E2 = = 1.052 В, что хорошо совпадает с табличными значением E2 = 1.0503 [34]. Также с помощью уравнений (4), (5) из значений $E_{1}^{^\circ }$ и $E_{2}^{^\circ }$ может быть вычислена термодинамическая константа образования трибромид-аниона K = [${\text{Br}}_{3}^{ - }$]/[Br2][Br–]. Принимая E1 = 1.0874 В, а E2 = 1.052 В, получим $K = {{10}^{{2A\left( {E_{1}^{^\circ } - E_{2}^{^\circ }} \right)}}}$ = 16 М–1, что неплохо согласуется с литературными значением K = 18 М–1 [35, 36]. В дальнейшем для термодинамических расчетов использовались $E_{1}^{^\circ }$ = 1.0874 В, а $E_{2}^{^\circ }$ = 1.052 В. Из рис. 4б видно, что предсказанные на основе таких констант зависимости также согласуются с экспериментальными: концентрация трибромид-аниона ${\text{Br}}_{3}^{ - }$ достигает максимума в 0.2 мМ в районе Y = Q/Qэкв = 0.45–0.5, а затем убывает до нуля при Y = 1.0.
На рис. 4б приведены теоретические и экспериментальные зависимости концентрации молекулярного брома от прошедшего во время электролиза заряда. Видно, что концентрация молекулярного брома Br2 постепенно растет по мере протекания электролиза вплоть до точки Y = 0.97, после прохождения которой эта величина начинает убывать. На графике можно выделить три основных области. В первой из них – от Y = 0 до Y = 0.1 экспериментальная концентрация брома по данным спектрофотометрии (точки) несколько меньше, чем концентрация, полученная из термодинамических предсказаний (сплошная линия). При этом 0.1 эквивалентного заряда проходит через систему уже за первые 50 с электролиза (при общей продолжительности электролиза в 5400 с). Такое отклонение экспериментальных данных от предсказаний может объясняться несколькими факторами. Во-первых, на таких маленьких временных масштабах в системе еще не устанавливаются равновесные значения концентраций – бром, образовавшийся во время электролиза в приэлектродном слое не успевает распределиться по объему системы и попасть в луч спектрофотометра. А во-вторых, в начальные моменты электролиза значительная доля энергии идет на образование поверхностных оксидов платины и, возможно, накопление адсорбированного брома.
На этапе электролиза от Y = 0.1 до Y = 0.8 экспериментально (спектрофотометрически) наблюдаемая концентрация молекулярного брома постепенно начинает превышать термодинамические предсказания и сравнивается с предельно возможной концентрацией, рассчитанной при условии отсутствия в системе трибромид-аниона (пунктирная линия). Такое отклонение экспериментальных результатов от предсказаний в свою очередь может объясняться систематическими экспериментальными погрешностями, связанными, например, с приготовлением раствора необходимой концентраций.
Наконец, на заключительном этапе электролиза – от Y = 0.8 до Y = 1.0 прирост концентрации молекулярного брома в анолите снова начинает постепенно отставать от термодинамических предсказаний, что может объясняться постепенным испарением летучего брома из спектрокюветы, которое в этой области уже оказывает существенное влияние на состав системы из-за сравнительно большей длительности заключительного этапа процесса: электролиз от Y = 0.8 до 1.0 эквивалентных заряда занимает 4700 с, в то время электролиз от Y = 0 до 0.8 – только 700 с.
В районе Y = 0.97 концентрация молекулярного брома достигает максимума в 4.5 мМ, что близко к максимальной концентрации в 5 мМ, достижимой при полном одноэлектронном окислении 10 мМ бромид-аниона до молекулярного брома Br2. После прохождения Y = 0.97 наблюдается резкий спад концентрации, который можно объяснить тем, что все бромид-анионы в системе уже окислены до брома и в системе Br2 начинает постепенно переходить в HOBr.
Оценка выхода по току для реакции электроокисления бромид-аниона Br– до молекулярного брома Br2
Выход по току для реакции электроокисления бромид-аниона Br до молекулярного брома Br2 за период от начала процесса до произвольного момента электролиза (Neff) может быть оценен по формуле:
(11)
${{N}_{{{\text{eff}}}}} = {{2\left[ {{\text{B}}{{{\text{r}}}_{{\text{2}}}}} \right]FV} \mathord{\left/ {\vphantom {{2\left[ {{\text{B}}{{{\text{r}}}_{{\text{2}}}}} \right]FV} Q}} \right. \kern-0em} Q}.$На рис. 5 представлена экспериментальная зависимость выхода по току от количества прошедшего заряда в эквивалентных единицах в сравнении с аналогичной зависимостью, построенной на основе предсказаний термодинамики.
Рис. 5.
Зависимость выхода по току Neff для реакции электроокисления бромид-аниона до молекулярного брома Br2 от относительного заряда: точки – результат обработки спектрофотометрических данных, сплошная линия – расчет согласно системе уравнений (4)–(7) при $E_{2}^{^\circ }$ = 1.052 В.

Для теоретической зависимости видно, что с протеканием электролиза интегральный выход по току постепенно увеличивается и при Y = 1.0 достигает 100% – то есть все прошедшие заряды пошли на электроокисление бромид-аниона до молекулярного брома и в системе не остается каких-либо значимых количеств трибромид-аниона. При этом в начале электролиза (вплоть до Y = 0.1) теоретический выход по току составляет не более 90%, то есть значительная часть прошедшего заряда – не менее 10% уходит на образование трибромид-аниона.
В экспериментальной зависимости можно выделить те же самые три области, что и на рис. 4а. В области от Y = 0 до Y = 0.1 эквивалентных заряда наблюдается резкий рост величины выхода по току от 0 до примерно 0.85. При этом экспериментальный выход по току везде в пределах этого участка остается меньше термодинамических предсказаний, что можно объяснить неравномерным распределением образовавшегося во время гетерогенной реакции брома по объему системы, а также образованием поверхностного оксида платины и адсорбцией брома.
Во второй области – от Y = 0.1 до Y = 0.8 – экспериментальное значение выхода по току начинает превышать аналогичную величину, рассчитанную на основе термодинамических предсказаний, и постепенно достигает максимально возможного значения, равного 1.0. Такое аномальное поведение может объясняться экспериментальными погрешностями, связанными с приготовлением исходных растворов – то есть исходно в системе была несколько большая концентрация атомов брома, чем 10 мМ используемая в термодинамических расчетах.
Наконец, в третьей области от Y = 0.8 до Y = 1.0 экспериментальные значения выхода по току начинают отставать от термодинамических предсказаний и постепенно уменьшаются до Neff = 0.9, а в точке Y = 0.97Neff резко уменьшается примерно до 0.8. Такое поведение может объясняться сочетанием двух факторов – возросшей на больших временных масштабах ролью процесса испарения брома, а также появлением в системе HOBr после полного электроокисления бромид-анионов до брома.
Зависимость потенциала индикаторного электрода от прошедшего заряда. Сопоставление с результатами расчета
Из термодинамических предсказаний также можно получить зависимость равновесного потенциала в системе бром–вода при электроокислении бромид-аниона от прошедшего заряда. На рис. 6 она дается в сравнении с экспериментальной зависимостью потенциала индикаторного электрода, введенного в контакт с раствором внутри спектроэлектрохимической ячейки. Видно, что представленные зависимости хорошо согласуются между собой, что подтверждает возможность использования индикаторного электрода для определения равновесного потенциала системы при проведении окислительного электролиза бромид-ионов.
Рис. 6.
Зависимость потенциала индикаторного электрода от относительного заряда. Точки – результат измерения в ходе электролиза, сплошная линия – расчет согласно системе уравнений (4)–(7) при $E_{2}^{^\circ }$ = = 1.052 В.

Таким образом, можно построить экспериментальные зависимости концентраций атомов брома в составе различных соединений брома (т.е. с учетом их стехиометрических коэффициентов: Br–, 2Br2 и 3${\text{Br}}_{3}^{ - }$) от потенциала индикаторного электрода и сравнить их с термодинамическими предсказаниями, как это сделано на рис. 7. Видно, что экспериментальная зависимость для ${\text{Br}}_{3}^{ - }$ повторяет теоретическую, как на качественном, так и на количественном уровне (за исключением небольшого сдвига максимума концентрации трибромид-аниона в область более низких потенциалов).
Рис. 7.
Распределительная диаграмма атомов брома по различным соединениям брома с учетом их стехиометрических коэффициентов: 1 – [Br–], 2 – 2[Br2], 3 – 3[${\text{Br}}_{3}^{ - }$] от равновесного потенциала системы: точки – результат обработки спектрофотометрических данных, сплошная линия – расчет согласно системе уравнений (4)–(7) при $E_{2}^{^\circ }$ = 1.052 В.

На кривой для [Br2] наблюдается анлогичное по сравнению с рис. 5 и 6 разбиение на характерные области: до E = 0.89 В экспериментальная зависимость отстает от теоретической, после она начинает ее опережать и такое поведение сохраняется вплоть до E = 0.95 В. При этом примерно в районе E = 0.97 В экспериментальная зависимость уже начинает сильно отставать от термодинамических предсказаний, и после выхода на плато 2[Br2] = 0.9 М в районе E = 1.04 В начинает резко спадать, что снова можно объяснить постепенным накоплением в системе HOBr после исчерпания бромид-анионов Br–, которые до этого вступали с молекулами HOBr в реакцию конпропорционирования (4).
Более точное выяснение природы этого отклонения, а также изучение возможностей образования HOBr и соединений брома в более высоких степенях окисления при электролизном окислении бромид-анионов в кислых средах – это тема отдельного исследования, особенно актуального в контексте вопроса регенерации отработанного топлива водородно-броматной проточной батареи [25]. В рамках данной работы показано, что метод спектроэлектрохимии с последующими алгоритмическим разложением оптических спектров поглощения на составляющие подходит для эффективного определения состава бромсодержащей среды во время электролизного окисления бромид-аниона до молекулярного брома.
ВЫВОДЫ
В работе проведен окислительный электролиз кислых бромидсодержащих растворов с синхронной регистрацией спектров поглощения, а также равновесного потенциала системы с помощью введенного индикаторного электрода. С помощью процедуры разложения спектров оценены зависимости концентраций различных соединений брома от прошедшего заряда. Полученные результаты хорошо совпадают с термодинамическими предсказаниями, что показывает возможность использования спектров поглощения для количественной оценки состава системы.
Показано, что зависимость равновесного потенциала системы, измеренного с помощью индикаторного электрода, от количества прошедшего заряда, совпадает с термодинамическими предсказаниями. Это демонстрирует возможность использования метода потенциометрии для мониторинга состава системы.
Оценена величина выхода по току для полного электроокисления бромид-аниона до молекулярного брома: Neff = 0.85.
Оценена термодинамическая константа образования трибромид-аниона K = 16 M–1. Ее величина хорошо согласуется с литературными данными.
Получены косвенные свидетельства (данные ЦВА, данные спектроэлектрохимии) возможности образования HOBr при окислительном электролизе бромид-аниона в кислой среде.
Список литературы
Alaee, M. et al., An overview of commercially used brominated flame retardants, their applications, their use patterns in different countries/regions and possible modes of release, Environment international, 2003, vol. 29, no. 6, pp. 683–689.
Ristaino, J.B. and Thomas, W., Agriculture, methyl bromide, and the ozone hole: can we fill the gaps?, Plant Disease, 1997, vol. 81, no. 9, pp. 964–977.
Jenner, H.A. et al., Cooling water management in European power stations: biology and control of fouling. Paris: Electricité de France, 1998, vol. 1.
Tolmachev, Y.V., Hydrogen-halogen electrochemical cells: A review of applications and technologies, Russian J. of Electrochemistry, 2014, vol. 50, no. 4, pp. 301–316.
Cho, K.T., Tucker, M.C., and Weber, A.Z. A review of hydrogen/halogen flow cells, Energy Technology, 2016, vol. 4, no. 6, pp. 655–678.
Tolmachev, Y.V. et al., Energy cycle based on a high specific energy aqueous flow battery and its potential use for fully electric vehicles and for direct solar-to-chemical energy conversion, J. of Solid State Electrochemistry, 2015, vol. 19, no. 9, pp. 2711–2722.
Vorotyntsev, M.A., Konev, D.V., and Tolmachev, Y.V., Electroreduction of halogen oxoanions via autocatalytic redox mediation by halide anions: novel EC” mechanism. Theory for stationary 1D regime, Electrochimica Acta, 2015, vol. 173, pp. 779–795.
Modestov, A.D. et al., A Hydrogen–Bromate Flow Battery for Air-Deficient Environments, Energy Technology, 2018, vol. 6, no. 2, pp. 242–245.
Kelsall, G.H., Welham, N.J., and Diaz, M.A., Thermodynamics of Cl–H2O, Br–H2O, I–H2O, Au–Cl–H2O, Au–Br–H2O and Au–I–H2O systems at 298 K, J. of Electroanalytical Chemistry, 1993, vol. 361, nos. 1–2, pp. 13–24.
Huang, X., Gao, N., and Deng, Y., Bromate ion formation in dark chlorination and ultraviolet/chlorination processes for bromide-containing water, J. of Environmental Sciences, 2008, vol. 20, no. 2, pp. 246–251.
Fang, J. et al., Bromate formation from the oxidation of bromide in the UV/chlorine process with low pressure and medium pressure UV lamps, Chemosphere, 2017, vol. 183, pp. 582–588.
Von Gunten, U. and Oliveras, Y., Advanced oxidation of bromide-containing waters: bromate formation mechanisms, Environmental Science & Technology, 1998, vol. 32, no. 1, pp. 63–70.
Ferro, S., The bromine electrode Part III: reaction kinetics at highly boron-doped diamond electrodes, J. of Applied Electrochemistry, 2005, vol. 35, no. 3, pp. 279–283.
Bergmann, M.E.H., Iourtchouk, T., and Rollin J., The occurrence of bromate and perbromate on BDD anodes during electrolysis of aqueous systems containing bromide: first systematic experimental studies, J. of Applied Electrochemistry, 2011, vol. 41, no. 9, pp. 1109.
Osuga, T. and Sugino, K., Electrolytic production of bromates, J. of The Electrochemical Society, 1957, vol. 104, no. 7, pp. 448–451.
Vacca, A. et al., On the formation of bromate and chlorate ions during electrolysis with boron doped diamond anode for seawater treatment, J. of Chemical Technology & Biotechnology, 2013, vol. 88, no. 12, pp. 2244–2251.
Cettou, P., Robertson, P.M., and Ibl, N., On the electrolysis of aqueous bromide solutions to bromate, Electrochimica Acta, 1984, vol. 29, no. 7, pp. 875–885.
Pavlović, O.Ž., Krstajić, N.V., and Spasojević, M.D., Formation of bromates at a RuO2TiO2 titanium anode, Surface and Coatings Technology, 1988, vol. 34, no. 2, pp. 177–183.
Conway, B.E., Phillips, Y., and Qian, S.Y., Surface electrochemistry and kinetics of anodic bromine formation at platinum, J. of the Chemical Society, Faraday Transactions, 1995, vol. 91, no. 2, pp. 283–293.
Ferro, S. and De Battisti, A., The bromine electrode. Part I: Adsorption phenomena at polycrystalline platinum electrodes, J. of Applied Electrochemistry, 2004, vol. 34, no. 10, pp. 981–987.
Ferro, S., Orsan, C., and De Battisti, A., The bromine electrode Part II: reaction kinetics at polycrystalline Pt, J. of Applied Electrochemistry, 2005, vol. 35, no. 3, pp. 273–278.
Xu, J., Georgescu, N.S., and Scherson, D.A., The Oxidation of Bromide on Platinum Electrodes in Aqueous Acidic Solutions: Electrochemical and In Situ Spectroscopic Studies, J. of The Electrochemical Society, 2014, vol. 161, no. 6, pp. H392–H398.
Kshirsagar, G. and Field, R.J., A kinetic and thermodynamic study of component processes in the equilibrium 5HOBr = 2Br2 + ${\text{BrO}}_{3}^{ - }$+ 2H2O + H+, The J. of Physical Chemistry, 1988, vol. 92, no. 25, pp. 7074–7079.
Chang, J., Bennett, B., and Bard, A.J., Detection of an Unstable Intermediate in Br− Electro-oxidation to ${\text{Br}}_{3}^{ - }$ on a Platinum Electrode in Nitrobenzene by Scanning Electrochemical Microscopy, Electrochimica Acta, 2017, vol. 238, pp. 74–80.
Allen, G.D. et al., A mechanistic study of the electro-oxidation of bromide in acetonitrile and the room temperature ionic liquid, 1-butyl-3-methylimidazolium bis (trifluoromethylsulfonyl) imide at platinum electrodes, J. of Electroanalytical Chemistry, 2005, vol. 575, no. 2, pp. 311–320.
Konev, D.V. et al., In situ UV-visible spectroelectrochemistry in the course of oxidative monomer electrolysis, Electrochimica Acta, 2015, vol. 179, pp. 315–325.
Michalowski, T., Calculation of pH and potential E for bromine aqueous solution, J. of Chemical Education, 1994, vol. 71, no. 7, pp. 560.
Clavilier, J. et al., Preparation of monocrystalline Pt microelectrodes and electrochemical study of the plane surfaces cut in the direction of the {111} and {110} planes, J. of Electroanalytical Chemistry and Interfacial Electrochemistry, 1980, vol. 107, no. 1, pp. 205–209.
Johnson, D.C. and Bruckenstein, S.A., Ring-Disk Study of HOBr Formation at Platinum Electrodes in 1.0 M H2SO4, J. of The Electrochemical Society, 1970, vol. 117, no. 4, pp. 460–467.
Beckwith, R.C., Characteristics of aqueous bromine and hypobromous acid and the kinetics and mechanism of the reaction of bromine and hydroxylamine, 1997.
Beckwith, R.C. and Margerum, D.W., Kinetics of hypobromous acid disproportionation, Inorganic chemistry, 1997, vol. 36, no. 17, pp. 3754–3760.
Betts, R.H. and Mackenzie, A.N., Formation and stability of hypobromous acid in perchloric acid solutions of bromine and bromate ions, Canadian J. of Chemistry. 1951, vol. 29, no. 8, pp. 666–677.
Citri, O. and Epstein, I.R., Systematic design of chemical oscillators. 43. Mechanistic study of a coupled chemical oscillator: the bromate-chlorite-iodide reaction, The J. of Physical Chemistry, 1988, vol. 92, no. 7, pp. 1865–1871.
Bard, A.J., Standard potentials in aqueous solution. Routledge, 2017.
Griffith, R.O., McKeown, A., and Winn, A.G., The bromine-bromide-tribromide equilibrium, Transactions of the Faraday Society, 1932, vol. 28, pp. 101–107.
Liebhafsky, H. A., The equilibrium constant of the bromine hydrolysis and its variation with temperature, J. of the American Chemical Society, 1934, vol. 56, no. 7, pp. 1500–1505.
Дополнительные материалы отсутствуют.