

Электрохимия, 2019, T. 55, № 8, стр. 1011-1017
Электрохимические свойства N-метил- и N-фенил-2,4,6-трифенилпиридиний перхлората
А. В. Долганов a, *, Б. С. Танасейчук a, О. В. Тарасова a, О. Ю. Черняева a, Ю. М. Селиванова a, А. Д. Юдина a, К. А. Григорьян a, А. В. Баландина a, В. Ю. Юрова a
a Национальный исследовательский Мордовский государственный университет им. Н.П. Огарева, Институт физики и химии
430005 Саранск, ул. Большевистская, 68, Россия
* E-mail: dolganov_sasha@mail.ru
Поступила в редакцию 04.09.2018
После доработки 24.01.2019
Принята к публикации 06.02.2019
Аннотация
С использованием метода циклической вольтамперометрии (ЦВА) изучены электрохимические свойства N-метил- и N-фенил-2,4,6-трифенилпиридиний перхлоратов. Показано, что природа заместителя у атома азота оказывает сильное влияние как на значения потенциалов восстановления солей, так и на электрохимическую обратимость процессов переноса второго электрона.
ВВЕДЕНИЕ
В настоящее время большое внимание уделяется переходу от традиционных технологий получения и переработки тепла и электричества к новой водородной энергетике: использование молекулярного водорода является одним из возможных путей рационального, экологически безопасного и устойчивого развития современной энергетики [1, 2]. Одним из бурно развивающихся в настоящее время направлений этой энергетики является поиск высокопроизводительных каталитических систем [3, 4]. Однако дизайн и синтез оптимальных электрокатализаторов остается весьма сложной задачей [5, 6]. Критически важными параметрами любой электрокаталитической системы являются значения потенциалов восстановления и устойчивость редокс-форм [7–10]. “Настраивать” значения редокс-потенциалов необходимо для осуществления эффективного процесса генерирования молекулярного водорода без перенапряжения либо при минимальных значениях [10]. “Молекулярные платформы” на основе гетероциклических органических солей представляют в настоящее время одну из перспективных “платформ” для создания катализаторов нового поколения [11, 12]. Ранее советскими учеными был выполнен большой цикл работ, посвященный изучению процесса электрокаталитического получения водорода в присутствии пиридина, а затем и в присутствии других органических систем [13–18]. В работах было показано, что органические системы имеют большой потенциал для их использования в электрокаталитическом процессе. Данная статья является логическим продолжением развитых в выше названных работах идей. “Молекулярные платформы” на основе пиридина способны легко трансформироваться путем точечной функционализации, что позволит “настраивать” потенциал в зависимости от типа используемого источника H+ [19]. Ранее нами представлены данные по проявлению электрокаталитической активности в реакции получения молекулярного водорода у нового класса катализаторов – органической гетероциклической соли N-метил-9-фенилакридиния иодида [11, 12]. В представленной статье описываются электрохимические свойства гетероциклических солей на основе 2,4,6-триарилпиридинильных N-метил (I)- и N-фенил-2,4,6-трифенилпиридиний перхлората (II) (схема 1 ).
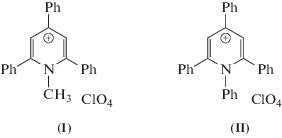
Схема 1 . N-метил (I)- и N-фенил-2,4,6- трифенилпиридиний перхлората (II).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Методы исследования
Электрохимические данные были получены методом циклической вольтамперометрии в растворе ацетонитрила (0.1 М фонового электролита ((н-C4H9)4N)BF4) с использованием потенциостата Gamry (Канада) в электрохимической ячейке объемом 5 мл. В качестве рабочего электрода использовался стеклоуглеродный (CУ) электрод S2 = 0.125 см2. Электрод тщательно отполировали и промыли перед измерениями. Вспомогательным электродом выступал платиновый, а электродом сравнения – стандартный хлоридсеребряный электрод (E0 = 0.33 В в CH3CN отн. Fc/Fc+). Все растворы были полностью деаэрированы путем продувания через них аргона.
Полная оптимизация геометрии и вычисление полной энергии исследуемых модельных систем проводилось методом функционала плотности (DFT) при использовании гибридного функционала B3LYP [20] и стандартного базисного набора 6-31G [21] из пакета прикладных программ Firefly [22]. Полная оптимизация молекулярной геометрии проводилась без каких-либо ограничений по симметрии с учетом влияния растворителя (ацетонитрила) в рамках модели поляризованного континуума РСМ [20], в которой полость, содержащая растворенную частицу, строится из совокупности пересекающихся атомных сфер определенного радиуса. Внутри полости диэлектрическая проницаемость такая же, как в вакууме, снаружи она принимает значение растворителя [23]. Данные для учета растворителя: диэлектрическая проницаемость, радиус молекулы взяты с сайта https://www.scm.com/doc/ADF/Input/COSMO.html. Для подтверждения достижения истинного минимума в ходе оптимизации геометрии рассчитывались частоты нормальных колебаний. Отсутствие мнимых частот в колебательном спектре оптимизированной структуры означает, что полученная структура отвечает минимуму на полной поверхности потенциальной энергии.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Наличие объемных заместителей в 2,4,6-положении у производных пиридина (I и II) необходимо для экранирования образующихся в ходе электрокаталитического процесса реакционноспособных интермедиатов. Отсутствие заместителей в представленных положениях приводит к быстрой потере каталитической активности катализатора в связи с протеканием побочных химических процессов. Присутствие ароматических фрагментов позволяет влиять на электрокаталитическую активность соединений за счет дополнительной делокализации электронов.
Электрохимические свойства соединений I и II были изучены методом ЦВА (рис. 1).
Рис. 1.
ЦВА N-метил-2,4,6-трифенилпиридиний перхлорат (а) и N-фенил-2,4,6-трифенилпиридиний перхлорат (б) (c = 1 мМ, СУ, 0.2 В/с, CH3CN, 0.1 M Bu4NBF4, Ag/AgCl/KClaq). Вставка: зависимость ∆E для первой (1) и второй (2) волны восстановления ΔE от скорости развертки потенциала.

Все наблюдаемые на вольтамперограммах волны соответствуют процессам с диффузионным ограничением тока. В анодной области ЦВА всех изученных соединений наблюдаются необратимые волны окисления, типичные для окисления перхлорат-иона (на рис. 1 не приведены). В катодной области в случае соединений I и II наблюдаются по две одноэлектронных волны при потенциалах Е1 = –(0.89/0.83), Е2 = –(1.067/0.97) и Е1 = = –(0.93/0.87), Е2 = –(1.4/1.33) соответственно, при скорости развертки потенциала 0.2 В/с. Отношение токов прямого к обратному равно 1, а величины ΔE = $E_{{\text{p}}}^{{\text{a}}}$ – $E_{{\text{p}}}^{{\text{c}}}$, характеризующие обратимый процесс, равны 60 и 70 мВ как для соединения I, так и для соединения II. Из рис. 1 видно, что оба соединения претерпевают первый редокс-переход при близких значениях потенциала с образованием устойчивого радикала, следовательно, можно предположить, что заместитель у атома азота слабо влияет на данный потенциал. Это может быть объяснено лишь тем, что положительный заряд (т.е., место атаки электрона) локализован на атоме С4, а заместители, расположенные у атома азота, слабо на него влияют.
Наиболее интересными оказались влияние природы заместителя у атома азота на значение второй волны восстановления, поскольку изменение природы заместителя в случае N-метил-2,4,6-трифенилпиридиний перхлората на N-Ph приводит к смещению потенциала восстановления в катодную область на 330 мВ. На наш взгляд, столь необычное влияние заместителей связано с тем, что в случае восстановления N-фенил-2,4,6-трифенилпиридиний перхлората образующийся радикал может делокализоваться по фенильному заместителю, находящемуся у атома азота. Тогда как в случае N-метил-2,4,6-трифенилпиридиний перхлората, образующийся радикал не имеет дополнительных мест делокализации, и, кроме того, образующийся из него анион будет дестабилизироваться индуктивным эффектом метильной группы у атома азота (схема 2 ).

Схема 2 .
Для подтверждения предположения о различной степени делокализации электронов в образующихся после электрохимического восстановления соединений I и II радикалов, были проведены квантовохимические расчеты с использованием метода DFT (см. Экспериментальную часть) (рис. 2).

Как видно из рис. 2, в случае соединения I метильный заместитель, расположенный у атома азота, не участвует в делокализации неспаренного электрона, тогда как фенильный заместитель в соединении II участвует в делокализации, что и приводит к увеличению устойчивости системы. Кроме того, интересно отметить, что длина связи C–NPh при переходе от катиона к радикалу существенно уменьшается, тогда как связь C‒NСH3 в том же ряду практически не меняется (см. ниже рис. 3). Укорочение связи в данном случае также может свидетельствовать о возможности делокализации неспаренного электрона по фенильному заместителю.
Рис. 3.
Равновесные геометрии катиона, радикала и аниона для соединений I и II, а также их свободные энергии, ккал/моль.

При изучении зависимости ΔE от скорости развертки потенциала были выявлены следующие интересные зависимости: ΔE первых волн восстановления не зависят от скорости развертки потенциала, что характерно для обратимых электрохимических систем, тогда как для вторых волн восстановления с увеличением скорости развертки потенциала ΔE увеличивается. Такое поведение типично для квазиобратимого электрохимического процесса [25]. Можно предположить, поскольку положительный заряд в перхлоратах N‑метил- и N-фенил-2,4,6-трифенилпиридиний главным образом локализован на 4-м атоме углерода, имеющем sp2-гибридизацию, перенос на него одного электрона существенно не влияет на изменение гибридизации, так как для радикалов также характерна sp2-гибридизация. Тогда как перенос второго электрона на SOMO-орбиталь образующихся радикалов приводит к изменению гибридизации с sp2 на sp3 и образованию малостабильного 8-электронного антиароматического остова, что способствует увеличению энергии орбитали, и как следствие этого электрохимический процесс приобретает квазиобратимый характер [26].
С целью выявления основных мотивов, влияющих на различное электрохимическое поведение при последовательном переходе редокс-форм от катиона до аниона, для соединений I и II были проведены квантовохимические расчеты. Как уже отмечалось выше, делокализация неспаренного электрона в случае соединения II заметно выше, чем для соединения I, однако электрохимическое восстановление обоих соединений до аниона происходит квазиобратимо. Таким образом, можно предположить, что эффект делокализации не является основным мотивом, влияющим на квазибратимость процесса. Как видно из рис. 3, при последовательном переносе двух электронов для обоих соединений сильно изменяется геометрия остова и, соответственно, длины связей. На основе этого, можно предположить, что реорганизация сольватной оболочки при переходе от радикала к аниону будет сильно влиять на обратимость редокс-перехода. Действительно, как видно из представленных на рис. 3 данных, в случае обоих соединений электрохимическое образование аниона из радикала сопровождается сильными структурными искажениями. Как следствие этого, данный процесс должен протекать с высокой энергией реорганизации сольватной оболочки при переходе от радикала к аниону: радикал – электронейтральное соединение, тогда как анион – заряженное. Таким образом, можно предположить, что именно высокая энергия реорганизации сольватной оболочки при электрохимическом восстановлении радикалов до аниона является “ответственной” за квазиобратимость редокс-процесса.
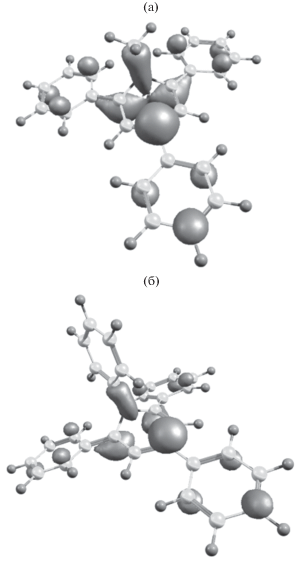
Кроме того, как видно из рис. 3 присоединение второго электрона приводит к сильным структурным искажениям: в обоих случаях наблюдается частичное нарушение планарности ароматического цикла вследствие выхода из него атома азота гетероцикла и “вхождения” фенильных заместителей, расположенных у 2, 4 и 6-го атома углерода, в сопряжение с ароматической системой (рис. 3 и 4).
Рис. 4.
Распределение электронной плотности электронов для НОМО орбиталей для анионов соединения I (а) и соединения II (б).
Распределение электронной плотности на НОМО-орбиталях обоих соединений показывает участие в делокализации отрицательного заряда фенильных заместителей, расположенных в 2, 4 и 6-м положениях, тогда как заместители у атома азота не участвуют в делокализации (рис. 4). Таким образом, при схожей геометрии анионов, а также схожей карты распределения электронной плотности на НОМО-орбиталях соединений I и II, основным фактором, влияющим на различное электрохимическое поведение радикалов для соединений I и II, является распределение электронной плотности в образующихся радикалах и как следствие этого отличия в значениях вторых потенциалов восстановления. Для подтверждения предположения нами были рассчитаны, окислительно-восстановительные потенциалы для исходных соединений I и II и продуктов их последовательного электрохимического восстановления. Расчет был произведен с использованием уравнения [27]:
(1)
$E_{{{\text{abc}}}}^{0} = - \left( {{{\Delta G_{{{\text{abc}}}}^{0}} \mathord{\left/ {\vphantom {{\Delta G_{{{\text{abc}}}}^{0}} {nF}}} \right. \kern-0em} {nF}}} \right),$На основе полученных свободных энергий нами были рассчитаны теоретические значения потенциалов для наблюдаемых редокс-процессов (табл. 1)
Таблица 1.
Сравнение экспериментальных и теоретических катодных редокс-потенциалов для соединений I и II
| Соединение | Эксперимент, *E0,В отн. | Расчет | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| **Fc/Fc+ | Ag/AgCl/KCl | $\Delta G_{{{\text{abs}}}}^{0}$, кДж/моль | $E_{{{\text{abc}}}}^{{\text{0}}}$, В Fc+/Fc = 4.4 В |
E0 отн. Fc+/Fc Fc+/Fc = 0 В |
||||||
| Е1 | Е2 | Е1 | Е2 | Е1 | Е2 | Е1 | Е2 | Е1 | Е2 | |
| I | –1.19 | –1.35 | –0.86 | –1.02 | –312.66 | –300.12 | 3.24 | 3.11 | –1.16 | –1.29 |
| II | –1.23 | –1.70 | –0.9 | –1.37 | –310.73 | –265.38 | 3.22 | 2.75 | –1.18 | –1.65 |
Как видно из таблицы, рассчитанные показатели находятся в хорошем соответствии с экспериментальными значениями. Кроме того, как следует из данных, представленных на рис. 2, в случае соединения II как исходный катион, так радикал и анион более стабильны по сравнению с аналогами для соединения I. На основе полученных результатов можно однозначно утверждать, что именно разница в термодинамической стабильности при переходе от катиона к аниону является ответственной за наблюдаемые нетривиальные редокс-свойства.
ЗАКЛЮЧЕНИЕ
Таким образом, показано, что природа заместителя у атома азота в замещенных трифенилпиридинильных солях оказывает сильное влияние на значение второго редокс-перехода процесса восстановления. Полученные результаты в дальнейшем будут полезны при конструировании молекулярных катализаторов процесса получения молекулярного водорода в присутствии органических солей, так как близость значений потенциалов восстановления по двум стадиям может приводить к деактивации катализатора за счет образования 4–H продукта.
Список литературы
Turner, J.A., Sustainable Hydrogen Production, Science, 2004, vol. 305, p. 972.
Lewis, N.S. and Nocera, D.G., Powering the planet: chemical challenges in solar energy utilization, Proc. Natl. Acad. Sci. U.S.A., 2006, vol. 103, p. 15729.
Helm, M.L., Stewart, M.P., Bullock, R.M., Rakowski, DuBois M., and DuBois, D.L., A Synthetic Nickel Electrocatalyst with a Turnover Frequency Above 100,000 s−1 for H2 Production, Science, 2011, vol. 333, p. 863.
Karunadasa, H.I., Chang, C.J., and Long, J.R., A molecular molybdenum-oxo catalyst for generating hydrogen from water, Nature, 2010, vol. 464, p. 1329.
McKone, J.R., Marinescu, S.C., Brunschwig, B.S., Winkler, J.R., and Gray, H.B., Earth-abundant hydrogen evolution electrocatalysts, Chemical Science, 2014, vol. 5, p. 865.
Afgan, N.H., Veziroglu, A., and Carvalho, M.G., Multi-criteria evaluation of hydrogen system options, Int. J. Hydrogen Energy, 2007, vol. 32, p. 3183.
Wang, M., Chen, L., and Sun, L., Recent progress in electrochemical hydrogen production with earth-abundant metal complexes as catalysts, Energy Environ Sci., 2012, vol. 5, p. 6763.
Artero, V., Chavarot-Kerlidou, M., and Fontecave, M., Splitting water with cobalt, Angew. Chem. Int. Ed., 2011, vol. 50, p. 7238.
Belaya, I.G., Svidlov, S.V., Dolganov, A.V., Zelinskii, G.E., Potapova, T.V., Vologzhanina, A.V., Varzatskii, O.A., Bubnov, Y.N., and Voloshin, Y.Z., Apically linked iron(II) dioximate and oximehydrazonate bis-clathrochelates with hydrocarbon spacer substituents and their semi- and monoclathrochelate precursors and analogs: synthetic strategy, structure, redox and electrocatalytic properties, Dalton Trans., 2013, vol. 42, p. 13667.
Lebed, E.G., Belov, A.S., Dolganov, A.V., Vologzhanina, A.V., Szebesczyk, A., Gumienna-Kontecka, E., Kozlowski, H., Bubnov, Y.N., Dubey, I.Y., and Voloshin, Y.Z., First clathrochelate iron and cobalt(II) tris-dioximates with reactive apical substituents, Inorg. Chem. Comm., 2013, vol. 30, p. 53.
Dolganov, A.V., Tanaseichuk, B.S., Moiseeva, D.N., Yurova, V.Y., Sakanyan, J.R., Shmelkova, N.M., and Lobanov, V.V., Acridinium salts as metal-free electrocatalyst for hydrogen evolution reaction, Electrochem Commun., 2016, vol. 68, p. 59.
Dolganov, A.V., Tanaseichuk, B.S., Ivantsova, P.M., Tsebulaeva, Y.V., Kostrukov, S.G., Moiseeva, D.N., Shmelkova, N.M., Yurova, V.Y., Balakireva, O.I., Nagaeva, I.G., and Trushkova, N.N. Metal-free electrocatalyst for hydrogen production from water, Intern. J. Electrochem. Sci., 2016, vol. 11, p. 9559.
Майрановский, С.Г. Каталитические и кинетические волны в полярографии. М.: Наука, 1966. 288 с.
Майрановский, С.Г. Теория каталитических волн водорода в полярографии органических соединений. Успехи химии, 1964, Т. 33. С. 75. [Mairanovskii, S.G., Theory of catalytic hydrogen waves in organic polarography Russ. Chem. Rev., 1964, vol. 33, p. 38.]
Майрановский, С.Г. Зависимость полярографических каталитических волн водорода от строения органических катализаторов. Докл. АН СССР. 1962. Т. 42. С. 1327. [Mairanovskii, S.G., Polarographic catalytic hydrogen waves as dependent on the structure of the organic catalyst, Dokl. Akad. Nauk SSSR, 1962, vol. 142, p. 1327.]
Майрановский, С.Г. Реакции электровосстановления органических веществ в присутствии катализаторов, вызывающих каталитическое выделение водорода и электросинтез хиральных соединений. Успехи Химии. 1991. Т. 60. С. 2113. [Mairanovskii, S.G., The electroreduction of organic compounds in the presence of catalysts causing catalytic evolution of hydrogen and the electrosynthesis of chiral compounds, Russian Chem. Rev., 1991, vol. 60, p. 1085.]
Leibson, V.N., Churilina, A.P., Mendkovic, A.S., and Gultyai, V.P., New ideas of the mechanism of catalytic hydrogen evolution in the buffer solutions of organic compounds, J. Electroanal. Chem., 1989, vol. 261, p. 165.
Лейбзон, В.Н., Чурилина, А.П., Мендкович, А.С., Гултяй, В.П. Природа полярографических каталитических волн водорода в присутствии органических соединений Изв. Акад. АН СССР, сер. Хим. 1986. № 8. С. 1914. [Leibzon, V.N., Churilina, A.P., Mendkovich, A.S., and Gultyai, V.P., Nature of the polarographic catalytic hydrogen waves due to organic compounds, Russ. Chem. Bull., 1986, vol. 35, p. 1773.]
Дорофеенко, Г.Н., Садекова, Е.И., Кузнецова, Е.В. Препаративная химия пирилиевых солей, Ростов на Дону: Изд-во Ростов. ун-та, 1979. 233 c. [Dorofeenko, G.N., Sadenkova, E.I., Kuznetsova, E.V., Preparative chemistry prelievi salts (in Russian), Rostov-on-Don: Publishing house Rostov University, 1979. 233 p.]
Stephens, P.J., Devlin, F.J., Chablowski, C.F., and Frisch, M.J., Ab initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields, J. Phys. Chem., 1994, vol. 98, p. 11623.
Ditchfield, R., Hehre, W.J., and Pople, J.A., Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules, J. Chem. Phys., 1971, vol. 54, p. 724.
Schmidt, M.W., Baldridge, K.K., Boatz, J.A., Elbert, S.T., Gordon, M.S., Jensen, J.J., Koseki, S., Matsunaga, N., Nguyen, K.A., Su, S., Windus, T.L., Dupuis, M., and Montgomery, J.A., General Atomic and Molecular Electronic Structure System, J. Comput. Chem., 2003, vol. 14, p. 1347.
Granovsky, A.A., Firefly version 8.0, http://classic.chem.msu.su/gran/firefly/index.html.
Tomasi, J., Mennucci, B., and Cammi, R., Quantum Mechanical Continuum Solvation Models, 2005, Chem. Rev., vol. 105, p. 2999.
Bard, A.J. and Faulkner, L.R., Electrochemical Methods: Fundamentals and Applications, John Wiley: New York, 2001. 850 p.
Hogan, D.T. and Sutherland, T.C., Modern Spin on the Electrochemical Persistence of Heteroatom-bridged Triphenylmethyl-type Radicals, J. Phys. Chem. Lett, 2018, vol. 9, p. 2825.
Baik, M. and Friesner, R.A., Computing Redox Potentials in Solution: Density Functional Theory as A Tool for Rational Design of Redox Agents, J. Phys. Chem. A, 2002, vol. 106, p. 7407.
Дополнительные материалы отсутствуют.