

Электрохимия, 2020, T. 56, № 11, стр. 1002-1023
Ионный транспорт в литиевых электрохимических системах: проблемы и решения
А. В. Иванищев a, *, И. А. Иванищева a
a Институт химии, Саратовский национальный исследовательский государственный университет
им. Н.Г. Чернышевского
410012 Саратов, ул. Астраханская, 83, Россия
* E-mail: ivanischevav@inbox.ru
Поступила в редакцию 21.09.2019
После доработки 18.01.2020
Принята к публикации 20.02.2020
Аннотация
Создание новых электродных материалов и модификация существующих являются важными тенденциями в развитии литий-ионных аккумуляторов. Особое значение имеет оценка их диффузионной способности, т.е. способности обеспечивать перенос электроактивного компонента. Для этой цели используются такие электрохимические методы, как циклическая вольтамперометрия, электрохимическая импедансная спектроскопия, метод потенциостатического прерывистого титрования и метод гальваностатического прерывистого титрования. Показано, что значения химического коэффициента диффузии D, оцененные в аналогичных материалах электродов, имеют разброс в несколько порядков. Обсуждаются основные причины этой довольно значительной дисперсии, в том числе неопределенность оценок диффузионной поверхности и использование различных подходов к получению уравнений для расчета D. Наши выводы иллюстрируются примерами оценок D в электродных материалах на основе LixC6, LixSn, LixTiO2, LixWO3, LiMyMn2 –yO4 и LiFePO4.
ВВЕДЕНИЕ
Литиевые электрохимические системы отличаются от систем классической электрохимии тем, что стадия массопереноса в них маскирует стадию электрохимической реакции. Наиболее медленным, а потому основным процессом, определяющим массоперенос в литиевых системах, является твердотельная диффузия. В результате, электрохимическая кинетика литиевых систем сводится к диффузионной кинетике, подходы к исследованию которой впервые применялись к системам с металлическим литиевым электродом, а в дальнейшем получили значительное развитие применительно к интеркаляционным литиевым системам. Наилучшим, на взгляд авторов настоящей статьи, способом представления теоретического и практического инструментария современной электрохимии литиевых систем является хронология его развития. По этой причине авторы предлагают взглянуть на эту проблему через призму тенденций в российской и мировой электрохимической науке.
Основателем и идейным вдохновителем развития электрохимии литиевых систем в России был В.С. Багоцкий. Это научное направление появилось в России практически одновременно с его зарождением в мировой электрохимической науке. Подходы, которые были выбраны первоначально для исследования электрохимической кинетики литиевых систем, оказались нетипичными для общемировых тенденций: применялись методы фотоэлектрохимии. Пионерские работы в этом русле были выполнены под руководством Ю.М. Поварова [1–3]. Была надежно установлена фоточувствительность металлического литиевого электрода в жидких апротонных электролитных средах, которая была интерпретирована с позиций существования на поверхности лития тонких пассивирующих слоев (Solid Electrolyte Interphase, SEI), обладающих свойствами ионного полупроводника. Далее подключились исследования с использованием электрохимических методов, в частности электродного импеданса (Electrochemical Impedance Spectroscopy, EIS) [4–7]. В результате такого комплексного исследования были подтверждены как установленные в мировой электрохимической науке факты, например о сложной структуре SEI, состоящей из первичного и вторичного слоев, о замедленной кинетике ее роста, так и совершенно новые данные, например представление о сосуществовании ионной и дырочной проводимости SEI. Была также предложена оригинальная электрическая эквивалентная схема для интерпретации EIS-спектров. Накопленные данные получили новый уровень обобщения в работе В.С. Багоцкого [8]. В эти же годы начала развиваться тематика интеркаляционных литиевых систем: появились работы по электрохимическому сплавообразованию лития с алюминием [9, 10]. Это стало прологом последующих исследований материалов литий-ионного аккумулятора.
Вдохновленный успехами коллег из коллектива В.С. Багоцкого, к исследованиям в области электрохимии литиевых систем подключился коллектив кафедры физической химии Саратовского государственного университета, который имел многолетний опыт плодотворного сотрудничества с Институтом электрохимии АН СССР. Коллектив из Саратова под руководством А.Л. Львова и Е.С. Нимона в ряде своих работ [11, 12] существенно расширил и детализировал исследования, ранее проведенные коллегами из Москвы. Было обнаружено и детально изучено новое явление электронной и ионной инжекции в SEI на литии [13–17].
Дальнейшая история развития электрохимии литиевых систем в России пошла по пути исследований интеркаляционных соединений лития, в авангарде которых выступил коллектив ближайшего соратника В.С. Багоцкого – А.М. Скундина [18–20]. Глубокая проработка фундаментальных проблем исследования интеркаляционных процессов с развитием теоретических основ использования электрохимических методов велась в Саратове под руководством А.В. Чурикова [21–23].
Как было показано в представленном кратком обзоре, функционирование интеркаляционных электродных материалов определяется кинетикой литиевой интеркаляции в твердотельные структуры. Основной задачей электрохимии литиевых систем становится корректная оценка диффузионных характеристик интеркаляционного процесса. Как правило, эта способность оценивается путем экспериментальных измерений с последующим расчетом химического коэффициента диффузии D; такие расчеты обычно преследуют как фундаментальные, так и практические цели. Работы, где D измеряется в чисто химическом эксперименте с наведением градиента концентрации и использованием решений уравнений Фика, довольно редки. Напротив, почти всегда D оценивается с помощью электрохимических методов. Сводка измеренных значений D, которые отождествляются с коэффициентом диффузии ионов лития DLi в ряде анодных и катодных материалов литий-ионного аккумулятора, по данным различных авторов [21, 23–83 ] приведены в табл. 1–3.
Таблица 1.
Химический коэффициент диффузии лития в различных электродных материалах литий-ионного аккумулятора: LixC6, LixFePO4
| Материал и способ получения | Ссылка | Найденное значение D (см2 с–1) | Содержание лития в формульной единице: x в LixC6, LixFePO4 (или электродный потенциал E, В отн. Li/Li+) | Метод определения D |
|---|---|---|---|---|
| LixC6 | [24] | 3.9 × 10–10 | ||
| Аморфный углерод | [25] | 2.5 × 10–15 | EIS | |
| Нефтяной кокс | [26] | 2 × 10–7–10–8 | 0.02–0.6 | GITT |
| Углеродный материал | [27] | 10–6–10–9 | 0.05–1 | EIS |
| Разупорядоченный углерод | [28] | 5 × 10–10–3 × 10–12 | 0.13–2 | EIS |
| Дисперсный природный графит | [29] | 10–5–10–9 | (0.4–0.04) | EIS |
| Графит | [30] | 10–9–10–12 | 0–1 | EIS, PITT |
| Мезофазные микрогранулы графита | [31] [32] | 5 × 10–10–6 × 10–11 | (0.2–0.05) | EIS |
| Природный графит | [33] | 7 × 10–10 | 0.2 | EIS |
| Искусственный графит | [34] | 10–9–10–10 | EIS | |
| Графит | [35] | 1 × 10–10–6 × 10–11 | 0–0.35 | EIS |
| 10–12–10–13 | 0.1–0.6 | PITT | ||
| Мезофазные микрогранулы графита | [36] | 10–9–10–11 | (0.25–0.05) | PITT |
| Мезофазные микрогранулы графита | [37] | 10–7–10–10 | 0–0.8 | Релаксация потенциала |
| Графитизированные мезофазные микрогранулы | [38] | 10–6–10–10 | (0.3–0.02) | EIS |
| Разупорядоченные мезофазные микрогранулы | [39] | 10–6–10–12 | (1.2–0.05) | EIS |
| Продукт пиролиза толуола | [21] [23] | 10–8–10–11 | 0–1 | PITT, GITT |
| LiFePO4, пленка магнетронного напыления | [40] | 10–12–10–14 | (3.4–3.6) | PITT |
| 10–12–10–14 | (3.4–3.6) | EIS | ||
| 1.93 × 10–14 | Ток пика | CV | ||
| LiFePO4, пленка импульсного лазерного напыления | [41] | 2.1 × 10–14 (anodic), | Ток пика | CV |
| 1.8 × 10–14 (cathodic) | 0–0.8 | |||
| 3.5×10–15–1.4×10–19 | 0–0.8 | GITT | ||
| 4.0×10–14–9.0×10–18 | EIS | |||
| FePO4, микроэмульсионный синтез с термообработкой 380°C |
[42] | 8.09 × 10–14 | EIS | |
| 6.71 × 10–14 | Ток пика | CV | ||
| 460°C | 1.06 × 10–13 | EIS | ||
| 8.28 × 10–14 | Ток пика | CV | ||
| 550°C | 4.88 × 10–14 | EIS | ||
| 4.89 × 10–14 | Ток пика | CV | ||
| 650°C | 2.59 × 10–14 | EIS | ||
| 1.60 × 10–14 | Ток пика | CV | ||
| LiFePO4/C, твердотельный синтез 5% C |
[43] | 1.01 × 10–13 | EIS | |
| 10% C | 9.20 × 10–13 | |||
| 20% C | 4.89 × 10–12 | |||
| LiFePO4/C, микроэмульсионный синтез с термообработкой | [44] | 1.26×10–16–8.82×10–18 | 0.1–1 | EIS |
| LiFePO4/C, гидротермальный синтез, pH 8.19 |
[45] | 2.56 × 10–13 | EIS | |
| pH 7.02 | 1.19 × 10–13 | |||
| pH 6.30 | 5.83 × 10–14 | |||
| LiFePO4/C, сольвотермальный синтез | [46] | 5.86×10–11–2.06×10–15 | 0–1 | EIS |
| LiFePO4/C, 0.9% C |
[47] | 1.47 × 10–12 | EIS | |
| 5.5% C | 3.96 × 10–12 | |||
| 8.6% C | 1.01 × 10–11 | |||
| 14.6% C | 8.74 × 10–12 | |||
| 26.6% C | 3.49 × 10–12 | |||
| LiFePO4/углеродные нанотрубки | [48] | 2.74 × 10–9 | Ток пика | CV |
| 4.17 × 10–9 | ||||
| LiFePO4/графен | 5.9 × 10–9 | |||
| LiFePO4/C, карботермальный синтез | [49] | 2.90 × 10–11 | EIS | |
| V-допированный | 1.86 × 10–10 | |||
| LiFePO4/C, твердотельный синтез с механоактивацией | [50] | 10–10–10–16 | 0–1 | PITT, GITT |
Таблица 2.
Химический коэффициент диффузии лития в различных электродных материалах литий-ионного аккумулятора: LiMyMn2 –yO4
| Материал и способ получения | Параметры электрода (толщина, мкм) | Средний размер частиц материала, нм | Метод определения D | Найденное значение D, см2 с–1 | Ссылка |
|---|---|---|---|---|---|
| LiMn2O4, синтез в растворе | Тонкопленочный (0.18, |
20–50 | EIS | 6.3 × 10–11 | [51] |
| 0.26, | 5.2 × 10–11 | ||||
| 0.63, | 5.5 × 10–11 | ||||
| 1.04) | 5.1 × 10–11 | ||||
| LiMn2O4, пленка импульсного лазерного напыления | Тонкопленочный | CV | 10–8–10–10 | [52] | |
| LiMn2O4 | Композитный | EIS | 10–7–10–9 | [53] | |
| LiMn2O4 | Композитный | 250 | PS | 2.8×10–13 | [54] |
| LiMn2O4, твердотельный синтез | Тонкопленочный | PITT | 10–9–10–11 | [55] | |
| LiMn2O4, электроосаждение аэрозоля | Тонкопленочный (10) | 40 000 | PITT | 10–9 | [56] |
| LiMn2O4, электроосаждение аэрозоля | Композитный | 8000 | EIS | 6 × 10–15–1 × 10–17 | [57] |
| Тонкопленочный (0.2–1) | 3 × 10–15–1 × 10–16 | ||||
| LiMn2O4, электроосаждение аэрозоля | Тонкопленочный (0.1) | EIS | 10–8–10–10 | [58] | |
| LiMn2O4, LiCo1/6Mn11/6O4, LiFe1/6Mn11/6O4, LiNi1/6Mn11/6O4, золь–гель-синтез | Композитный | GITT | 10–8–10–12 | [59] | |
| LiNi0.5Mn1.5O4, электроосаждение аэрозоля | Тонкопленочный (200) | EIS | 10–10–10–12 | [60] | |
| LiMn2O4, синтез насыщения расплава | Композитный | Кубические: 5000 | PITT | 10–9.5–10–11.5 | [61] |
| сферические: 2000, 3500, 8000 | CITT | 10–8.0–10–10.5 | |||
| LiMn2O4, топохимический синтез | Композитный | 1000 | CV | 4.61 × 10–10 | [62] |
| LiMn2-1.667xNbxO4,
твердотельный синтез x =0, |
Композитный | 1000 | EIS | 1.24 × 10–16 | [63] |
| x =0.01, | 1.15 × 10–17 | ||||
| x =0.02, | 6.26 × 10–17 | ||||
| x =0.03, | 2.61 × 10–16 | ||||
| x =0.04 | 1.81 × 10–16 | ||||
| LiMn2O4, твердотельный синтез | Композитный | 14 800 | CITT | 10–9–10–11 | [64] |
| LiMn2O4, пленка импульсного лазерного напыления | Тонкопленочный (0.1) | CV | 10–13 | [65] | |
| PITT | 10–13 | ||||
| EIS | 10–11–10–13 | ||||
| GITT | 10–11–10–14 | ||||
| LiMn2O4, пленка магнетронного напыления | Тонкопленочный (0.6) | CV | 10–10 | [66] | |
| PITT | 10–11 | ||||
| EIS | 10–12 | ||||
| LC | 10–9–10–11 | ||||
| LiMn2O4, пленка импульсного лазерного напыления | Тонкопленочный (0.33) | CV | 10–11–10–12 | [67] | |
| PITT | 8 × 10–11–1.9 × 10–12 | ||||
| EIS | 10–10–10–12 | ||||
| Li0.967Mn1.97O4 | Композитный | 675, 800 | EIS | 3.49 × 10–11 | [68] |
| Li0.974Mn1.96O4(PO4)0.005 | 1.39 × 10–8 | ||||
| Li1.017Mn1.97O4(PO4)0.0015, гидротермальный синтез | 5.38 × 10–8 | ||||
| LiMn2O4, электроосаждение аэрозоля | Тонкопленочный (0.01–0.02) | 51.3, 55.7, 52.3 | CV | 2.516 × 10–9 | [69] |
| EIS | 1.49 × 10–11 | ||||
| LiMn2O4, синтез под высоким давлением | Композитный | 10–8–10–10 | [70] | ||
| LixMn2O4 | Композитный | 4000–20 000 | PITT | 10–10–10–13 | [71] |
| LixMn1.95Cr0.05O4, | GITT | 10–10–10–12 | |||
| Золь–гель-синтез, синтез насыщением расплава | EIS | 10–9–10–12 |
Таблица 3.
Химический коэффициент диффузии лития в различных электродных материалах литий-ионного аккумулятора: LixWO3, LixTiO2, LixSn
| Материал и способ получения | Параметры электрода (толщина, мкм) | Средний размер частиц материала, нм | Метод определения D | Найденное значение D , см2 с–1 | Ссылка |
|---|---|---|---|---|---|
| LixWO3, гидротермальный | Тонкопленочный | 200–500 | EIS | 6.41 × 10–12–8 × 10–13 | [72] |
| LixWO3, гидротермальный | Тонкопленочный | EIS | 10–8–10–12 | [73] | |
| LixWO3, золь–гель | Тонкопленочный | GITT | 10–8–10–12 | [74] | |
| LixWO3, магнетронное напыление | Тонкопленочный | EIS | 10–11–10–13 | [75] | |
| LixWO3, вакуумное напыление | Тонкопленочный | EIS | 10–10–10–11 | [76] | |
| LixWO3, магнетронное напыление | Тонкопленочный | EIS, GITT, PITT | 10–11–10–14 | [77] | |
| LixWO3, вакуумное напыление | Тонкопленочный | EIS | 10–10–10–11 | [78] | |
| LixTiO2, гидротермальный | Тонкопленочный | CV | 10–13 | [79] | |
| LixTiO2, осаждение из раствора | Тонкопленочный | EIS | 10–15–10–17 | [80] | |
| LixTiO2, вакуумное напыление | Тонкопленочный | PITT | 10–13 | [81] | |
| LixSn, осаждение из раствора | Композитный | EIS | 10–13 | [82] | |
| LixSn, электроосаждение из раствора | Тонкопленочный | GITT | 10–9–10–11 | [83] |
Основными электрохимическими методами, теория и экспериментальная реализация которых интенсивно развивались до недавнего времени в приложении к интеркаляционным материалам литий-ионного аккумулятора, следует считать электрохимическую импедансную спектроскопию (EIS), циклическую вольтамперометрию (CV), а также хроноамперометрию и хронопотенциометрию в вариантах потенциостатического прерывистого титрования (PITT) и гальваностатического прерывистого титрования (GITT) соответственно [84–96]. Теоретическая основа всех этих методов является общей и основана на совместном решении диффузионного уравнения второго закона Фика, термодинамического уравнения Нернста и уравнения Батлера–Фольмера для описания электрохимической кинетики с введением граничных и начальных условий, свойственных каждому методу. Как правило, рассмотрение задачи начинается с простейших случаев неограниченного массопереноса в полубесконечной среде, после чего начинают добавлять различные геометрические ограничения диффузионного потока, а также факторы поверхностной кинетики и взаимодействия между частицами внедренного компонента с матрицей-хозяином и между собой. На рис. 1 схематично показаны варианты граничных условий, применявшихся при разработке моделей диффузии для интеркалируемых литием электродных материалов в порядке “от простого к сложному”: полубесконечная среда, линейный перенос в ограниченной области, сферическая модель, ограниченная модель с движущейся межфазной границей. Математические затруднения в поиске решения задачи возрастают по мере усложнения условий, однако улучшается и соответствие модели экспериментальным данным. Принимая во внимание общую теоретическую основу, все упомянутые методы должны давать один и тот же результат при определении D, а любое сравнение результатов различных методов имеет смысл только для проверки корректности постановки эксперимента. Тем не менее результаты часто довольно сильно различаются, иногда на несколько порядков. Значения коэффициента диффузии лития в интеркаляционных материалах, полученные разными авторами, зависят от многих факторов и показывают значительный разброс. Например, согласно табл. 1, значения коэффициента диффузии лития, измеренные в разнообразных углеродных материалах, по данным различных авторов, варьируются в диапазоне от 10–5 до 10–15 см2 с–1. Поскольку D представляет собой коэффициент пропорциональности между направленным потоком частиц (электрический ток) и градиентом концентрации, всегда возникает разумный вопрос: действительно ли эти материалы настолько сильно отличаются друг от друга, что электрический ток, проходящий через них в одинаковых условиях, отличается на несколько порядков, или это просто артефакт? Кроме того, в табл. 1 и 2 приведен обзор найденных значений коэффициента диффузии лития D в распространенных в настоящее время катодных материалах, таких как литий–железо(II) фосфат LiFePO4 и литий–марганцевая шпинель LiMyMn2 –yO4 (М является металлом-заместителем). Можно наблюдать аналогичный разброс значений D на несколько порядков величины: от 10–9 до 10–18 см2 с–1 (как правило, более низкие значения коэффициента диффузии не удается в принципе измерить экспериментально) для LiFePO4 и от 10–7 до 10–17 см2 с–1 для LiMyMn2 –yO4. Низкий коэффициент диффузии лития в оливине на начальном этапе разработки был основным препятствием для его использования. Данная проблема была успешно решена с помощью нанотехнологий и дизайна проводящего углеродного покрытия на частицах оливина. Однако возникает также обоснованное сомнение: были ли эти подходы настолько эффективными, что электрический ток вырос на 8–10 порядков в аналогичных условиях?
Рис. 1.
Диффузионные модели для описания ионного транспорта в электродных материалах ЛИА: а – диффузия в полубесконечной среде, б – линейная диффузия в ограниченном пространстве с поглощающей или блокирующей границами, в – диффузия в сферическом пространстве, г – линейная диффузия в ограниченной среде с движущейся межфазной границей.
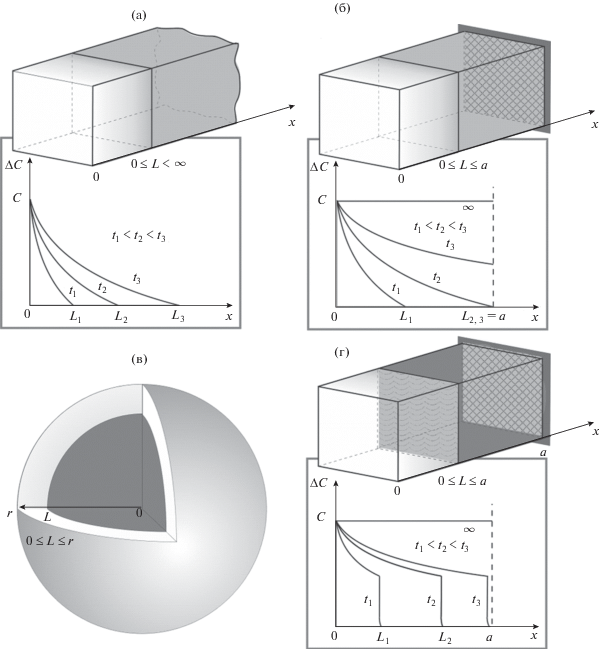
Исследование кинетики транспорта ионов лития имеет существенное значение не только для нашедших практическое применение электродных материалов, но и тех, которые имеют определенные перспективы использования в будущем. В табл. 3 приведена сводка значений коэффициента диффузии ионов лития в интеркаляционных соединениях LixWO3, LixTiO2, LixSn. Видно, что попытки определения этой величины в указанных соединениях также дают результаты с довольно заметным разбросом.
Правильность определения D, несомненно, важна. Знание D позволяет оценить возможности материала электрода по простому принципу: чем выше D, тем шире возможности материала данного электрода. Кроме того, значения D используются в математических моделях, описывающих работу литий-ионного аккумулятора. В настоящей работе освещается проблема корректности расчета D в электродных материалах с использованием экспериментальных данных современных электрохимических методов и нахождение основных источников ошибок такого определения.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве рассматриваемых в настоящей работе объектов исследования диффузионной кинетики были выбраны электроды состава LixSn, LixTiO2, LixC6, LixWO3, LiMn2O4 и LiFePO4. Электроды на основе олова, диоксида титана, графита и триоксида вольфрама были изготовлены в виде однородных пленок без связующего вещества на металлической подложке и насыщены литием путем катодного внедрения после сборки электрохимических ячеек. Электроды на основе LiMn2O4 и LiFePO4 были композитными, изготовлены путем смешивания 80% активного вещества, 15% ацетиленовой сажи и 5% PVDF; активную массу наносили на алюминиевую подложку, затем электроды в атмосферном сушильном шкафу при 110–120°С в течение 12 ч до достижения постоянного веса.
Тонкопленочные электроды на основе олова были изготовлены металлургическим способом (нанесением жидкого олова на специально подготовленную железную подложку). Наличие интерметаллида FeSn2 на границе раздела Fe/Sn обеспечивает высокую морфологическую стабильность такого электрода при циклировании. Исследование кинетики было выполнено в ходе варьирования содержания лития в электроде LixSn после 40 предварительных циклов заряда–разряда, в соответствии с методикой, описанной в [97]. Тонкопленочные электроды на основе TiO2 полиморфной кристаллической модификации рутил толщиной около 20 мкм были получены термическим окислением титановой фольги. Титан отжигали в шахтной печи на воздухе при 800°С в течение 1.5 ч в соответствии с методикой, описанной в [98]. Процедура анодного окисления металлического вольфрама в кислых растворах описана в [99]. Для изготовления электродов на основе WO3 использовалась вольфрамовая проволока диаметром 1 мм, предварительно подвергнутая механической полировке и затем электрополировке в 2%-ном растворе NaOH. Анодное окисление металла проводилось в 0.1 M растворе H2SO4. Пленки анодного оксида, сформированные на металле, дополнительно подвергали отжигу на воздухе при 420–450°С в течение нескольких часов для образования кристаллической фазы WO3.
Тонкопленочные графитовые электроды без связующего были получены каталитическим пиролизом паров толуола на никелевой пластине при 950°C в атмосфере аргона, методика которого была подробно описана в нашей ранней работе [100]. Способ низкотемпературного синтеза графитовых пленок на каталитически активной подложке в результате разложения паров углеводородов впервые был упомянут в патенте [101]. В конце реакции пиролиза подложка покрывалась равномерным слоем графита толщиной ~1 мкм. Рентгенограмма продукта показывает интенсивную (002) линию базальных плоскостей графита (рис. 2). Согласно проведенному анализу рентгенограммы, расстояние между плоскостями d002 составляет 0.3346 нм, размер кристаллитов графита находится в интервале 50–100 нм, что соответствует хорошо окристаллизованному пирографиту. При сборке электрохимической ячейки графитовые электроды подвергались заряду до состава интеркалята LiC6. Для достижения воспроизводимых показаний электрод LixC6 был предварительно подвергнут нескольким циклам интеркаляции–деинтеркаляции, что способствовало образованию и стабилизации SEI. Методы, использованные при подготовке и испытании образцов, были подробно описаны в наших ранних работах [21, 23, 102].
Рис. 2.
Дифрактограмма рентгеновского FeKα-излучения отдельной линии (002) базальных плоскостей тонкой графитовой пленки, полученной пиролизом на Ni-подложке.

Шпинель LiMn2O4 была синтезирована методом насыщения расплава по методике, описанной ранее [71, 103]. Синтез включал насыщение оксидом γ-MnO2 расплавленного гидроксида лития LiOH при 460°С с последующей термообработкой промежуточного продукта при 600–700°С. Подробное описание методики и механизма твердофазного синтеза фосфата железа–лития LiFePO4 с использованием оксалата железа, карбоната лития и дигидрофосфата аммония можно найти в работах [50, 104]. Синтез включает стадии механохимической активации в шаровой мельнице-активаторе АГО-2 (Новиц, Россия) при комнатной температуре и 560 об мин–1 в течение 20 мин и термической обработки в трубчатой печи с продувкой аргоном со скоростью нагрева 10°C мин–1 и последующей выдержкой при фиксированной температуре 800°C в течение 3 ч. Усредненные параметры решетки образца LiFePO4 (a = 10.327 Å; b = 6.006 Å; c = 4.693 Å) и средний размер наночастиц LiFePO4 ❬D❭ = 60 ± 15 нм были найдены с использованием рентгеноструктурного анализа и являются оптимальными для электродного материала (значения ❬D❭, находящиеся в пределах от 15 до 90 нм, были получены для различных режимов синтеза и постепенно увеличивались с ростом температуры термообработки, как ранее сообщалось в работе [105]). Микроструктура порошкового материала была образована агломератами наночастиц. Эти агломераты создавали дисперсию с максимумом распределения около 2 мкм, согласно гранулометрическим кривым, полученным методом дифракции монохроматического излучения.
Для определения коэффициента диффузии лития D в вышеуказанных электродных материалах применялись методы PITT, GITT, EIS и CV. Наши способы приложения этих методов к интеркалируемым литием материалам было описано в работах [21, 23, 50, 97–99, 106–108]. В рамках настоящей работы PITT и GITT не использовались в их классической версии. Следующая последовательная процедура PITT, GITT и EIS была использована как более быстрая и оптимальная. После слабого и короткого возмущения PITT с регистрацией и интегрированием пропущенного электрического тока, ячейка отключалась, а потенциал электрода стабилизировался в условиях разомкнутой цепи (до конца выраженного дрейфа потенциала и его стабилизации с варьированием ±0.5 мВ). Это было критерием достаточной релаксации системы после единичного возмущения. После релаксации появлялась возможность сделать аналогичное измерение методом GITT, а после окончания повторной релаксации – методом EIS. В результате был получен массив данных различных методов, измеренных в точках вдоль оси концентраций ионов лития в электроде. В такой процедуре количество точек измерения вдоль оси концентраций определяется зарядом, пропущенным в ходе каждого измерения; теоретически, величина такого шага может быть любой, а потому любым может быть количество точек на оси концентраций ионов лития. Длительность единичного PITT измерения выбиралась по одному из следующих критериев: либо небольшое дозированное количество пропущенного заряда (по аналогии с GITT), либо снижение тока до предварительно установленного уровня (что означает квазистабилизацию процесса). В обоих случаях переход интеркалированного электрода в новое квазиравновесное состояние происходил при релаксации в условиях разомкнутой цепи, а не в потенциостатических условиях. Сравнительно небольшая длительность измерений, широкое варьирование концентрации лития в электроде, а также малые отклонения концентрации при измерительном воздействии, следствием чего была близость состояний электрода при измерениях различными методами, были преимуществами применяемой процедуры.
Электрохимические измерения проводились в герметичных электрохимических ячейках, содержащих исследуемый электрод, литиевый противоэлектрод и литиевый электрод сравнения, с помощью модульного сильноточного потенциостата–гальваностата Autolab/PGSTAT302N (ECO CHEMIE, Нидерланды). В качестве электролита использовали 1 M раствор LiPF6 в смеси этиленкарбонат–диметилкарбонат–диэтилкарбонат = = 1 : 1 : 1 по объему (BASF Corporation, Германия). Сборка электрохимических ячеек осуществлялась в перчаточном боксе (MBRAUN UNIlabpro Modular Inert Glove box, M. BRAUN Inc., США) в атмосфере сухого аргона (содержание паров воды не превышало 1 ppm). Все измерения проводились при 25°C в режиме компенсации iROhm. Здесь и далее потенциалы электродов приведены относительно литиевого электрода сравнения в том же растворе.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Теоретические основы анализа экспериментальных данных
В случае, если диффузионная среда имеет плоскую геометрию или толщина диффузионного слоя L мала по сравнению с радиусом частицы, феноменологическая задача сводится к решению уравнений Фика для одномерной диффузии в однородном твердом теле с торможением Rs на внешней границе. В результате были получены теоретические уравнения PITT, т.е. зависимость плотности тока j от поляризации электрода ΔE [21, 23]:
(1)
$\begin{gathered} j = \frac{{2nFD\Delta E}}{L}\frac{{{\text{d}}{{c}_{{\text{s}}}}}}{{{\text{d}}E}} \times \\ \times \,\,\sum\limits_{k = 1}^\infty {\frac{{\left( {\alpha _{k}^{{\text{2}}} + {{{\left( {hL} \right)}}^{2}}} \right){{{\sin }}^{2}}{{\alpha }_{k}}}}{{\alpha _{k}^{{\text{2}}} + hL + {{{\left( {hL} \right)}}^{2}}}}\exp \left( { - \frac{{\alpha _{k}^{{\text{2}}}Dt}}{{{{L}^{2}}}}} \right)} , \\ \end{gathered} $(2)
$\begin{gathered} j = \frac{{\Delta E}}{{{{R}_{{\text{s}}}}}}\exp \left( {{{h}^{2}}Dt} \right){\text{erfc}}\left( {h\sqrt {Dt} } \right)\,\,\,\,{\text{при}}\,\,\,\,t \ll {{\tau }_{{\text{d}}}}, \\ j = \frac{{\Delta E}}{{{{R}_{{\text{s}}}}}}\left( {1 - 2\frac{{\left| {{{{\text{d}}E} \mathord{\left/ {\vphantom {{{\text{d}}E} {{\text{d}}{{c}_{{\text{s}}}}}}} \right. \kern-0em} {{\text{d}}{{c}_{{\text{s}}}}}}} \right|}}{{nF{{R}_{{\text{s}}}}}}\sqrt {\frac{t}{{\pi D}}} } \right)\,\,\,\,{\text{при}}\,\,\,\,t \ll {{\left( {D{{h}^{{\text{2}}}}} \right)}^{{ - {\text{1}}}}}, \\ \end{gathered} $(3)
$\begin{gathered} \Delta E = \Delta {{E}_{{\text{s}}}} - \frac{j}{{nF}}\frac{{{\text{d}}E}}{{{\text{d}}{{c}_{{\text{s}}}}}} \times \\ \times \,\,\left[ {\frac{t}{L} - \frac{L}{D}\left\{ {\frac{1}{3} - \frac{2}{{{{\pi }^{2}}}}\sum\limits_{k = 1}^\infty {\frac{1}{{{{k}^{2}}}}\exp \left( { - \frac{{{{k}^{2}}{{\pi }^{2}}Dt}}{{{{L}^{2}}}}} \right)} } \right\}} \right], \\ \end{gathered} $(4)
$\Delta E = \Delta {{E}_{{\text{s}}}} - \frac{{2j}}{{nF}}\frac{{dE}}{{d{{c}_{{\text{s}}}}}}\sqrt {\frac{t}{{\pi D}}} ,$(5)
$D = \frac{1}{2}{{\left( {\frac{{{{{\left| {{{{\text{d}}E} \mathord{\left/ {\vphantom {{{\text{d}}E} {{\text{d}}c}}} \right. \kern-0em} {{\text{d}}c}}} \right|}}_{{c = {{c}_{{\text{0}}}}}}}}}{{nFS{{W}_{{\text{D}}}}}}} \right)}^{2}}$(6)
$D = {{T}_{{\text{D}}}}{{\left( {\frac{{{{{\left| {{{{\text{d}}E} \mathord{\left/ {\vphantom {{{\text{d}}E} {{\text{d}}c}}} \right. \kern-0em} {{\text{d}}c}}} \right|}}_{{c = {{c}_{{\text{0}}}}}}}}}{{nFS{{R}_{{\text{D}}}}}}} \right)}^{2}},$Если диффузионная среда представляет собой составное (двухслойное) твердое тело с разными коэффициентами диффузии D1 и D2 в каждом слое и разными параметрами проницаемости для двух границ R1 и R2, эту задачу можно упростить. Так как в электрохимическом эксперименте потенциал электрода определяется поверхностной концентрацией cs, для ее нахождения достаточно рассмотреть только задачу переноса во внешнем слое, ограниченном двумя границами. Предполагая, что R1 = R2 = Rs, были получены теоретические уравнения PITT и GITT [50]:
(7)
$\begin{gathered} j(t) = nFDh\Delta E\left| {\frac{{{\text{d}}{{c}_{{\text{s}}}}}}{{{\text{d}}E}}} \right| \times \\ \times \left( {\frac{1}{{2 + hL}} + \,\,4\sum\limits_{k = 1}^\infty {\left[ {\frac{{\exp \left( { - \alpha _{k}^{2}\frac{{4Dt}}{{{{L}^{2}}}}} \right)}}{{2 + hL + \frac{{4\alpha _{k}^{2}}}{{hL}}}} + \frac{{\exp \left( { - \beta _{k}^{2}\frac{{4Dt}}{{{{L}^{2}}}}} \right)}}{{2 + hL + \frac{{4\beta _{k}^{2}}}{{hL}}}}} \right]} } \right), \\ \end{gathered} $(8)
$\begin{gathered} \Delta E = \Delta {{E}_{{\text{s}}}} + \frac{j}{{nFDh}}\left| {\frac{{{\text{d}}E}}{{{\text{d}}{{c}_{{\text{s}}}}}}} \right| \times \\ \times \,\,\left( {1 + hL - \sum\limits_{k = 1}^\infty {\frac{{2hL\left( {\alpha _{k}^{2} + {{{\left( {hL} \right)}}^{2}}} \right)}}{{\alpha _{k}^{2}\left( {hL + {{{\left( {hL} \right)}}^{2}} + \alpha _{k}^{2}} \right)}}} \exp \left[ { - \frac{{\alpha _{k}^{2}Dt}}{{{{L}^{2}}}}} \right]} \right), \\ \end{gathered} $Для описания зависимости E(c) с использованием изотермы интеркаляции Фрумкина в рамках модели решеточного газа [84–90] используется дифференциальная емкость Cdif(x) в соответствии с выражением:
(9)
$\begin{gathered} {{C}_{{{\text{dif}}}}}(x) = \left( {g + \frac{1}{x} + \frac{1}{{1 - x}} - 0.5\gamma \alpha \times } \right. \\ {{\left. { \times \,\,\left[ {1 - \operatorname{th} {{{\left( {\alpha \left( {x - 0.5} \right)} \right)}}^{2}}} \right]} \right)}^{{ - 1}}}, \\ \end{gathered} $(10)
$\frac{D}{{{{D}_{0}}}} = \frac{{Fx\left( {1 - x} \right){{Q}_{m}}}}{{RT{{C}_{{{\text{dif}}}}}}},$(11)
$\begin{gathered} {{C}_{{{\text{dif}}}}}\left( x \right) = \left( {g + \frac{1}{x} + \frac{1}{{1 - x}} - 2\gamma \frac{{{\text{d}}p}}{{{\text{d}}x}} + } \right. \\ {{\left. { + \,\,\lambda {{{\left( {\frac{{{\text{d}}p}}{{{\text{d}}x}}} \right)}}^{2}} + \left( {\lambda p - \gamma x} \right)\frac{{{{{\text{d}}}^{2}}p}}{{{\text{d}}{{x}^{2}}}}} \right)}^{{ - 1}}} \\ \end{gathered} $Подходы к корректному определению термодинамических и кинетических характеристик интеркалируемых литием электродных материалов
Электродный потенциал или электрический ток являются непосредственно измеряемыми величинами в электрохимических методах, в то время как диффузионная теория имеет дело с концентрациями веществ (компонентов, частиц и т.д.). Как известно, поверхностная концентрация cs потенциалопределяющих частиц задает потенциал электрода E [112]. Зависимость E(c) или E(x) всегда задается для связи электродного потенциала E с объемной концентрацией частиц c или их относительным содержанием в электроде x = c/cmax. Использование равновесной зависимости E(c) предполагает электрохимическое равновесие на каждой границе раздела. Как известно, термодинамическое уравнение Нернста должно содержать активности компонентов aOx и aRed, а не их концентрации cOx и cRed:
(14)
$E = E^\circ + \frac{{RT}}{{nF}}\ln \frac{{\prod {{{a}_{{{\text{Ox}}}}}} }}{{\prod {{{a}_{{{\text{Red}}}}}} }},$(16)
${{\left| {\frac{{{\text{d}}E}}{{{\text{d}}c}}} \right|}_{{c = {{c}_{{\text{0}}}}}}} = \frac{{RT}}{{nF}}{{\left( {\frac{1}{c} + \frac{1}{f}\frac{{{\text{d}}f}}{{{\text{d}}c}}} \right)}_{{c = {{c}_{{\text{0}}}}}}}.$Между тем, многие уравнения электроаналитических методов содержат множитель RT/nFc0, происхождение которого обусловлено необходимостью соотнесения диффузионной теории с электрохимией, чтобы связать известный измеряемый или заданный потенциал электрода с неизвестным распределением концентраций частиц в его объеме [112]. Это соотношение задается уравнением
(17)
${{\left| {\frac{{{\text{d}}E}}{{{\text{d}}c}}} \right|}_{{c = {{c}_{{\text{0}}}}}}} = \frac{{RT}}{{nF{{c}_{0}}}}$Этот упрощенный подход предполагает f ≡ 1. Однако для твердых интеркаляционных фаз характерны сильные межчастичные взаимодействия, и хорошо известно, что равновесная зависимость E(c) (изотерма интеркаляции или изотерма сорбции) твердых электродов переменного состава в общем случае не подчиняется уравнению (18). Использование упрощенной зависимости между потенциалом электрода и концентрацией потенциалопределяющих частиц является наиболее слабым местом электрохимии литиевых систем. Пренебрежение различием между активностью и концентрацией оправдано для разбавленных растворов электроактивных компонентов, которые удовлетворяют классическому теоретическому рассмотрению, представленному в [100], однако оно достигает границы своей применимости, когда готовые уравнения теории используются для описания электрохимических свойств твердых электродов переменного состава. Возможным решением является использование подходящих модельных представлений для теоретического описания зависимости f(c), или же экспериментальное определение зависимости E(c) с ее последующим дифференцированием. Веппнер и Хаггинс [113] впервые предложили использовать методику GITT применительно к интеркаляционным электродам, в ходе которой наряду с хронопотенциометрическими измерениями при постоянном токе проводилась одновременная регистрация экспериментальной изотермы интеркаляции. Фактически, такой подход предусматривает использование в расчетах экспериментального значения производной dE/dc (или dE/dx, dE/dδ), измеренной в каждой отдельной точке изотермы интеркаляции. Этому подходу также присущи свои затруднения, если, например, материал претерпевает фазовые переходы и его изотерма интеркаляции имеет ступенчатую форму [50]. Однако он, очевидно, применим и к исследованиям диффузии в любых твердых электродных материалах.
В настоящее время подход, основанный на применении изотермы интеркаляции Фрумкина и на концепции модели решеточного газа с взаимодействиями притяжения (ближнего и дальнего действия), развит в работах Леви с соавт. [84–90] и считается наиболее оправданным для описания E(c)-зависимости интеркаляционных электродов. Этот подход учитывает взаимодействия внедренных частиц и реакцию матрицы хозяина на их присутствие в структуре материала. Использование изотермы интеркаляции Фрумкина вместо уравнения (18) позволяет не только исключить экспериментальные ошибки в оценке производной dE/dc, но также составить термодинамически обоснованный прогноз хода зависимости D(E).
По мере разработки новых электродных материалов и иссдедования их транспортных свойств возникла достаточно интересная ситуация. Из анализа публикаций становится ясно, что авторы используют различные подходы к расчету D для разных электродных материалов, включая классические формулы (основанные на уравнениях (17) и (18)) [40, 42–45, 47], подход Веппнера [41, 46, 49], изотерма Фрумкина и модель решеточного газа [84–90]. Логично предположить, что основной причиной огромного разброса значений D являются не свойства материалов, а различные способы оценки диффузионной поверхности электродов. Напрашивается совершенно неожиданный вывод, а именно бессмысленность любого сравнения диффузионных оценок схожих материалов, представленных в различных работах, поскольку различие результатов расчетов может быть значительным. На самом деле, перед сравнением данных, необходимо проанализировать, как был определен коэффициент диффузии в каждом случае.
На рис. 3–5 представлены результаты определения коэффициента диффузии лития в электродах на основе LixSn, LixTiO2, LixC6, LixWO3, LiMn2O4 и LiFePO4 с использованием методов EIS, PITT и GITT. Значения D для построения зависимости на рис. 5 были рассчитаны по начальным участкам транзиентов с использованием уравнения (2). В каждом случае расчет D проводился двумя способами: с использованием классического подхода, основанного на уравнениях (17) и (18), а также с использованием экспериментальных значений производной dE/dc, найденных в различных точках изотермы интеркаляции, полученной с помощью кулонометрического титрования. Имеет место очень существенная разница (в несколько порядков) в значениях D, рассчитанных двумя методами, а также в форме зависимости D(E).
Рис. 3.
Зависимости коэффициента диффузии лития от равновесного потенциала электрода, измеренные методом EIS на электродах: а – LixC6, б – LixTiO2, в – LixWO3 и г – LiMn2O4. Значения D рассчитаны с использованием экспериментальных значений dE/dc (кривая 1) и уравнения (17) (кривая 2).

Рис. 4.
Зависимости коэффициента диффузии лития от равновесного потенциала электрода, измеренные методом GITT на электродах: а – LixC6, б – LixSn, в – LixWO3 и г – LiMn2O4. Значения D рассчитаны с использованием экспериментальных значений dE/dc (кривая 1) и уравнения (17) (кривая 2).

Рис. 5.
Зависимости коэффициента диффузии лития от равновесного потенциала электрода, измеренные методом PITT на электродах: а – LixC6, б – LiMn2O4, в – LiFePO4. Значения D рассчитаны с использованием экспериментальных значений dE/dc (кривая 1) и уравнения (17) (кривая 2).

В некоторых случаях (рис. 3г, 4б–4г и 5б, 5в) близость экспериментальных и рассчитанных по уравнению (17) значений dE/dc была подтверждена экспериментально, что нашло отражение в близости значений D, определенных двумя методами. Чаще всего это происходит при наиболее положительных потенциалах электродов, т.е. при минимальной концентрации лития в материале, что согласуется с общим термодинамическим принципом: f → 1 при c → 0. Как видно из сравнения уравнений (16) и (17), возможны две ситуации, при которых различие между ними пренебрежимо мало: это область малых концентраций c → 0 и df/dc ≈ 0, т.е. вблизи экстремума зависимости f(c).
В этой связи коэффициент химической диффузии является, пожалуй, наименее воспроизводимой характеристикой многих электродных материалов. Основными источниками неопределенности при оценке коэффициента химической диффузии с помощью электрохимических методов являются: неучтенные взаимодействия в твердом теле, приводящие к значительной разнице активности и концентрации и, как следствие, изотерме интеркаляции, отличной от задаваемой уравнением Нернста [84]; невозможность корректной оценки диффузионной поверхности; игнорирование роли SEI для литиевых электрохимических систем, а также их возможная нелинейность [50]. Нужно иметь в виду, что простые диффузионные уравнения хорошо соответствуют только некоторым специфическим электродам, характеристики, структура и геометрия которых максимально удовлетворяют начальным и граничным условиям диффузионных задач. Например, они хорошо применимы к тонкопленочным электродам без связующих и проводящих добавок. Такая требовательность к условиям приводит к трудностям в оценке D даже с точностью до порядка.
При анализе рис. 3–5 привлекает внимание в некоторых случаях параллельный ход зависимостей D(E), рассчитанных двумя методами (рис. 3a–3в и 5a). Это не означает близость экспериментального значения dE/dc к рассчитанному по уравнению (17), а указывает на их различие на постоянный множитель. Здесь уместно упомянуть метод CV, который часто используется не только для определения емкостных характеристик электродов и проверки обратимости литиевой интеркаляции, но также для быстрой оценки коэффициента диффузии D электроактивных частиц по линейной зависимости плотности тока в пике jp от квадратного корня из скорости сканирования потенциала v. В отличие от гораздо более точных методов PITT и GITT, которые позволяют измерять зависимость D(E), CV дает усредненную оценку коэффициента диффузии $\hat {D}$, который должен быть постоянным в пределах диапазона потенциалов, в котором проводится измерение. Это предположение, вероятно, является “наиболее слабым местом” метода CV. Как правило, для интеркаляционных материалов характерна выраженная концентрационная зависимость коэффициента диффузии (рис. 3–5). CV не может измерить зависимости D(E), D(c) или D(x) и даже не обнаруживает их существование; CV дает определенное среднее значение $\hat {D}$.
В то же время CV привлекает исследователей своей экспрессностью и простотой как в плане эксперимента, так и расчета по сравнению с более длительными и трудоемкими PITT и GITT, поэтому рассмотрим примеры применения нами этого метода к исследованию электрохимических свойств электродных материалов ЛИА на основе LixWO3, LixTiO2 и LiFePO4. Помимо закона Фика и постоянства $\hat {D},$ уравнение (18) является формулировкой типичного граничного условия в электроаналитической теории CV при использовании его в растворах с фоновым электролитом и постоянной ионной силой [112]. Неприменимость, в общем случае, уравнений (17) и (18) для равновесных условий в твердых интеркаляционных фазах требует использование более сложных эмпирических формул или модельных представлений, например изотермы интеркаляции Фрумкина. Это, в свою очередь, значительно усложняет конечные выражения [84, 85].
Однако существует значительно более простой способ. Как показано в [99] с электродом LixWO3 в качестве примера, самый простой способ – аппроксимировать ограниченный диапазон кривой E(c) электрода, включающий в себя потенциал пика на CV, с помощью “модифицированного” уравнения Нернста, которое отличается от уравнения (18) включением эмпирического подгоночного параметра z:
(Значения E° в уравнениях (18) и (19) могут различаться). Физический смысл параметра z можно прояснить, сравнив эмпирическое уравнение (19) с термодинамическим уравнением (15). Они оба дают один и тот же результат при(20)
$f\left( c \right) = {\text{const}} \times {{c}^{{{{(1 - z)} \mathord{\left/ {\vphantom {{(1 - z)} z}} \right. \kern-0em} z}}}}.$На рис. 6 показаны фрагменты E(c)-кривых для электродов LixWO3 и LixTiO2, измеренные при ступенчатом изменении концентрации ионов лития в электроде в ходе процедуры GITT. Действительно, хотя кривые имеют некоторые особенности, они приближенно линеаризуются в координатах E ~ ln c, что необходимо для использования уравнения (19). Значения параметра z были определены отдельно для анодной и катодной кривых LixWO3 и составили 0.091 и 0.056 соответственно. Для электрода LixTiO2, извлечение лития из которого весьма затруднено, воспроизводимая кривая E(c)-зависимости получается при катодном литировании предварительно делитированного TiO2. Линеаризация зависимости в диапазоне потенциалов 1.1–2.4 В (что соответствует диапазону значений x в LixTiO2 от 0.01 до 0.3; рис. 6б) дает z, равное 0.067.
Рис. 6.
Фрагменты анодной и катодной зависимостей E(ln c), измеренные методом GITT для электродов: а – LixWO3 и б – LixTiO2.

Уравнение (19) имеет преимущество перед другими эмпирическими уравнениями, связывающими потенциал с концентрацией лития в электроде, поскольку оно позволяет использовать классические решения диффузионной теории [112]. В результате, при интегрировании второго закона Фика с постоянной $\hat {D},$ уравнением (19) в качестве граничного условия и постоянной скоростью сканирования потенциала v, было получено [87] следующее уравнение, которое справедливо в области потенциалов, соответствующей применимости уравнения (19):
(21)
${{j}_{{\text{p}}}} = {\text{0}}{\text{.4463}}{{v}^{{{{\text{1}} \mathord{\left/ {\vphantom {{\text{1}} {\text{2}}}} \right. \kern-0em} {\text{2}}}}}}\frac{{{{z}^{{{{\text{1}} \mathord{\left/ {\vphantom {{\text{1}} {\text{2}}}} \right. \kern-0em} {\text{2}}}}}}{{n}^{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}}{{F}^{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}}}}{{{{R}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}{{T}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}}}{{c}_{0}}{{\hat {D}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}.$Для 25°C и n = 1 можно записать в единицах СИ:
Так как в любом случае CV позволяет только оценить усредненный коэффициент $\hat {D},$ дальнейшее усложнение модели с повышением точности расчета может не дать практического эффекта. Легко увидеть из уравнения (20), что введение коэффициента z в уравнение (19) приводит к значениям $\hat {D}$, более чем на порядок отличающимся от классических уравнений для электродов LixWO3 и LixTiO2. Циклические вольтамперограммы электродов LixWO3 и LixTiO2 (не показаны на рисунке) имеют максимумы тока при потенциалах около Е ≈ 2.4 В и Е ≈2.0 В, соответственно. Найденные усредненные значения коэффициента диффузии лития, рассчитанные по формуле (22), составили 1.4 × 10–12 и 1.8 × 10–12 см2 с–1 в LixWO3, а также 1.4 × 10–13 и 0.8 × 10–13 см2 с–1 в LixTiO2 для анодного и катодного процессов соответственно. Представляет значительный интерес сравнить эти значения со значениями, полученными с использованием других электрохимических методов. Усредненный коэффициент диффузии лития в LixWO3 хорошо согласуется с результатами, полученными в этом диапазоне потенциалов с использованием методов EIS и GITT и рассчитанными с использованием экспериментально определенных значений dE/dc (рис. 3в и 4в). Для электрода LixTiO2$\hat {D}$, рассчитанный по данным CV, находится вблизи верхней границы значений, определенных методом EIS и рассчитанных с использованием экспериментального значения dE/dc (рис. 3б).
В научной литературе значительное внимание уделено синтезу и исследованию свойств катодного материала на основе оливина LiFePO4 [40‒50], который претерпевает фазовый переход в ходе токообразующего процесса
(23)
${\text{LiFeP}}{{{\text{O}}}_{4}} \leftrightarrow {\text{L}}{{{\text{i}}}^{ + }} + {\bar {e}} + {\text{FeP}}{{{\text{O}}}_{4}}.$Зависимости потенциала E электрода на основе LiFePO4 от молярной концентрации Li+ (или E от относительного содержания лития в электроде x) были получены в ходе заряда и разряда в квазиравновесных условиях, соответствующих электрохимической реакции (23) с использованием PITT и GITT. На рис. 7а представлены зависимости E(x) отдельно для литированной (Li1 –xFePO4) и делитированной (LixFePO4) форм фосфата железа в координатах, соответствующих линеаризованному уравнению Нернста. Кривые можно условно разделить на несколько характерных фрагментов, соответствующих стехиометрическим состояниям LiFePO4 и FePO4: областям гомогенных твердых растворов Li1– xFePO4 и LixFePO4 и двухфазной области сосуществования Li1– xFePO4 и LixFePO4, ограничивающей твердые растворы. Состав этих ограниченных твердых растворов при 25°C близок к Li0.985FePO4 и Li0.025FePO4. Таким образом, процесс (23) фактически разделяется на три реакции, каждая из которых происходит при определенных составах электрода: в пределах монофазного литированного состава Li1– xFePO4 происходит истощение–обогащение твердого раствора
(24)
${\text{LiFeP}}{{{\text{O}}}_{4}} \leftrightarrow {\text{L}}{{{\text{i}}}_{{1 - x}}}{\text{FeP}}{{{\text{O}}}_{4}} + x{\text{L}}{{{\text{i}}}^{ + }} + x\bar {e},$(25)
${\text{L}}{{{\text{i}}}_{{{\text{0}}{\text{.025}}}}}{\text{FeP}}{{{\text{O}}}_{4}} + 0.96{\text{L}}{{{\text{i}}}^{ + }} + 0.96\bar {e}{\text{ }} \leftrightarrow {\text{L}}{{{\text{i}}}_{{{\text{0}}{\text{.985}}}}}{\text{FeP}}{{{\text{O}}}_{4}},$(26)
${\text{FeP}}{{{\text{O}}}_{4}} + x{\text{L}}{{{\text{i}}}^{ + }} + x{\bar {e} } \leftrightarrow {\text{L}}{{{\text{i}}}_{x}}{\text{FeP}}{{{\text{O}}}_{4}}.$Рис. 7.
Зависимости потенциал–концентрация интеркаляционного электрода, измеренные в ходе процедуры PITT: а – для литированной (Li1 –xFePO4) и делитированной (LixFePO4) форм фосфата железа; б – для монофазных областей Li1– xFePO4 и LixFePO4, аппроксимированные уравнением (19).
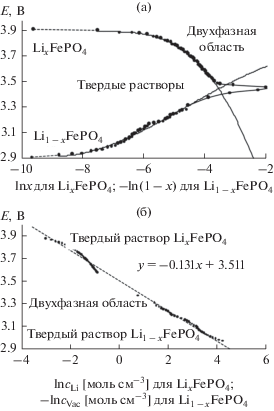
Уравнение (19) очевидно применимо только к равновесиям (24) и (26). На рис. 7б показано, что фрагменты зависимости E(c), соответствующие двум областям твердых растворов Li1– xFePO4 и LixFePO4, могут быть приблизительно аппроксимированы одной прямой линией, соответствующей уравнению (19) с параметром z = 0.195. Все измеренные экспериментальные точки лежат вблизи этой прямой линии. Это позволяет использовать уравнение (21) для расчета усредненного коэффициента диффузии лития в оливине.
На рис. 8а приведен набор циклических вольтамперограмм электрода LiFePO4, измеренных при нескольких скоростях v, а на рис. 8б представлено подтверждение диффузионного характера лимитирующей стадии процесса интеркаляции–деинтеркаляции лития для электрода LiFePO4. Однако, неполное согласие с уравнением (21) выражается в том, что линейная зависимость jp(v0.5) не проходит через начало координат. Кроме того, смысл начальной концентрации c0 в уравнении (21) различен для разных полупериодов вольтамперограммы. Для катодного процесса c0 = cvac – начальная концентрация свободных мест в LixFePO4, доступных для заполнения литием. Для анодного процесса c0 = cLi – начальная концентрация лития, способного растворяться в Li1 –xFePO4 (стехиометрический состав LiFePO4 соответствует расчетному значению cLi = 22.8 М). Невозможно гарантировать, что в экспериментальных условиях CV будет выполнено равенство cvac = cLi, поэтому, вообще говоря, наклоны анодной и катодной зависимостей jp(v0.5), которые соответствуют линеаризации уравнения (21), могут отличаться, как это видно на рис. 8б.
Рис. 8.
а – Циклические вольтамперограммы электрода LiFePO4, измеренные при нескольких скоростях сканирования потенциала; б – зависимости тока пика CV от скорости сканирования потенциала.

При условии, что ${{c}_{{{\text{Li}}}}} = c{{v}_{{{\text{ac}}}}}\sqrt 2 ,$ анодные и катодные ветви циклических вольтамперограмм для LixFePO4 дают одинаковый усредненный коэффициент диффузии лития $\hat {D}$ = 3 × 10–12 см2 с–1. Это значение находится вблизи верхней границы диапазона значений D, которые приводились в большинстве экспериментальных работ (табл. 1). Согласно теоретическим расчетам [114], может быть достигнута значительно более высокая скорость диффузии лития в LixFePO4. Зависимость коэффициента диффузии от потенциала электрода, измеренная с использованием PITT, показывает его приблизительное постоянство в пределах диапазона потенциалов существования твердых растворов (кривая 1 на рис. 5в) со средним значением $\hat {D}$ ≈ (2 ± 1) × 10–12 см2 с–1. Поэтому в данном случае PITT и CV дали очень близкие результаты при условии использования предложенной методики расчета. В нашей работе [50] мы обсуждали вопрос о том, как ошибка в определении различных параметров влияет на корректность расчета D. Было показано, что параметр A (диффузионная поверхность) не влияет на результаты оценки D по PITT и GITT, если обработке подвергается полный транзиент (фронт диффузии при движении от внешней границы достигает внутренней границы диффузионного слоя), корректная оценка толщины L диффузионного слоя становится решающей. Однако, если используется только начальный участок транзиента в пределах условий полубесконечной диффузии, L не участвует в таких расчетах, и правильная оценка диффузионной поверхности становится решающей: 2-кратная ошибка в величине поверхности изменит D в 4 раза. Вместо фактической диффузионной поверхности A обычно используется площадь внешней поверхности, которая зависит от метода измерения и предполагаемого физического смысла. В различных работах в качестве таковой используются геометрическая (видимая) поверхность электрода [23, 58], удельная площадь по БЭТ [44], совокупная внешняя поверхность частиц из предположения их сферической формы [50, 115] и т.д. Такие величины отличаются на несколько порядков, что существенно повлияет на значения D.
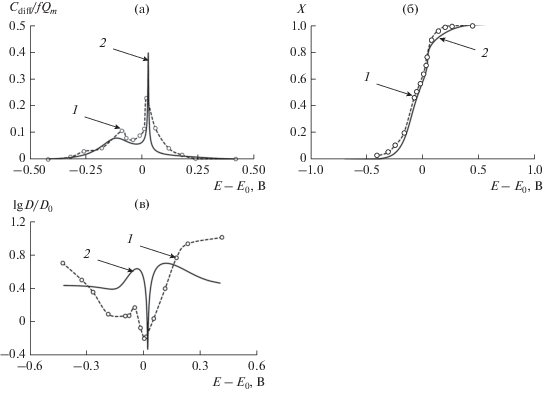
Значительный интерес представляет анализ D(E)-зависимостей рассмотренных электродов с точки зрения применяемой изотермы интеркаляции. Экстремальные зависимости D(E) были выбраны в качестве примеров, которые позволяют наилучшим образом проиллюстрировать функциональность используемой модели. Это D(E)-зависимости LiMn2O4-электрода. Параметры модели (межчастичного взаимодействия) на основе изотермы интеркаляции Фрумкина и модели решеточного газа рассчитывались путем подгонки теоретически рассчитанной изотермы интеркаляции к экспериментально измеренной кривой. Последний был представлен как зависимости x(E) и CDiff(E) с предварительным масштабированием вдоль оси потенциала для соответствия диапазону потенциалов расчетной кривой. Результат моделирования представлен на рис. 9 (найденные модельные параметры приведены в подписи к рисунку). Сравнение расчетных и экспериментальных зависимостей D(E) также представлено на рисунке. Поскольку модель предполагает сравнение форм обеих зависимостей D(E), но данных для теоретического расчета значений D недостаточно, экспериментальные зависимости D(E) были соответствующим образом масштабированы. Можно заключить, что формы обеих зависимостей имеют общие черты.
Рис. 9.
Зависимости: а – дифференциальной емкости, б – степени литирования электродного материала и в – коэффициента диффузии лития в LiMn2O4-электроде от электродного потенциала: экспериментальные данные (кривая 1), рассчитанные в соответствии с уравнениями (9)–(13) со следующим набором параметров (кривая 2): g = –1, γ = 2, λ = 2.3, α1 = –5, x1 = 0.8, p1 = 0.7, α2 = –1, x2 = 0.1, p2 = 0.3.
ЗАКЛЮЧЕНИЕ
Представленная работа обращает внимание читателей на значительный разброс (несколько порядков) значений химического коэффициента диффузии D, определенных в аналогичных электродных материалах ЛИА, по данным различных авторов. Основной причиной этого является использование нескольких различных подходов при выводе уравнений для расчета D: (1) классический подход, основанный на совместном решении диффузионного уравнения второго закона Фика и уравнения зависимости потенциала от концентрации Нернста, и (2) различные варианты подходов, учитывающих существенное различие между активностью и концентрацией потенциалопределяющих частиц из-за сильных взаимодействий в твердых электродах переменного состава. Несомненно, последние из упомянутых подходов концептуально лучше обоснованы. Тем не менее наш литературный анализ показывает, что различные подходы и модели примерно поровну используются исследователями при разработке новых электродных материалов. Поэтому при сравнительном анализе данных для аналогичных электродных материалов представляется правильным сравнить значения химического коэффициента диффузии, рассчитанные только по идентичным формулам, поскольку различие результатов расчета может быть огромным, как показано в настоящей работе.
Помимо игнорируемых твердотельных взаимодействий, являющихся причиной значительного различия активности и концентрации, и ненернстовской изотермы интеркаляции, основными источниками неопределенности в определении химического коэффициента диффузии D по электроаналитическим данным являются: неверные оценки площади и длины диффузии; несоответствие между структурой и геометрией реального электрода и математическими условиями диффузионной задачи (плохая реализация теоретических ограничений, начальных и граничных условий в экспериментальной практике); для литиевых систем – игнорируемая роль SEI, а также возможная нелинейность системы.
Список литературы
Поваров, Ю.М., Бекетаева, Л.А. О механизме пассивации лития в тионилхлориде. Электрохимия. 1980. Т. 16. С. 1252.
Чахов, Н.И., Поваров, Ю.М., Плесков, Ю.В. Влияние освещения на процесс электровосстановления тионилхлорида на углеродных материалах. Электрохимия. 1980. Т. 16. С. 1445.
Поваров, Ю.М., Ситнина, Е.Н. Влияние освещения на поляризационные характеристики литиевого электрода в пропиленкарбонатных растворах. Электрохимия. 1981. Т. 17. С. 633.
Поваров, Ю.М., Бекетаева, Л.А., Астахов, И.И., Суриков, В.В., Моштев, Р., Пурешева, Б. Исследование пассивации лития в растворах двуокиси серы в нитрометане электрохимическим и электронно-микроскопическими методами. Электрохимия. 1982. Т. 18. С. 1160.
Поваров, Ю.М., Бекетаева, Л.А., Пурешева, Б.К. Импеданс литиевого электрода в растворах окислителей. Электрохимия. 1982. Т. 18. С. 1340.
Поваров, Ю.М., Воробьева, И.В. Импеданс литиевого электрода в тионилхлоридных электролитах. Электрохимия. 1982. Т. 18. С. 1693.
Поваров, Ю.М., Бекетаева, Л.А., Воробьева, И.В. Импеданс литиевого электрода в среде апротонных органических растворителей. Электрохимия. 1983. Т. 19. С. 586.
Kazarinov, V.E. and Bagotzky, V.S. Properties of a passivating film on the surface of lithium electrodes. J. Power Sources, 1987, vol. 20, p. 259.
Алексеева, Л.А., Астахов, И.И., Попова, С.С., Киселева, И.Г., Суриков, В.В. Электронно-микроскопическое исследование поверхности алюминиевого электрода в процессе катодного внедрения лития в алюминий. Электрохимия. 1985. Т. 21. С. 1116.
Попова, С.С., Алексеева, Л.А., Белова, О.В., Петрова, Л.Н., Киселева, И.Г., Кабанов, Б.Н. Влияние оксидных слоев на процесс катодного внедрения лития в алюминий. Электрохимия. 1986. Т. 22. С. 1497.
Нимон, Е.С., Чуриков, А.В., Сенотов, А.А., Львов, А.Л., Придатко, И.А. Ионный транспорт в пассивирующих слоях на литиевом электроде. Доклады АН СССР. 1988. Т. 303. С. 1180.
Нимон, Е.С., Чуриков, А.В., Сенотов, А.А., Львов, А.Л., Чувашкин, А.Н. Ионные токи, ограниченные пространственным зарядом, в твердоэлектролитных пленках на поверхности. Физика твердого тела. 1989. Т. 31. С. 278.
Nimon, E.S., Churikov, A.V., Gamayunova, I.M., and Lvov, A.L., Photoelectrochemistry of lithium, J. Power Sources, 1993, vol. 43, p. 157.
Nimon, E.S., Churikov, A.V., Shirokov, A.V., Lvov, A.L., and Chuvashkin, A.N., Ionic transport in passivating layers on the lithium electrode, J. Power Sources, 1993, vol. 44, p. 365.
Модестов, А.Д., Нимон, Е.С., Ротенберг, З.А., Чуриков, А.В. Исследование фотоэлектрохимического поведения лития и его сплавов методом модулированного освещения. Электрохимия. 1996. Т. 32. С. 764. [Modestov, A.D., Nimon, E.S., Rotenberg, Z.A., and Churikov, A.V., A light-intensity-modulation study of photoelectrochemical behavior of lithium and its alloys, Russ. J. Electrochem., 1996, vol. 32, p. 705.]
Nimon, E.S., Churikov, A.V., and Kharkats, Yu.I., Relaxation photocurrent at the electronic emission from lithium into the surface passivating film, J. Electroanal. Chem., 1997, vol. 420, p. 135.
Churikov, A.V., Kharkats, Yu.I., Gamayunova, I.M., Nimon, E.S., and Shirokov, A.V., Diffusion processes at photoemission from lithium into its passivating layer, Electrochim. Acta, 2001, vol. 46, p. 2929.
Багоцкий, В.С., Скундин, А.М. Основные научные проблемы создания перезаряжаемых источников тока. Электрохимия. 1998. Т. 34. С. 741. [Bagotzky, V.S. and Skundin, A.M., Fundamental scientific problems related to the development of rechargeable lithium batteries, Russ. J. Electrochem., 1998, vol. 34, p. 654.]
Skundin, A.M., Efimov, O.N., and Yarmolenko, O.V., The state-of-the-art and prospects for the development of rechargeable lithium batteries, Russ. Chem. Rev., 2002, vol. 71, p. 329.
Kulova, T.L. and Skundin, A.M., Decreasing irreversible capacity of graphite electrodes in lithium-ion batteries by direct contact of graphite with metallic lithium, Russ. J. Electrochem., 2002, vol. 38, p. 1319.
Churikov, A.V., Volgin, M.A. and Pridatko, K.I., On the determination of kinetic characteristics of lithium intercalation into carbon, Electrochim. Acta, 2002, vol. 47, p. 2857.
Чуриков, А.В., Волгин, М.А., Придатко, К.И., Иванищев, А.В., Гридина, Н.А., Львов, А.Л. Электрохимическая интеркаляция лития в углерод: исследование релаксационными методами. Электрохимия. 2003. Т. 39. С. 591. [Churikov, A.V., Volgin, M.A., Pridatko, K.I., Ivanishchev, A.V., Gridina, N.A., and L’vov, A.L., Electrochemical Intercalation of Lithium into Carbon: A Relaxation Study, Russ. J. Electrochem., 2003, vol. 39, p. 531.]
Churikov, A.V. and Ivanischev, A.V., Application of pulse methods to the determination of the electrochemical characteristics of lithium intercalates, Electrochim. Acta, 2003, vol. 48, p. 3677.
Doyle, M., Newman, J., Gozdz, A.S., Schmutz, C.N., and Tarascon. J.-M., Comparison of Modeling Predictions with Experimental Data from Plastic Lithium Ion Cells, J. Electrochem. Soc., 1996, vol. 142, p. 1890.
Sandí, G., Winans, R.E., and Carrado, K.A., New Carbon Electrodes for Secondary Lithium Batteries, J. Electrochem. Soc., 1996, vol. 143, p. L95.
Jean, M., Desnoyer, C., Tranchant, A., and Messina, R., Electrochemical and Structural Studies of Petroleum Coke in Carbonate-Based Electrolytes, J. Electrochem. Soc., 1995, vol. 142, p. 2122.
Takami, N., Satoh, A., Hara, M., and Ohsaki, T., Structural and Kinetic Characterization of Lithium Intercalation into Carbon Anodes for Secondary Lithium Batteries, J. Electrochem. Soc., 1995, vol. 142, p. 371.
Takami, N., Satoh, A., Ohsaki, T., and Kanda, M., Large Hysteresis during Lithium Insertion into and Extraction from High-Capacity Disordered Carbons, J. Electrochem. Soc., 1998, vol. 145, p. 478.
Funabiki, A., Inaba, M., Ogumi, Z., Yuasa, S.-I., Otsuji, J., and Tasaka, A., Impedance Study on the Electrochemical Lithium Intercalation into Natural Graphite Powder, J. Electrochem. Soc., 1998, vol. 145, p. 172.
Piao, T., Park, S.-M., Doh, C.-H., and Moon, S.-I., Intercalation of Lithium Ions into Graphite Electrodes Studied by AC Impedance Measurements, J. Electrochem. Soc., 1999, vol. 146, p. 2794.
Chang, Y.-C. and Sohn, H.-J., Electrochemical Impedance Analysis for Lithium Ion Intercalation into Graphitized Carbons, J. Electrochem. Soc., 2000, vol. 147, p. 50.
Chang, Y.-C., Jong, J.-H., and Fey, G.T.-K., Kinetic Characterization of the Electrochemical Intercalation of Lithium Ions into Graphite Electrodes, J. Electrochem. Soc., 2000, vol. 147, p. 2033.
Ong, T. S. and Yang, H., Symmetrical Cell for Electrochemical AC Impedance Studies of Lithium Intercalation into Graphite, Electrochem. Solid St., 2001, vol. 4, p. A89.
Dollé, M., Orsini, F., Gozdz, A.S., and Tarascon, J.-M., Development of Reliable Three-Electrode Impedance Measurements in Plastic Li-Ion Batteries, J. Electrochem. Soc., 2001, vol. 148, p. A851.
Yu, P., Popov, B.N., Ritter, J.A., and White, R.E., Determination of the Lithium Ion Diffusion Coefficient in Graphite, J. Electrochem. Soc., 1999, vol. 146, p. 8.
Nishizawa, M., Hashitani, R., Itoh, T., Matsue, T., and Uchida, I., Measurements of Chemical Diffusion Coefficient of Lithium Ion in Graphitized Mesocarbon Microbeads Using a Microelectrode, Electrochem. Solid St., 1998, vol. 1, p. 10.
Wang, Q., Li, H., Huang, X., and Chen, L., Determination of Chemical Diffusion Coefficient of Lithium Ion in Graphitized Mesocarbon Microbeads with Potential Relaxation Technique, J. Electrochem. Soc., 2001, vol. 148, p. A737.
Umeda, M., Dokko, K., Fujita, Y., Mohamedi, M., Uchida, I., and Selman, J.R., Electrochemical impedance study of Li-ion insertion into mesocarbon microbead single particle electrode: Part I. Graphitized carbon, Electrochim. Acta, 2001, vol. 47, p. 885.
Dokko, K., Fujita, Y., Mohamedi, M., Umeda, M., Uchida, I., and Selman, J.R., Electrochemical impedance study of Li-ion insertion into mesocarbon microbead single particle electrode: Part II. Disordered carbon, Electrochim. Acta, 2001, vol. 47, p. 933.
Xie, J., Imanishi, N., Zhang, T., Hirano, A., Takeda, Y., and Yamamoto O., Li-ion diffusion kinetics in LiFePO4 thin film prepared by radio frequency magnetron sputtering, Electrochim. Acta, 2009, vol. 54, p. 4631.
Tang, K., Yu, X., Sun, J., Li, H., and Huang, X., Kinetic analysis on LiFePO4 thin films by CV, GITT, and EIS, Electrochim. Acta, 2011, vol. 56, p. 4869.
Zhang, S.M., Zhang, J.X., Xu, S.J., Yuan, X.J., and He, B.C., Li ion diffusivity and electrochemical properties of FePO4 nanoparticles acted directly as cathode materials in lithium ion rechargeable batteries, Electrochim. Acta, 2013, vol. 88, p. 287.
Liu, H., Li, C., Zhang, H.P., Fu, L.J., Wu, Y.P., and Wu, H.Q., Kinetic study on LiFePO4/C nanocomposites synthesized by solid state technique, J. Power Sources, 2006, vol. 159, p. 717.
Gao, F. and Tang, Z., Kinetic behavior of LiFePO4/C cathode material for lithium-ion batteries, Electrochim. Acta, 2008, vol. 53, p. 5071.
Liu, J., Jiang, R., Wang, X., Huang, T., and Yu, A., The defect chemistry of LiFePO4 prepared by hydrothermal method at different pH values, J. Power Sources, 2009, vol. 194, p. 536.
Li, L., Tang, X., Liu, H., Qu, Y., and Lu, Z., Morphological solution for enhancement of electrochemical kinetic performance of LiFePO4, Electrochim. Acta, 2010, vol. 56, p. 995.
Jiang, Z. and Jiang, Z.-J., Effects of carbon content on the electrochemical performance of LiFePO4/C core/shell nanocomposites fabricated using FePO4/polyaniline as an iron source, J. Alloy. Compd., 2012, vol. 537, p. 308.
Zhao, D., Feng, Y.-L., Wang, Y.-G., and Xia, Y.-Y., Electrochemical performance comparison of LiFePO4 supported by various carbon materials, Electrochim. Acta, 2013, vol. 88, p. 632.
Sun, C.S., Zhou, Z., Xu, Z.G., Wang, D.G., Wei, J.P., Bian, X.K., and Yan, J., Improved high-rate charge/discharge performances of LiFePO4/C via V-doping, J. Power Sources, 2009, vol. 193, p. 841.
Churikov, A.V., Ivanishchev, A.V., Ivanishcheva, I.A., Sycheva, V.O., Khasanova, N.R., and Antipov, E.V., Determination of lithium diffusion coefficient in LiFePO4 electrode by galvanostatic and potentiostatic intermittent titration techniques, Electrochim. Acta, 2010, vol. 55, p. 2939.
Wu, X., Chen, S., He, Z., Ma, M., and Liu, J., Influence of thickness on the properties of solution-derived LiMn2O4 thin films, J. Wuhan. Univ. Technol., 2009, vol. 24, p. 706.
Singh, D., Kim, W.-S., Craciun, V., Hofmann, H., and Singh, R.K., Microstructural and electrochemical properties of lithium manganese oxide thin films grown by pulsed laser deposition, Appl. Surf. Sci., 2002, vols. 197–198, p. 516.
Yamada, O., Ishikawa, M., and Morita, M., Charge/discharge cycling and impedance response of LiMn2O4 electrode in organic electrolyte solutions with different compositions, Electrochim. Acta, 2000, vol. 45, p. 2197.
Deiss, E., Haringer, D., Novak, P., and Haas, O., Modeling of the charge–discharge dynamics of lithium manganese oxide electrodes for lithium-ion batteries, Electrochim. Acta, 2001, vol. 46, p. 4185.
Eftekhari, A., Electrochemical behavior of thin-film LiMn2O4 electrode in aqueous media, Electrochim. Acta, 2001, vol. 47, p. 495.
Cao, F. and Prakash, J., A comparative electrochemical study of LiMn2O4 spinel thin-film and porous laminate, Electrochim. Acta, 2002, vol. 47, p. 1607.
Hjelm, A.-K. and Lindbergh, G., Experimental and theoretical analysis of LiMn2O4 cathodes for use in rechargeable lithium batteries by electrochemical impedance spectroscopy (EIS), Electrochim. Acta, 2002, vol. 47, p. 1747.
Mohamedi, M., Takahashi, D., and Uchida, T., Electrochemical stability of thin film LiMn2O4 cathode in organic electrolyte solutions with different compositions at 55°C, Electrochim. Acta, 2002, vol. 47, p. 3483.
Bang, H.J., Donepudi, V.S., and Prakash, J., Preparation and characterization of partially substituted LiMyMn2 –yO4 (M = Ni, Co, Fe) spinel cathodes for Li-ion batteries, Electrochim. Acta, 2002, vol. 48, p. 443.
Mohamedi, M., Makino, M., Dokko, K., and Uchida, T., Electrochemical investigation of LiNi0.5Mn1.5O4 thin film intercalation electrodes, Electrochim. Acta, 2002, vol. 48, p. 79.
Xiao, L., Guo, Y., Qu, D., Deng, B., Liu, H., and Tang, D., Influence of particle sizes and morphologies on the electrochemical performances of spinel LiMn2O4 cathode materials, J. Power Sources, 2013, vol. 225, p. 286.
Wang, Y.Z., Shao, X., Xu, H.Y., Xie, M., Deng, S.X., Wang, H., Liu, J.B., and Yan, H., Facile synthesis of porous LiMn2O4 spheres as cathode materials for high-power lithium ion batteries, J. Power Sources, 2013, vol. 226, p. 140.
Yi, T.-F., Yin, L.-C., Ma, Y.-Q., Shen, H.-Y., Zhu, Y.-R., and Zhu, R.-S., Lithium-ion insertion kinetics of Nb-doped LiMn2O4 positive-electrode material, Ceram. Int., 2013, vol. 39, p. 4673.
Tang, X.-C., Song, X.-W., Shen, P.-Z., and Jia, D.-Z., Capacity intermittent titration technique (CITT): A novel technique for determination of Li+ solid diffusion coefficient of LiMn2O4, Electrochim. Acta, 2005, vol. 50, p. 5581.
Xie, J., Kohno, K., Matsumura, T., Imanishi, N., Hirano, A., Takeda, Y., and Yamamoto, O., Li-ion diffusion kinetics in LiMn2O4 thin films prepared by pulsed laser deposition, Electrochim. Acta, 2008, vol. 54, p. 376.
Xie, J., Tanaka, T., Imanishi, N., Matsumura, T., Hirano, A., Takeda, Y., and Yamamoto, O., Li-ion transport kinetics in LiMn2O4 thin films prepared by radio frequency magnetron sputtering, J. Power Sources, 2008, vol. 180, p. 576.
Tang, S.B., Lai, M.O., and Lu, L., Study on Li+-ion diffusion in nano-crystalline LiMn2O4 thin film cathode grown by pulsed laser deposition using CV, EIS and PITT techniques, Mater. Chem. Phys., 2008, vol. 111, p. 149.
Ye, S.H., Bo, J.K., Li, C.Z., Cao, J.S., Sun, Q.L., and Wang, Y.L., Improvement of the high-rate discharge capability of phosphate-doped spinel LiMn2O4 by a hydrothermal method, Electrochim. Acta, 2010, vol. 55, p. 2972.
Quan, Z., Ohguchi, S., Kawase, M., Tanimura, H., and Sonoyama, N., Preparation of nanocrystalline LiMn2O4 thin film by electrodeposition method and its electrochemical performance for lithium battery, J. Power Sources, 2013, vol. 244, p. 375.
Manjunatha, H., Mahesh, K.C., Suresh, G.S., and Venkatesha, T.V., The study of lithium ion de-insertion/insertion in LiMn2O4 and determination of kinetic parameters in aqueous Li2SO4 solution using electrochemical impedance spectroscopy, Electrochim. Acta, 2011, vol. 56, p. 1439.
Чуриков, А.В., Качибая, Э.И., Сычева, В.О., Иванищева, И.А., Имнадзе, Р.И., Паикидзе, Т.В., Иванищев, А.В. Электрохимические свойства шпинелей LiMn2 –yMeyO4 (Me = Cr, Co, Ni) как катодных материалов для литий-ионного аккумулятора, Электрохимия. 2009. Т. 45. С. 185. [Churikov, A.V., Kachibaya, E.I., Sycheva, V.O., Ivanishcheva, I.A., Imnadze, R.I., Paikidze, T.V., and Ivanishchev, A.V., Electrochemical properties of LiMn2 –yMeyO4 (Me = Cr, Co, Ni) spinels as cathodic materials for lithium-ion batteries, Russ. J. Electrochem., 2009, vol. 45, p. 175.]
Yu, A., Kumagai, N. and Yashiro H., Synthesis of pyrochlore tungsten trioxide thin film and electrochemical lithium intercalation, Solid State lonics, 1997, vol. 100, p. 267.
Yu, A., Kumagai, N., Liu, Z., and Lee, J.-Y., Electrochemical lithium intercalation into WO3 and lithium tungstates LixWO3 +x/2 of various structures, J. Solid State Electr., 1998, vol. 2, p. 394.
Avellaneda, C.O. and Bulhoes, L.O.S., Intercalation in WO3 and WO3:Li films, Solid State Ionics, 2003, vol. 165, p. 59.
Pyun, S.-I. and Bae, J.-S., Lithium ion transport in r.f.-magnetron sputtered WO3 film as a function of lithium content, J. Alloy Compd., 1996, vol. 245, p. L1.
Kim, J.J., Tryk, D.A., Amemiya, T., Hashimoto, K., and Fujishima, A., Color impedance and electrochemical impedance studies of WO3 thin films: H+ and Li+ transport, J. Electroanal. Chem., 1997, vol. 435, p. 31.
Mattsson, M.S., Li insertion into WO3: introduction of a new electrochemical analysis method and comparison with impedance spectroscopy and the galvanostatic intermittent titration technique, Solid State Ionics, 2000, vol. 131, p. 261.
Lee, S.-H., Cheong, H.M., Tracy, C.E., Mascarenhas, A., Pitts, R., Jorgensen, G., and Deb, S.K., Influence of microstructure on the chemical diffusion of lithium ions in amorphous lithiated tungsten oxide films, Electrochim. Acta, 2001, vol. 46, p. 3415.
Kavan, L., Fattakhova, D., and Krtil, P., Lithium Insertion into Mesoscopic and Single-Crystal TiO2 (Rutile) Electrodes, J. Electrochem. Soc., 1999, vol. 146, p. 1375.
Krtil, P. and Fattakhova, D., Li Insertion into Li–Ti–O Spinels: Voltammetric and Electrochemical Impedance Spectroscopy Study, J. Electrochem. Soc., 2001, vol. 148, A1045.
Kanamura, K., Yuasa, K., and Takehara, Z., Diffusion of lithium in the tioz cathode of a lithium battery, J. Power Sources, 1987, vol. 20, p. 127.
Li, H., Huang, X., and Chen, L., Electrochemical impedance spectroscopy study of SnO and nano-SnO anodes in lithium rechargeable batteries, J. Power Sources, 1999, vol. 81–82, p. 340.
Besenhard, J.O., Wachtler, M., Winter, M., Andreaus, R., Rom, I., and Sitte, W., Kinetics of Li insertion into polycrystalline and nanocrystalline “SnSb” alloys investigated by transient and steady state techniques, J. Power Sources, 1999, vol. 81–82, p. 268.
Levi, M.D. and Aurbach, D., Frumkin intercalation isotherm D a tool for the description of lithium insertion into host materials: a review, Electrochim. Acta, 1999, vol. 45, p. 167.
Levi, M.D., Gamolsky, K., Aurbach, D., Heider, U., and Oesten, R., Determination of the Li-ion chemical diffusion coefficient for the topotactic solid-state reactions occurring via a two-phase or single-phase solid solution pathway, J. Electroanal. Chem., 1999, vol. 477, p. 32.
Levi, M.D., Salitra, G., Markovsky, B., Teller, H., Aurbach, D., Heider, U., and Heider, L., Solid-State Electrochemical Kinetics of Li-Ion Intercalation into Li1 –xCoO2: Simultaneous Application of Electroanalytical Techniques SSCV, PITT, and EIS, J. Electrochem. Soc., 1999, vol. 146, p. 1279.
Levi, M.D., Aurbach, D., and Vorotyntsev, M.A., Interpretation of potential intermittence titration technique experiments for various Li-intercalation electrodes, Condens. Matter Phys., 2002, vol. 5, p. 329.
Vorotyntsev, M.A., Levi, M.D., and Aurbach, D., Spatially Limited Diffusion Coupled with Ohmic Potential Drop and/or Slow Interfacial Exchange: A New Method to Determine the Diffusion Time Constant and External Resistance from Potential, J. Electroanal. Chem., 2004, vol. 572, p. 299.
Levi, M.D., Markevich, E., and Aurbach, D., The Effect of Slow Interfacial Kinetics on the Chronoamperometric Response of Composite Lithiated Graphite Electrodes and on the Calculation of the Chemical Diffusion Coefficient of Li Ions in Graphite, J. Phys. Chem. B, 2005, vol. 109, p. 7420.
Levi, M.D., Aurbach, D., and Maier, J., Electrochemically driven first-order phase transitions caused by elastic responses of ion-insertion electrodes under external kinetic control, J. Electroanal. Chem., 2008, vol. 624, p. 251.
Montella C., EIS study of hydrogen insertion under restricted diffusion conditions: I. Two-step insertion reaction, J. Electroanal. Chem., 2001, vol. 497, p. 3.
Montella, C., Discussion of the potential step method for the determination of the diffusion coefficients of guest species in host materials Part I. Influence of charge transfer kinetics and ohmic potential drop, J. Electroanal. Chem., 2002, vol. 518, p. 61.
Montella, C., Discussion of three models used for the investigation of insertion/extraction processes by the potential step chronoamperometry technique, Electrochim. Acta, 2005, vol. 50, p. 3746.
Montella, C., Apparent diffusion coefficient of intercalated species measured with PITT. A simple formulation, Electrochim. Acta, 2006, vol. 51, p. 3102.
Li, J., Yang, F., Xiao, X., Verbrugge, M.W., and Cheng, Y.-T., Potentiostatic intermittent titration technique (PITT) for spherical particles with finite interfacial kinetics, Electrochim. Acta, 2012, vol. 75, p. 56.
Li, J., Xiao, X., Yang, F., Verbrugge, M.W., and Cheng, Y.-T., Potentiostatic Intermittent Titration Technique for Electrodes Governed by Diffusion and Interfacial Reaction, J. Phys. Chem. C, 2012, vol. 116, p. 1472.
Чуриков, А.В., Придатко, К.И., Иванищев, А.В., Иванищева, И.А., Гамаюнова, И.М., Запсис, К.В., Сычева, В.О. Спектроскопия импеданса пленочных литий-оловянных электродов. Электрохимия. 2008, Т. 44. С. 594. [Churikov, A.V., Pridatko, K.I., Ivanishchev, A.V., Ivanishcheva, I.A., Gamayunova, I.M., Zapsis, K.V., and Sycheva, V.O., Impedance Spectroscopy of Lithium–Tin Film Electrodes, Russ. J. Electrochem., 2008, vol. 44, p. 550.]
Чуриков, А.В., Зобенкова, В.А., Придатко, К.И. Внедрение лития в пленки диоксида титана из пропиленкарбонатного раствора. Электрохимия. 2004. Т. 40. С. 74. [Churikov, A.V., Zobenkova, V.A., and Pridatko, K.I., Lithium Intercalation into Titanium Dioxide Films from a Propylene Carbonate Solution, Russ. J. Electrochem., 2004, vol. 40, p. 63.]
Чуриков, А.В., Иванищев, А.В., Иванищева, И.А., Запсис, К.В., Гамаюнова, И.М., Сычева, В.О. Кинетика электрохимического внедрения лития в тонкие слои оксида вольфрама(VI). Электрохимия. 2008. Т. 44. С. 574. [Churikov, A.V., Ivanishchev, A.V., Ivanishcheva, I.A., Zapsis, K.V., Gamayunova, I.M., and Sycheva, V.O., Kinetics of Electrochemical Lithium Intercalation into Thin Tungsten(VI) Oxide Layers, Russ. J. Electrochem., 2008, vol. 44, p. 530.]
Волгин, М.А., Чуриков, А.В., Коноплянцева, Н.А., Гридина, Н.А., Львов, А.Л. Электрохимическая интеркаляция лития в тонкие слои пироуглерода. Электрохимия. 1998. Т. 34. С. 761. [Volgin, M.A., Churikov, A.V., Konoplyantseva, N.A., Gridina, N.A., L’vov, A.L., Electrochemical intercalation of lithium into thin pyrocarbon layers, Russ. J. Electrochem., 1998, vol. 34, p. 681.]
Suzuki, T., WH, YM, Graphite electrode, United States Patent 5,169,508, Dec 8, 1992.
Иванищев, А.В., Чуриков, А.В., Иванищева, И.А., Запсис, К.В., Гамаюнова И.М. Импедансная спектроскопия литий-углеродных электродов. Электрохимия. 2008. Т. 44. С. 553. [Ivanishchev, A.V., Churikov, A.V., Ivanishcheva, I.A., Zapsis, K.V., and Gamayunova, I.M., Impedance Spectroscopy of Lithium–Carbon Electrodes, Russ. J. Electrochem., 2008, vol. 44, p. 510.]
Kachibaya, E.I., Imnadze, R.A., Paikidze, T.V., and Akhvlediani, R.A., Cathodic materials for lithium-ion batteries based on spinels LixMn2 –yMeyO4: Synthesis, phase composition, and structure of LixMn2 –yCryO4 at x = 1.0–1.2 and y = 0–0.5, Russ. J. Electrochem., 2006, vol. 42, p. 1224.
Churikov, A.V., Ivanishchev, A.V., Ushakov, A.V., Ga-mayunova, I.M., and Leenson, I.A., Thermodynamics of LiFePO4 Solid-Phase Synthesis Using Iron(II) Oxalate and Ammonium Dihydrophosphate as Precursors, J. Chem. Eng. Data, 2013, vol. 58, p. 1747.
Churikov, A., Gribov, A., Bobyl, A., Kamzin, A., and Terukov, E., Mechanism of LiFePO4 solid-phase synthesis using iron(II) oxalate and ammonium dihydrophosphate as precursors, Ionics, 2014, vol. 20, p. 1.
Иванищев, А.В., Чуриков, А.В., Иванищева, И.А., Ушаков, А.В., Снеха, М.Дж., Баббар, П., Диксит, А. Модели литиевого транспорта и их приложение к определению диффузионных характеристик интеркаляционных электродов. Электрохимия. 2017. Т. 53. С. 795. [Ivanishchev, A.V., Churikov, A.V., Ivanishcheva, I.A., Ushakov, A.V., Sneha, M.J., Babbar, P., and Dixit, A., Models of lithium transport as applied to determination of diffusion characteristics of intercalation electrodes, Russ. J. Electrochem., 2017, vol. 53, p. 706.]
Иванищев, А.В., Чуриков, А.В., Акмаев, А.С., Ушаков, А.В., Иванищева, И.А., Гамаюнова, И.М., Снеха, М.Дж., Диксит, А. Синтез, строение и электрохимические свойства литий-аккумулирующего электродного материала на основе Li2FeSiO4. Электрохимия. 2017. Т. 53. С. 340. [Ivanishchev, A.V., Churikov, A.V., Akmaev, A.S., Ushakov, A.V., Ivanishcheva, I.A., Gamayunova, I.M., Sneha, M.J., and Dixit, A., The synthesis, structure, and electrochemical properties of Li2FeSiO4-based lithium-accumulating electrode material, Russ. J. Electrochem., 2017, vol. 53, p. 302.]
Ivanishchev, A.V., Churikov, A.V., and Ushakov, A.V., Lithium transport processes in electrodes on the basis of Li3V2(PO4)3 by constant current chronopotentiometry, cyclic voltammetry and pulse chronoamperometry, Electrochim. Acta, 2014, vol. 122, p. 187.
Ivanishchev, A.V., Churikov, A.V., Ivanishcheva, I.A., and Ushakov, A.V., Lithium diffusion in Li3V2(PO4)3-based electrodes: a joint analysis of electrochemical impedance, cyclic voltammetry, pulse chronoamperometry, and chronopotentiometry data, Ionics, 2016, vol. 22, p. 483.
Ivanishchev, A.V., Ushakov, A.V., Ivanishcheva, I.A., Churikov, A.V., Mironov, A.V., Fedotov, S.S., Khasanova, N.R., and Antipov, E.V., Structural and electrochemical study of fast Li diffusion in Li3V2(PO4)3-based electrode material, Electrochim. Acta, 2017, vol. 230, p. 479.
Ivanishchev, A.V., Bobrikov, I.A., Ivanishcheva, I.A., and Ivanshina, O.Y., Study of structural and electrochemical characteristics of LiNi0.33Mn0.33Co0.33O2 electrode at lithium content variation, J. Electroanal. Chem., 2018, vol. 821, p. 140.
Galus, Z., Teoretyczne podstawy elektroanalizy chemicznej: polarografia, chronowoltamperometria, chronopotencjometria, metoda wirującego dysku, Państwowe Wydawnictwo Naukowe, 1971. 390 p.
Weppner, W. and Huggins, R.A., Determination of the Kinetic Parameters of Mixed-Conducting Electrodes and Application to the System Li3Sb, J. Electrochem. Soc., 1977, vol. 124, p. 1569.
Dathar, G.K.P., Sheppard, D., Stevenson, K.J., and Henkelman, G., Calculations of Li-Ion Diffusion in Olivine Phosphates, Chem. Mater., 2011, vol. 23, p. 4032.
Arora, P., Doyle, M., Gozdz, A.S., White, R.E., and Newman, J., Comparison between computer simulations and experimental data for high-rate discharges of plastic lithium-ion batteries, J. Power Sources, 2000, vol. 88, p. 219.
Дополнительные материалы отсутствуют.