

Электрохимия, 2020, T. 56, № 10, стр. 941-950
De-alloyed PtCu/C-катализаторы электроокисления метанола
В. С. Меньщиков a, *, В. Е. Гутерман a, С. В. Беленов a, О. А. Спиридонова a, Д. В. Резван a
a Южный федеральный университет, химический факультет
344090 Ростов-на-Дону, ул. Зорге, 7, Россия
* E-mail: men.vlad@mail.ru
Поступила в редакцию 26.09.2019
После доработки 29.01.2020
Принята к публикации 20.02.2020
Аннотация
Изучена активность PtCu/C-катализаторов в реакции электроокисления метанола в растворе HClO4. При этом проведено сравнение электрохимического поведения катализаторов, используемых в состояниях “как получено” и после предобработки в азотной кислоте, приводящей к уменьшению содержания меди в их составе. Установлено, что селективное растворение части легирующего компонента не оказывает негативного влияния на поведение биметаллических катализаторов. Полученные материалы проявляют большую толерантность к промежуточным продуктам окисления метанола, а их удельная активность в 5–7 раз превосходит активность коммерческого Pt/C-катализатора. Результаты проведенного исследования открывают возможность использования де-легированных (de-alloyed) платиномедных катализаторов в спиртовых топливных элементах, значительно снижая риск отравления полимерной мембраны.
ВВЕДЕНИЕ
Низкотемпературные топливные элементы (ТЭ) с полимерной протонпроводящей мембраной находят широкое применение в качестве источника энергии благодаря высоким удельным характеристикам, удобному для эксплуатации температурному интервалу и низкому уровню вредных выбросов в окружающую среду [1]. Прямые метанольные топливные элементы (МТЭ) весьма перспективны для применения в портативных электронных устройствах (стационарные электронные устройства, ноутбуки, резервные источники питания для мобильных телефонов, биосенсоры) [2–4]. МТЭ компактны, существующая инфраструктура вполне способна обеспечить хранение и транспортировку метанола. Наиболее активными электрокатализаторами для МТЭ являются платиносодержащие материалы, как правило, нанесенные на поверхность дисперсных углеродных носителей [1–4].
Широкому распространению МТЭ мешает высокая стоимость и малый срок службы электрокатализатора, обусловленный отравлением поверхности Pt продуктами окисления метанола (HCHO, HCOOH, COад), а также кроссовер метанола в катодную область [4–7]. Необходимость повышения активности и стабильности платиносодержащих катализаторов в реакциях электроокисления метанола (РОМ) и электровосстановления кислорода (РВК) привела к большому числу работ, посвященных использованию в МТЭ биметаллических материалов. Показано, что легирование платины неблагородными металлами, такими как Ni, Co, Cu, способствует повышению ее активности и стабильности токовых характеристик катализаторов в РОМ и РВК [8–10]. Для снижения содержания драгоценного металла при одновременном повышении активности катализаторов в РОМ некоторые исследователи предлагают использовать биметаллические системы с весьма высокой концентрацией легирующего компонента [11–13]. Увеличение активности биметаллических катализаторов в реакции электроокисления метанола по сравнению с платиновыми, как правило, объясняют бифункциональным механизмом катализа, при котором атомы легирующего компонента облегчают десорбцию промежуточных продуктов окисления метанола с поверхности Pt [14, 15].
К сожалению, состав биметаллических электрокатализаторов меняется в процессе их функционирования, а селективное растворение легирующего компонента из биметаллических наночастиц может вызывать отравление полимерной мембраны МТЭ [16]. Это приводит к ситуации замкнутого круга, при которой присутствие легирующего компонента с одной стороны необходимо, а с другой – вредно. Отметим, что селективное растворение легирующего компонента в значительной степени происходит на начальном этапе работы катализатора [17–19]. Далее, в процессе функционирования, изменение состава выражено гораздо слабее, и de-alloyed22 электрокатализаторы могут демонстрировать высокие стабильность и активность, например в РВК [17, 19]. При этом катионы легирующего компонента, перешедшие в раствор электролита на стадии предобработки, по-прежнему находятся в этом растворе, что неприемлемо в случае использования катализатора в мембранно-электродном блоке (МЭБ). Возникает вопрос – можно ли создать катализаторы, содержащие биметаллические наночастицы таких состава/структуры, которые обеспечивали бы сочетание высокой активности и стабильности электродных реакций с приемлемо низкой скоростью растворения легирующего компонента. Одним из подходов к решению такой задачи является предварительная обработка PtMx/C-материалов в агрессивной среде, в результате которой образуются de-alloyed PtMx – y/C-катализаторы, сохраняющие высокие функциональные характеристики, но устойчивые к селективному растворению атомов М. В работах [19–21] показано, что предобработка PtCux/C (x > 1)-катализаторов в кислотах приводит к растворению легирующего компонента до “безопасного” содержания. При относительно небольшой электрохимически активной площади поверхности de-alloyed PtCux – y/C-катализаторы продемонстрировали высокую активность в РВК как в электрохимической ячейке [17, 19, 21], так и в МЭБ водород-воздушного твердополимерного топливного элемента (ТПТЭ) [19]. Не ясно, однако, тождественны ли по своим составу, структуре и электрохимическому поведению материалы, деметаллизация (селективное растворение части меди) которых проведена электрохимически и посредством химической предобработки.
Ранее в [22] мы установили, что PtCu1.2/C-катализаторы в состоянии “как получено” и после термической обработки могут проявлять значительно более высокую активность в РОМ, по сравнению с коммерческим Pt/C-аналогом. Целью настоящей работы является сравнение активности в РОМ исходных PtCux/C-катализаторов и de-alloyed PtCux – y/C-образцов, полученных из них после обработки в азотной кислоте. Необходимо было: а) изучить влияние соотношения Pt:Cu в исходных материалах на состав, площадь электохимически активной поверхности и поведение соответствующих de-alloyed PtCux – y/C-катализаторов; б) проверить гипотезу о том, что предварительное “химическое” извлечение избыточной меди из PtCux/C (в состоянии “как получено”) катализаторов и их электрохимическая предобработка непосредственно в растворе электролита дают близкие результаты с точки зрения активности в РОМ образующихся при этом de-alloyed PtCux – y/C-катализаторов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Платиномедные катализаторы на углеродном носителе Vulcan XC-72 получали методом восстановления прекурсоров металлов боргидридом натрия в несколько стадий, как это описано в [23]. На первом этапе наночастицы меди осаждали в суспензии на углеродный носитель из раствора CuSO4. На второй и третьей стадии к суспензии добавляли расчетные количества прекурсоров меди (CuSO4) и платины (H2PtCl6), после чего восстанавливали их боргидридом натрия, взятым в избытке. Формирование платинового слоя на поверхности наночастиц на четвертой стадии проводили восстановлением Pt(IV) из раствора, не содержащего ионов меди. Суспензию PtCux/C фильтровали, катализатор высушивали над P2O5. Неоднородность распределения компонентов (рост концентрации платины в направлении от центра к поверхности) в платиномедных наночастицах, формирующихся при использовании описанной методики синтеза, была доказана в [23]. Полученные образцы расчетного состава PtCu1.5/C, PtCu2/C и PtCu3/C далее обозначены как A1, A2 и A3 соответственно. Их фактический состав несколько отличался от рассчитанного по загрузке прекурсоров (см. далее). Часть каждого из синтезированных катализаторов выдерживали в растворе 1 М азотной кислоты при 25°C в течение 2 часов при постоянном перемешивании. De-alloyed PtCux – y/C-материалы, полученные из A1, A2 и A3 в результате кислотной обработки, маркировали как A1K, A2K и A3K, соответственно. Коммерческий Pt/C-катализатор, используемый в качестве образца сравнения, обработке в азотной кислоте не подвергался.
Соотношение платины и меди в платиномедных катализаторах определяли методом рентгенофлуоресцентного анализа (РФлА) на спектрометре с полным внешним отражением рентгеновского излучения РФС-001 (НИИ физики ЮФУ). Массовая доля металлов была определена методом термогравиметрии по массе остатка, несгоревшего после 40-минутной выдержки при 800°С. При расчете полагали, что в результате высокотемпературного окисления вся медь превращается в CuO. Методом рентгенофазового анализа (напряжение на рентгеновской трубке – 50 кВ, ток – 150 мкА, время регистрации спектра – 300 с, молибденовый анод) был установлен фазовый состав катализаторов. Средний размер кристаллитов (область когерентного рассевания) определяли по полуширине отражения платины 111 с использованием формулы Шеррера [24].
При изучении электрохимического поведения электрокатализаторов каталитический слой формировали на торце вращающегося дискового стеклоуглеродного электрода. Для этого готовили суспензию, содержавшую 0.0060 г катализатора, 900 мкл изопропилового спирта и 100 мкл 0.5%‑ной водной эмульсии полимера Nafion®. После получения суспензию диспергировали в течение 15 мин. С помощью микродозатора при непрерывном перемешивании отбирали и наносили на торец дискового электрода 6 мкл суспензии. После сушки на воздухе в течение 20 мин на полученный слой наносили 7 мкл водной эмульсии Nafion 0.05% и сушили на воздухе еще в течение 15 мин.
Электрохимические измерения проводили в трехэлектродной ячейке: электролит – раствор 0.1 М HClO4 (х. ч.), хлоридсеребряный электрод сравнения (концентрация раствора KCl – 3.5 М), противоэлектрод – платиновая проволока. Исследуемый электрод предобрабатывали в течение 100 вольт-амперных циклов в диапазоне потенциалов от 0.04 до 1.2 В со скоростью развертки потенциала 200 мВ/с, затем, заменив раствор электролита на свежий, после 45-минутной продувки Ar проводили измерения площади электрохимически активной поверхности (ЭХАП) по адсорбции/десорбции атомарного водорода, а также по окислению монослоя хемосорбированного CO, как это описано в [25]. В последнем случае электрод в течение 20 мин выдерживали в электролите, насыщенным CO при потенциале 0.1 В. Затем 20 мин продували раствор аргоном, после чего, не прекращая продувку, регистрировали две циклические вольтамперограммы, по которым и проводили расчет. При изучении активности катализаторов в РОМ в качестве электролита использовали раствор 0.1 М HClO4 + 0.5 М CH3OH. Измерения проводили на стационарном дисковом электроде. Все потенциалы в работе приведены относительно обратимого водородного электрода.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Состав полученных PtCux/C-материалов, определенный по результатам термогравиметрии и рентгенофлуоресцентного анализа, незначительно отличается от ожидаемого по загрузке прекурсоров: PtCu1.5/C, PtCu2/C и PtCu3/C (табл. 1). Такая ситуация весьма характерна для жидкофазного синтеза биметаллических материалов и может быть связана как с неполной абсорбцией образовавшихся металлических наночастиц углеродом, так и с неполным восстановлением прекурсоров металлов.
Таблица 1.
Состав и структурные характеристики полученных PtCux/C- и коммерческого Pt/C-материалов
| Материал | Состав (РФлА) | Состав после электрохимической предобработки поверхности (РФлА) | ω(Pt), мас. % | 2θ (111), град | Dср, нм |
|---|---|---|---|---|---|
| А1 | Pt1Cu1.7 | Pt1Cu0.5 | 19.9 ± 0.4 | 41.2 | 3.0 ± 0.2 |
| А2 | Pt1Cu2.2 | Pt1Cu0.6 | 23.4 ± 0.4 | 41.3 | 3.0 ± 0.2 |
| А3 | Pt1Cu2.9 | Pt1Cu0.5 | 26.6 ± 0.4 | 41.2 | 2.8 ± 0.2 |
| А1К | Pt1Cu0.9 | Pt1Cu0.6 | 18.5 ± 0.4 | 40.9 | 2.7 ± 0.2 |
| А2К | Pt1Cu0.9 | Pt1Cu0.5 | 22.4 ± 0.4 | 41.0 | 3.0 ± 0.2 |
| А3К | Pt1Cu0. 8 | Pt1Cu0.5 | 26.4 ± 0.4 | 40.8 | 2.7 ± 0.2 |
| JM20 (Pt/C) | Pt | Pt | 20.0 ± 0.4 | 39.9 | 2.0 ± 0.2 |
На рентгеновских дифрактограммах PtCux/C-материалов присутствуют отражения, соответствующие фазам углерода и платины (рис. 1). При этом характерные для платины отражения смещены в сторону больших значений углов 2θ, что является следствием образования твердых растворов Pt–Cu. Отдельных отражений фазы меди или ее оксидов не обнаружено, что, однако, не является исчерпывающим подтверждением их полного отсутствия в полученных материалах [26]. Судя по положению максимума 111 на дифрактограммах, содержание металлической меди в PtCux-наночастицах уменьшается при переходе от исходных катализаторов в состоянии “как получено” к соответствующим de-alloyed образцам, что коррелирует с данными по определению их состава (рис. 1, табл. 1 ). В то же время для исходных катализаторов значения угла 2 тета максимума 111 практически не зависят от состава (содержания меди) (табл. 1 ). По-видимому, составы металлической компоненты в материалах А1–А3 близки, а часть меди в катализаторах содержится в виде рентгеноаморфного оксида, содержание которого растет в ряду А1 < A2 < A3. Присутствие рентгеноаморфной компоненты оксидов меди в платиномедных катализаторах ранее зафиксировано в [26]. Средние диаметры кристаллитов в исследуемых биметаллических материалах, рассчитанные по формуле Шеррера, составляют от 2.7 до 3.0 нм (табл. 1 ), что, с учетом точности измерений, говорит об отсутствии существенного влияния исходного состава и кислотной обработки на их размер.
Рис. 1.
Типичные рентгеновские дифрактограммы исходных А1, А2, А3 PtCu/C- и de-alloyed А1К, А2К, А3К PtCu/C-катализаторов.

Обработка PtCux/C-материалов в азотной кислоте приводит к растворению значительной части меди (табл. 1 ). Не исключена также потеря небольших количеств платины и углерода. Отметим, что, несмотря на существенное различие первоначального состава платиномедных катализаторов в состоянии “как получено”, составы металлической компоненты трех постобработанных образцов очень близки между собой. Это говорит о том, что количество “прочно связанной” меди, селективное растворение которой из платиномедных наночастиц затруднено, мало зависит от исходного состава материалов, полученных по описанной выше методике синтеза. Судя по смещению максимумов отражения металлической фазы PtCux – y/C-материалов A1K–A3K в сторону меньших значений 2 тета по сравнению с таковыми для исходных PtCux/C-материалов A1–A3, при кислотной обработке происходит растворение не только рентгеноаморфных оксидов меди, но и селективное растворение части меди из биметаллических наночастиц.
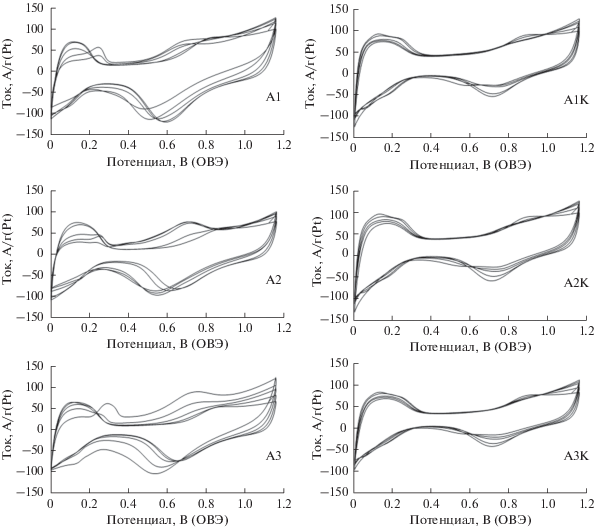
Электрохимическое поведение исходных и de-alloyed катализаторов на стадии предобработки имеет существенные различия. На циклических вольтамперограммах образцов A1–A3 (состояние “как получено”) (рис. 2) в течение нескольких первых циклов в диапазоне потенциалов 0.25–0.45 В наблюдались анодные максимумы, связанные с растворением меди из собственной фазы [27, 28]. Для образцов de-alloyed катализаторов A1K–A3K подобные максимумы на ЦВА не зафиксированы. Аналогичную ситуацию ранее наблюдали в [19].
Рис. 2.
ЦВА в процессе стандартизации (3-й, 5-й, 10-й, 30-й и 100-й циклы). Электролит раствор 0.1 М HClO4, насыщенный аргоном при атмосферном давлении. Скорость развертки потенциала 100 мВ/с.
Отметим, что после предобработки содержание меди уменьшилось как в катализаторах, взятых в состоянии “как получено” (А1–А3), так и для предобработанных в кислоте de-alloyed образцов А1К–А3К (табл. 1 ). В результате состав металлической компоненты всех образцов оказался примерно одинаковым – Pt1Cu0.55 ± 0.05 (табл. 1 ). При этом после завершения предобработки de-alloyed образцов в электролите содержалось значительно меньше катионов меди, поскольку часть ее была удалена ранее в процессе химической обработки. Для исключения влияния катионов Cu2+, находящихся в растворе после завершения процедуры предобработки, перед проведением последующих электрохимических исследований проводили замену электролита на свежеприготовленный 0.1 М раствор HClO4, который затем насыщали аргоном в течение 20 мин.
Циклические вольтамперограммы предобработанных катализаторов имеют вид характерный для Pt/C- и PtM/C-электрокатализаторов (рис. 3а, 3в). Значения ЭХАП платиномедных катализаторов, определенные по областям электрохимической адсорбции/десорбции атомарного водорода и пикам окисления монослоя хемосробированного СО, находятся в диапазоне от 30 до 40 м2/г(Pt) (табл. 2). Учитывая точность определения до 10% и различия в значениях ЭХАП, определенных двумя способами (табл. 2), можно с уверенностью констатировать только то, что ЭХАП всех платиномедных катализаторов значительно ниже, чем у коммерческого Pt/C-образца сравнения. Меньшие значения ЭХАП PtCu/C-материалов по сравнению с коммерческим Pt/C-катализатором в значительной степени обусловлены бóльшим размером кристаллитов (табл. 1 ).
Рис. 3.
Циклические вольтамперограммы электрокатализаторов в атмосфере аргона после завершения предобработки (а, в) и после пропускания СО (б, г). Коммерческий Pt/C(JM20)- и исходные PtCux/C-материалы – (а, б); коммерческий Pt/C(JM20)- и de-alloyed PtCux–y/C-материалы – (в, г).

Таблица 2.
Значения ЭХАП и потенциал начала окисления CO платиномедных и коммерческого Pt/C-электрокатализаторов
| Материал | ЭХАП (Hадс/дес), м2/г (Pt) | ЭХАП (CO), м2/г (Pt) | Eнач. окисления CO, В |
|---|---|---|---|
| A1 | 39 ± 4 | 31 ± 3 | 0.63 |
| A2 | 31 ± 3 | 33 ± 3 | 0.64 |
| A3 | 37 ± 4 | 29 ± 3 | 0.64 |
| A1K | 38 ± 4 | 31 ± 3 | 0.63 |
| A2K | 43 ± 4 | 37 ± 4 | 0.63 |
| A3K | 33 ± 3 | 32 ± 3 | 0.63 |
| JM20 | 81 ± 8 | 78 ± 8 | 0.68 |
Негативное влияние на величину ЭХАП может также оказывать агломерация (агрегация) наночастиц. Предобработка катализаторов в азотной кислоте не оказывает существенного влияния на значения ЭХАП, измеренные после предобработки. Таким образом, существенных различий, с точки зрения влияния химического или электрохимического способа селективного растворения “слабо связанной” меди из катализаторов, не обнаружено. Отметим, что окисление хемосорбированного СО на всех платиномедных катализаторах начинается при меньших потенциалах, чем на Pt/C и завершается в более узком диапазоне потенциалов: 0.63–0.88 В (рис. 3б, 3г). Бимодальные максимумы окисления СО (рис. 3б, 3г) могут быть связаны с неоднородностью состава и структуры биметаллических наночастиц [29, 30].
Активность катализаторов в РОМ рассчитывали по удельному количеству электричества ${{Q}_{{{\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{OH}}}}}$ (Кл/м2(Pt)), затраченному на окисление метанола при прямом ходе развертки потенциала ЦВА (рис. 4а, 4в, табл. 3). Для всех платиномедных катализаторов ${{Q}_{{{\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{OH}}}}}$ значительно больше, чем для Pt/C (табл. 3). То же самое характерно и для максимальных значений плотности тока окисления метанола на ЦВА: для биметаллических катализаторов они в 5–7 раз выше, чем для Pt/C (рис. 4 и табл. 3). Потенциалы начала окисления метанола, определенные методом касательных по соответствующим ЦВА прямого хода, для платиномедных катализаторов примерно на 50–100 мВ меньше, чем для коммерческого Pt/C. Несмотря на значительно более высокую скорость окисления метанола и, возможно, большую концентрацию промежуточных продуктов у поверхности электрода, платиномедные катализаторы продемонстрировали более высокие значения плотности тока во время регистрации хроноамперограмм при потенциале 0.87 В (рис. 4б, 4г), что в совокупности с меньшим потенциалом начала окисления монослоя хемосорбированного CO говорит о высокой толлерантности биметаллических катализаторов к промежуточным продуктам окисления метанола.
Рис. 4.
Циклические вольтамперограммы – (а, в) и хронопотенциограммы – (б, г) коммерческого Pt/C (JM20)- и полученных PtCu/C-катализаторов. Токи нормированы на ЭХАП. Скорость развертки потенциала 20 мВ/с. Электролит 0.1 М раствор HClO4 с добавкой 0.5 M CH3OH насыщенный Ar при атмосферном давлении.

Таблица 3.
Параметры, характеризующие поведение PtCu/C-материалов в РОМ относительно коммерческого Pt/C-катализатора
| Материал | ${{Q}_{{{\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{OH}}}}}$ × 102, Кл/г(Pt) | ${{Q}_{{{\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{OH}}}}}$ × 102, Кл/м2(Pt) | Iмакс. прямого пика окисления CH3OH, А/м2(Pt) | Eнач. окисления CH3OH, В | Iнач, А/м2(Pt) | Iконеч, А/м2(Pt) |
|---|---|---|---|---|---|---|
| PtCu/C A1 | 128 | 3.3 | 24.6 | 0.62 | 12.5 | 5.9 |
| PtCu/C A2 | 117 | 3.7 | 26.1 | 0.62 | 14.9 | 7.7 |
| PtCu/C A3 | 70 | 2.2 | 18.8 | 0.62 | 13.5 | 2.6 |
| PtCu/C A1K | 96 | 2.8 | 25.2 | 0.64 | 15.4 | 7.2 |
| PtCu/C A2K | 108 | 2.7 | 24.8 | 0.63 | 14.4 | 6.7 |
| PtCu/C A3K | 72 | 2.2 | 19.9 | 0.62 | 12.7 | 3.6 |
| Pt/C JM20 | 33 | 0.4 | 4.4 | 0.63 | 3.7 | 1.6 |
Важно, что de-alloyed PtCux – y/C-катализаторы не уступают по активности в РОМ электрокатализаторам, которые были нанесены на стеклографитовый электрод в состоянии “как получено”, и первоначально содержали значительно больше меди. Очевидно, что причиной их сходного поведения является близость состава/структуры платиномедных катализаторов после селективного растворения части меди вследствие электрохимической или химической предобработки исходных материалов (табл. 1 ). Аналогичные выводы при исследовании активности платиномедных катализаторов в РВК сделаны в [19].
Сравнительный анализ особенностей электрохимического поведения платиномедных катализаторов в РОМ (рис. 4, табл. 3) позволяет говорить о достоверно более высокой активности образцов А1, А1К, А2 и А2К по сравнению с А3 и А3К. Исходные и de-alloyed образцы А1 и А2 демонстрируют более высокие значения ${{Q}_{{{\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{OH}}}}}$ (табл. 3), тока “метанольного максимума” ЦВА (рис. 4а, 4в) и токов на хроноамперограммах окисления метанола (рис. 4б, 4г).
Такое отличие в поведении катализаторов отчасти может быть связано с возможными отличиями их состава после электрохимической предобработки: так, например, образец А3К содержит несколько меньше меди, чем А1К и А2К (см. табл. 1 ). При этом достоверного отличия величин ЭХАП разных платиномедных катализаторов установить не удалось (табл. 2). На наш взгляд, следует принимать во внимание возможные различия в характере локализации атомов “прочно связанной” меди в “предобработанных” наночастицах, поверхность которых обогащена платиной. Чем меньше толщина вторичной платиновой оболочки, тем более может быть выражено промотирующее влияние атомов легирующего компонента на активность платины в электрохимических реакциях.
Непосредственное сравнение полученных результатов с результатами других авторов осложняется тем, что методика приготовления каталитического слоя, содержание платины в катализаторе и в самом слое варьируются в разных исследованиях [31–33]. Стандартной практикой является сравнение активности полученных материалов с активностью коммерческого Pt/C при одинаковых условиях проведения эксперимента. В работе [31] PtRu/C-катализатор демонстрирует удельную активность в 4.2 раза выше, чем коммерческий Pt/C (20% Pt). В работе [33] удельная активность PtRu/C-материала в 8.8 раз больше по сравнению с коммерческим Pt/C (60% Pt). Как следует из результатов проведенных нами измерений (табл. 3), удельная активность PtCux – y/C-катализаторов А1К и А2К примерно в 6–7 раз превышает активность Pt/C. Таким образом, полученные de-alloyed платиномедные катализаторы близки по своей активности в РОМ к PtRu/C-катализаторам.
ЗАКЛЮЧЕНИЕ
Проведенное исследование показало, что нанесенные на углеродный носитель платиномедные катализаторы проявляют значительно более высокую удельную и масс-активность в РОМ по сравнению с коммерческим Pt/C-электрокатализатором, несмотря на существенно меньшую ЭХАП. Предобработка катализаторов в азотной кислоте способствует вымыванию значительной доли меди из наночастиц и не приводит к уменьшению активности катализаторов в РОМ. Следовательно, она может быть использована для снижения загрязнения МЭБ катионами меди. Состав исходных PtCux/C-катализаторов может оказывать некоторое влияние на изменение структуры наночастиц в ходе “кислотной” предобработки и, следовательно, может быть оптимизирован. Результаты проведенного исследования хорошо согласуются с полученными нами ранее [17–19] и известными литературными [20, 21] данными, и, на наш взгляд, подтверждают целесообразность проведения испытаний de-alloyed PtCux – y/C-катализаторов в МЭБ метанольных топливных элементов так же, как и необходимость исследования их поведения при более высоких температурах, когда процессы селективного растворения меди усиливаются [18].
Список литературы
Bagotsky, V.S., Fuel Cells: Problems and Solutions, 2nd Edition, John Wiley & Sons, 2012. 406 p.
Badwal, S.P.S., Giddey, S., Kulkarni, A., Goel, J., and Basu, S., Direct ethanol fuel cells for transport and stationary applications e a comprehensive review, Appl. Energy, 2015, vol. 145, p. 80.
Chen, Y., Bellini, M., Bevilacqua, M., Fornasiero, P., Lavacchi, A., Miller, H.A., Wang, L., and Vizza, F., Direct alcohol fuel cells: toward the power densities of hydrogen-fed proton exchange membrane fuel cells, ChemSusChem, 2015, vol. 8, p. 524.
Breeze, P., Chapter 8—Direct Methanol Fuel Cells, Fuel Cells, 2017, p. 78.
Martin, S., Martinez-Vazquez, B., Garcia-Ybarra, P.L., and Castillo, J.L., Peak utilization of catalyst with ultra-low Pt loaded PEM fuel cell electrodes prepared by the electrospray method, J. Power Sources, 2013, vol. 229, p. 179.
Decoopman, B., Vincent, R., Rosini, S., Paganelli, G., Thivel, P.-X., Proton exchange membrane fuel cell reversible performance loss induced by carbon monoxide produced during operation, J. Power Sources, 2016, vol. 324, p. 492.
Gwak, G., Kim, D., Lee, S., and Ju, H., Studies of the methanol crossover and cell performance behaviors of high temperature-direct methanol fuel cells (HT-DMFCs), Int. J. Hydrogen Energy, 2018, vol. 43, p. 13999.
Xu, C.X., Hou, J.G., Pang, X.H., Li, X.J., Zhu, M.L., and Tang, B.Y., Nanoporous PtCo and PtNi alloy ribbons for methanol electrooxidation, Int. J. Hydrogen Energy, 2012, vol. 37, p. 10489.
Cui, X.Z., Shi, J.L., Zhang, L.X., Ruan, M.L., and Gao, J.H., PtCo supported on ordered mesoporous carbon as an electrode catalyst for methanol oxidation, Carbon, 2009, vol. 47, p. 186.
Xu, C., Liu, Y., Wang, J., Geng, H., and Qiu, H., Fabrication of nanoporous Cu–Pt(Pd) core/shell structure by galvanic replacement and its application in electrocatalysis, ACS Appl. Mater. and Interfaces, 2011, vol. 3, p. 4626.
O Cui, C.H., Li, H.H., and Yu, S.H., Large Scale Restructuring of Porous Pt–Ni Nanoparticle Tubes for Methanol Oxidation: A Highly Reactive, Stable, and Restorable Fuel Cell Catalyst, Chem. Scie., 2011, vol. 2, p. 1611.
Yanga, S., Zhanga, F., Gaoa, C., Xiaa, J., Lub, L., and Wanga, Z., A sandwich-like PtCo-graphene/carbon dots/graphene catalyst for efficient methanol oxidation, J. Electroanal. Chem., 2017, vol. 802, p. 27.
Gao, S., Yang, X., Liang, S., Wang, Y., and Zang, H., Li, Y., One step synthesis of PtNi electrocatalyst for methanol oxidation, In. Chem. Com., 2019, vol. 106, p. 104.
Suntivich, J., Xu, Z.C., Carlton, C.E., Kim, J.Y., Han, B.H., Lee, S.W., Bonnet, N., Marzari, N., Allard, L.F., Gasteiger, H.A., Hamad-Schifferli, K., and Horn Y.S., Surface Composition Tuning of Au–Pt Bimetallic Nanoparticles for Enhanced Carbon Monoxide and Methanol Electro-oxidation, J. Am. Chem. Soc., 2013, vol. 135, p. 7985.
Park, K.W., Chemical and electronic effects of Ni in Pt/Ni and Pt/Ru/Ni alloy nanoparticles in methanol electrooxidation, J. Phys. Chem. B, 2002, vol. 106, p. 1869.
Khatib, F.N., Wilberforce, T., Ijaodola, O., Ogungbemi, E., El-Hassan, Z., Durrant, A., Thompson, J., and Olabi, A.G., Material degradation of components in polymer electrolyte membrane (PEM) electrolytic cell and mitigation mechanisms: A review, Renewable and Sustainable Energy Reviews, 2019, vol. 111, p.1.
Могучих, Е.А., Алексеенко, А.А., Гутерман, В.Е., Новиковский, Н.М., Табачкова, Н.Ю., Меньщиков, В.С. Влияние состава и структуры PtCu электрокатализаторов на их стабильность при различных условиях стресс-тестирования. Электрохимия. 2018. Т. 54. С. 976. [Moguchikh, E. A., Alekseenko, A.A., and Guterman, V.E., Novikovsky, N.M., Tabachkova, N.Yu., Menshchikov, V.S., Effect of the Composition and Structure of Pt(Cu)/C-Electrocatalysts on Their Stability under Different Stress Test Conditions, Russ. J. Eleсtrochem., 2018, vol. 54, p. 979.]
Alekseenko, A.A., Moguchikh, E.A., Safronenko, O.I., Guterman, V.E., Durability of de-alloyed PtCu/C-electrocatalysts, Int. J. Hydrogen Energy, 2018, vol. 43, p. 22885.
Киракосян, С.А., Алексеенко, А.А., Гутерман, В.Е., Новомлинский, И.Н., Меньщиков, В.С., Герасимова, Е.В., Никулин, А.Ю. Деметаллизированные PtCu/C-катализаторы электровосстановления кислорода. Электрохимия. 2019. Т. 55. C. 1532. [Kirakosyan, S.A., Alekseenko, A.A., Guterman, V.E., Novomlinskii, I.N., Men’shchikov, V.S., Gerasimova, E.V., and Nikulin A.Yu., Dealloyed PtCu/C Catalysts of Oxygen Electroreduction, Russ. J. of Electrochem., 2019, vol. 55, p. 1258.]
Sohn, Y., Park, J.H., Kim, P., and Joo, J.B., Dealloyed PtCu catalyst as an efficient electrocatalyst in oxygen reduction reaction, Current App. Phys., 2015, vol. 15, p. 993.
Gatalo, M., Moriau, L., Petek, U., Ruiz-Zepeda, F., Šala, M., Grom, M., Galun, T., Jovanovič, P., Pavlišič, A., Bele, M., Hodnik, N., and Gaberšček, M., CO-assisted ex-situ chemical activation of Pt–Cu/C oxygen reduction reaction electrocatalyst, Electrochim. Acta, 2019, vol. 306, p. 377.
Алексеенко, А.А., Гутерман, В.Е., Беленов, С.В., Меньшиков, В.С. Pt(Cu)/C-электрокатализаторы с пониженным содержанием платины. Электрохимия. 2018. Т. 54. С. 477. [Alekseenko, A.A., Belenov, S.V., Menshikov, V.S., and Guterman, V.E., Pt(Cu)/C-Electrocatalysts with Low Platinum Content, Russ. J. Eleсtrochem., 2018, vol. 54, p. 415.]
Alekseenko, A.A., Guterman, V.E., Belenov, S.V., Menshikov, V.S., Tabachkova, N.Yu., Safronenko, O.I., and Moguchikh, E.A., Pt/C-electrocatalysts based on the nanoparticles with the gradient structure, Int. J. Hydrogen Energy, 2018, vol. 43, p. 3676.
Langford, J.I., and Wilson, A.J.C., Scherrer after Sixty Years: A Survey and Some New Results in the Determination of Crystallite Size,” J. Appl. Crystallography, 1978, vol. 11, p. 102.
Rudi, S., Cui, C., Gan, L., and Strasser, P., Comparative Study of the Electrocatalytically Active Surface Areas (ECSAs) of Pt Alloy Nanoparticles Evaluated by Hupd and CO-stripping voltammetry, Electrocatalysis, 2014, vol. 5, p. 408.
Pryadchenko, V.V., Belenov, S.V., Shemet, D.B., Bulat, N.V., Srabionyan, V.V., Avakyan, L.A., Volochaev, V.A., Mikheykin, A.X., Bdoyan, K.E., Navikovskiy, N.X., Zizak, I., Guterman, V.E., and Bugaev, L.A., Effect of Thermal Treatment on the Atomic Structure and Electrochemical Characteristics of Bimetallic PtCu Core–Shell Nanoparticles in PtCu/C-Electrocatalysts, J. Phys. Chem. C, 2018, vol. 122, p. 17199.
Oezaslan, M. and Strasser, P., Activity of dealloyed PtCo and PtCu nanoparticle electrocatalyst for oxygen reduction reaction in polymer electrolyte membrane fuel cell, J. Power Sources, 2011, vol. 196, p. 5240.
Oezaslan, M., Hasche, F., and Strasser, P., PtCu3, PtCu and Pt3Cu Alloy Nanoparticle Electrocatalysts for Oxygen Reduction Reaction in Alkaline and Acidic Media, J. Electrochem. Soc., 2012, vol. 159, p. 444.
Rudi, S., Cui, C., Gan, L., and Strasser, P., Comparative Study of the Electrocatalytically Active Surface Areas (ECSAs) of Pt Alloy Nanoparticles Evaluated by Hupd and CO-stripping voltammetry, Electrocatalysis, 2014, vol. 5, p. 408.
Van der Vliet, D.F., Wang, C., Li, D., Paulikas, A.P., Greeley, J., Rankin, R.B., Strmcnik, D., Tripkovic, D., Markovic, N.M., and Stamenkovic, V.R., Unique Electrochemical Adsorption Properties of Pt-Skin Surfaces, Angewandte Chem. Int. Edition, 2012, vol. 51, p. 3139.
Zhao, W.Y., Ni, B., Yuan, Q., He, P.L., Gong, Y., Gu, L., and Wang, X., Highly Active and Durable Pt72Ru28 Porous Nanoalloy Assembled with Sub-4.0 nm Particles for Methanol Oxidation, Adv. Energy Mater, 2012, vol. 7, p. 1601593.
Cogenli, M.S. and Yurtcan, A.B., Catalytic activity, stability and impedance behavior of PtRu/C, PtPd/C and PtSn/C bimetallic catalysts toward methanol and formic acid oxidation, Int. J. Hydrogen Energy, 2018, vol. 7, p. 10698.
Zhang, J., Qu, X., Han, Y., Shen, L., Yin S., Li G., Jiang, Y., and Sun, S., Engineering PtRu bimetallic nanoparticles with adjustable alloying degree for methanol electrooxidation: enhanced catalytic performance, Applied Catalysis B: Environmental, 2019, vol. 263, p. 118345.
Дополнительные материалы отсутствуют.