

Электрохимия, 2020, T. 56, № 4, стр. 365-377
Электрокаталитическое окисление ароматических экополлютантов на композиционных анодных материалах
Т. А. Кенова a, *, Г. В. Корниенко a, b, В. Л. Корниенко a, **
a Федеральный исследовательский центр “Красноярский научный центр СО РАН”,
Институт химии и химической технологии СО РАН
660036 Красноярск, Академгородок, 50, стр. 24, Россия
b Сибирский государственный университет науки и техники имени акад. М.Ф. Решетнева
660049 Красноярск, просп. Мира, 82, Россия
* E-mail: kta@icct.ru
** E-mail: kvl@icct.ru
Поступила в редакцию 16.11.2018
После доработки 04.03.2019
Принята к публикации 31.07.2019
Аннотация
Исследован процесс электрокаталитического окисления ароматических поллютантов (анилин, метиловый оранжевый (МО), хромовый темно-синий (ХТС)) на оксидно-свинцовом (Pb/PbO2), допированном бором алмазном (ДБА) электродах и аноде на основе оксидов рутения и титана (ОРТА). С помощью циклической вольтамперометрии и гальваностатического электролиза изучены каталитические свойства исследуемых материалов. Показано, что активность электродов в процессе электрохимической конверсии органического субстрата увеличивается в ряду ОРТА < Pb/PbO2 < ДБА. Для всех электродов скорость окисления снижается в ряду ХТС > МО > анилин. Кинетика процесса окисления выбранных соединений описывается уравнением реакции псевдо-первого порядка. Кажущаяся константа скорости процесса увеличивается с ростом плотности тока и уменьшением исходной концентрации поллютанта. С использованием метода конкурирующих реакций подтверждено образование радикальных частиц •ОН и ${\text{SO}}_{4}^{{2\centerdot {\kern 1pt} - }}$ и оценен их вклад в процесс деструкции красителя ХТС.
ВВЕДЕНИЕ
Ароматические соединения, такие как анилин, синтетические красители находят широкое применение во многих отраслях промышленности, включая текстильную, бумажную, кожевенную индустрии, производство лекарственных и косметических средств, продуктов питания и т.д. Сточные воды, содержащие ароматические производные и/или продукты их деструкции, даже в незначительных количествах попадая в гидросферу, являются серьезной экологической проблемой, поскольку эти поллютанты большей частью относятся к трудно фото- и биоразлагаемым соединениям [1–3]. Обладая токсичными, канцерогенными и мутагенными свойствами, они оказывают пагубное влияние на природные экосистемы и здоровье человека.
Традиционные методы (биологические, физические, химические) водоподготовки зачастую оказываются неэффективными в случае очистки и утилизации сточных вод, содержащих ядовитые и трудно окисляемые загрязняющие вещества. В этой связи перспективными и наиболее востребованными являются технологии, в основе которых лежит использование сильных окислителей.
Инновационные электрохимические процессы (“electrochemical advanced oxidation processes” (EAOPs)) все чаще рассматриваются как альтернативные способы для обработки промышленных и муниципальных сточных вод, содержащих различные органические и неорганические поллютанты, очистки и дезинфекции питьевой воды, рекультивации загрязненных почв [4–10]. Растущий интерес фундаментальной и прикладной науки к таким процессам подтверждается значительным ростом количества публикаций в рецензируемых журналах, патентов и материалов международных конференций [3]. Следует, однако, отметить достаточно низкую публикационную активность по данной проблематике, наблюдаемую в российских научных изданиях.
Среди EAOPs электрокаталитическое окисление (ЭО) или анодное окисление (АО) является одним из наиболее перспективных методов для деградации токсичных, бионеразлагаемых органических субстратов [8, 11, 12].
Многочисленные исследования показали, что ЭО можно эффективно применять для минерализации таких устойчивых экополлютантов как ароматические (антрахинонные и азо красители, анилин, фенол, бензол и их производные) соединения, пестициды, гербициды, поверхностно-активные вещества, лекарственные препараты и фторорганические соединения [11–14].
В литературе рассматриваются два основных пути окисления органических субстратов: прямой электролиз, когда перенос электронов осуществляется непосредственно с поверхности анода на адсорбированные молекулы органики, и непрямое (медиаторное) окисление активными формами кислорода (•ОН, Н2О2/НО2•, О3), образующимися в качестве промежуточных частиц при разложении воды, в тонком слое, вблизи поверхности электрода. В отличие от прямого ЭО, в котором зачастую происходит необратимая адсорбция органики, сопровождающаяся полимеризацией исходного субстрата и/или продуктов окисления и приводящая к снижению электрокаталитической активности (“отравлению”) электрода, при непрямом окислении подобные проблемы не возникают [15].
При непрямом окислении прослеживаются два характерных поведения органических поллютантов в ходе их деградации – селективное окисление с образованием устойчивых конечных продуктов, и практически полная минерализация, т.е. их конверсия до СO2, H2O и неорганических ионов. Наиболее приемлемая теоретическая модель для объяснения такого поведения была предложена Comninellis [16], усовершенствованная впоследствии Marselli, et al. [17]. В соответствии с предложенной моделью, в зависимости от характера взаимодействия •ОН-радикалов, образующихся при окислении воды (1), с поверхностью анода, электродные материалы делят на два типа: так называемые “активные” электроды, такие как Pt, аноды на основе оксидов металлов MOx (TiO2, RuO2, IrO2), графит и другие sp2 углеродсодержащие электроды и “неактивные”: PbO2 и SnO2, MnO2, допированный бором алмазный электрод (ДБА) [17].
(1)
${\text{M}}{{{\text{O}}}_{x}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \to {\text{M}}{{{\text{O}}}_{x}}{{(}^{\centerdot }}{\text{OH}}) + {{{\text{H}}}^{ + }} + {\text{e}}.$В случае “неактивных” анодов, слабое взаимодействие гидроксил-радикалов с поверхностью электрода и высокое перенапряжение выделения кислорода способствует минерализации органических поллютантов:
(2)
$\begin{gathered} {\text{M}}{{{\text{O}}}_{x}}{{(}^{\centerdot }}{\text{OH}}) + {\text{R }} \to \\ \to {\text{M}}{{{\text{O}}}_{x}} + m{\text{C}}{{{\text{O}}}_{2}} + n{{{\text{H}}}_{{\text{2}}}}{\text{O}} + {{{\text{H}}}^{ + }} + {\text{e}}{\text{.}} \\ \end{gathered} $На “активных” электродах сильная адсорбция •ОН-радикалов (хемосорбция) может приводить к образованию высших оксидов MOx+1 (3), когда такое состояние возможно, при потенциалах выше термодинамического потенциала выделения кислорода (Е°Н2О/О2 = 1.23 В отн. НВЭ):
(3)
${\text{M}}{{{\text{O}}}_{x}}{{(}^{\centerdot }}{\text{OH}}) \to {\text{M}}{{{\text{O}}}_{x}}_{{ + 1}} + {{{\text{H}}}^{ + }} + {\text{e}}.$Реакция окисления органических молекул редокс-парой MOx + 1/MOx (4) является значительно более селективной по сравнению с реакцией, включающей •ОН-радикалы (2), и конкурирует с побочной реакцией выделения О2 (5), (6), особенно при высоких плотностях тока [18]:
(4)
${\text{M}}{{{\text{O}}}_{x}}_{{ + 1}} + {\text{R}} \to {\text{RO}} + {\text{M}}{{{\text{O}}}_{x}},$(5)
${\text{M}}{{{\text{O}}}_{x}}_{{ + 1}} \to {\text{M}}{{{\text{O}}}_{x}} + {1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}{{{\text{O}}}_{2}},$(6)
${\text{M}}{{{\text{O}}}_{x}}{{(}^{\centerdot }}{\text{OH}}) \to {\text{M}}{{{\text{O}}}_{x}} + {1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}{{{\text{O}}}_{2}} + {{{\text{H}}}^{ + }} + {\text{e}}{\text{.}}$Кроме того, в зависимости от состава фонового электролита, возможно образование стабильных окислителей, таких как пероксидикарбонаты (${{{\text{C}}}_{2}}{\text{O}}_{6}^{{2 - }}$), пероксидифосфаты (${{{\text{P}}}_{2}}{\text{O}}_{8}^{{4 - }}$), пероксидисульфаты (${{{\text{S}}}_{2}}{\text{O}}_{8}^{{2 - }}$), активный хлор (Cl2) и кислородные соединения хлора (ClO–, ${\text{ClO}}_{2}^{ - },$ ${\text{ClO}}_{3}^{ - }$) которые могут диффундировать в объем электролита, увеличивая общую скорость окисления.
Так, например, в [19] показано, что электролиз сульфатных растворов на допированном бором алмазном электроде сопровождается образованием пероксидисульфат-аниона, концентрация которого зависит от плотности тока
(7)
${{{\text{Н}}}_{2}}{\text{О}} \to {{\,}^{\centerdot }}{\text{ОН}} + {{{\text{Н}}}^{ + }} + {\text{e}},$(8)
${\text{НSO}}_{4}^{ - } + {{\,}^{\centerdot }}{\text{ОН}} \to {\text{SO}}_{4}^{{ - {\kern 1pt} \centerdot }} + {{{\text{Н}}}_{{\text{2}}}}{\text{О}},$(9)
${\text{SO}}_{4}^{{ - {\kern 1pt} \centerdot }} + {\text{SO}}_{4}^{{ - {\kern 1pt} \centerdot }} \to {{{\text{S}}}_{2}}{\text{O}}_{8}^{{2 - }}.$Электрохимическому окислению ароматических аминов и красителей посвящено достаточно много работ [5, 7, 10]. Интерес к таким исследованиям связан со сложным механизмом протекания процесса и возможным образованием ряда интермедиатов, обладающих бóльшим сопротивлением к дальнейшей деструкции, чем исходное вещество, влиянием значительного числа параметров на кинетику и механизм электролиза. Сравнительные результаты, полученные для различных типов анодов, показали, что применимость того или иного электрокатализатора существенно зависит от структуры окисляемого субстрата, хотя условия электролиза также оказывают немаловажное влияние на кинетику и селективность процесса.
В данной работе исследовано электрокаталитическое окисление ароматических экополлютантов (анилин, азокрасители: хромовый темно-синий (ХТС) и метиловый оранжевый (МО)) на композиционных анодных материалах (Pb/PbO2, ДБА и аноде на основе оксидов рутения и титана (ОРТА)). Изучено влияние природы материала анода, плотности тока электролиза и исходной концентрации субстрата на кинетику процесса окисления. С помощью метода конкурирующих реакций оценен вклад •ОН- и ${\text{SO}}_{4}^{{ - {\kern 1pt} \centerdot }}$-радикалов в общий процесс окисления.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Модельные растворы анилина (C6H7N), ХТС [3-(5-хлор-2-гидроксифенил)азо-4,5-дигидроксинафтален-2,7-дисульфоновой кислоты динатриевая соль, С16H9ClN2Na2O4S2] и МО [4-(4-диметиламинофенилазо)-бензолсульфонат натрия, C14H14N3O3SNa] готовили добавлением фонового электролита к аликвотной части раствора субстрата известной концентрации. Исходное содержание поллютантов в исследуемых растворах составляло от 50 до 200 мг л–1.
Эксперименты проводили в трехэлектродной однокамерной ячейке, содержащей 150 мл раствора, с перемешиванием на магнитной мешалке при температуре 25–30°С. В качестве фонового электролита использовали 0.5 М Na2SO4 с добавлением 1 М H2SO4 до рН 2. Электролиз проводили в гальваностатическом режиме с использованием потенциостата ПИ 50–1.1 (ЗИП, г. Гомель, Россия) при плотностях тока 25–125 мА см–2. ОРТА (Ti/Ru0.3Ti0.7O2), Pb/PbO2 или ДБАЭ с геометрической площадью поверхности 4 см2 использовали как рабочий электрод (анод), вспомогательным электродом (катодом) служил графитовый стержень. Электродный потенциал анода измеряли относительно насыщенного хлоридсеребряного электрода сравнения (ХСЭ). Перед каждым экспериментом электроды активировали для стабилизации электродной поверхности и получения воспроизводимых результатов по методикам, использованным в [20].
Остаточную концентрацию красителя определяли фотометрическим методом на спектрофотометре LEKI SS1103 (LEKI Instruments) [21]. В ряде экспериментов растворы после электролиза дополнительно анализировали методом УФ-спектроскопии на приборе Cary 100 Scan фирмы Varian Inc. с целью подтверждения результатов измерения оптической плотности (D), идентификации и определения структуры образующихся промежуточных продуктов реакции.
Циклические (ЦВА) и потенциодинамические вольт-амперные характеристики исследуемых электродов получали с помощью потенциостата ПИ 50–1.1 (ЗИП, г. Гомель, Россия) в растворе фонового электролита 0.5 М Na2SO4 (рН 2) без добавления и с добавлением 200 мг л–1 модельного поллютанта при скорости развертки потенциала 20, 50 и 100 мВ с–1. Область сканирования потенциала находилась в интервале 0–2.0 В для электродов ОРТА и Pb/PbO2 и 0–3.8 В – на ДБАЭ.
Для метода конкурирующих реакций в качестве “ловушки” радикалов использовали спирты: этанол (EtOH) (0.6 M, С2Н5ОН) и трет-бутиловый спирт (ТБА) (0.5 M, (СН3)3СОН). Все химические реагенты были марки “ч. д. а.”, серная кислота – марки “х. ч.” и использовались без дальнейшей очистки.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
При выборе анодного материала для процессов деструктивного окисления органических соединений важно оценить его электрокаталитическую активность как по отношению к самому субстрату, так и в реакции выделения кислорода. Электроокисление органики без пассивации электрода может быть проведено в области потенциалов разряда воды с участием активных форм кислорода (АФК), при этом, однако, снижается токовая эффективность за счет конкурирующего процесса выделения кислорода. Поэтому, чем выше перенапряжение этой реакции, тем эффективнее процесс деструкции органического субстрата [12].
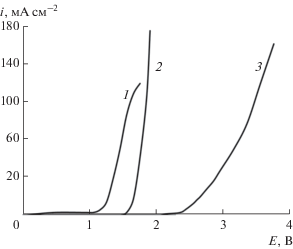
На рис. 1 представлены поляризационные кривые ОРТА-, Pb/PbO2- и ДБА-электродов, полученные в растворе фонового электролита при скорости развертки потенциала 50 мВ с–1. Анализ полученных i,Е-зависимостей показывает, что электроды довольно существенно различаются по своей электрокаталитической активности в реакции выделения кислорода. Потенциал начала выделения кислорода увеличивается в ряду: ОРТА < Pb/PbO2$ \ll $ ДБА и составляет 1.25, 1.65 и 2.4 В (относительно ХСЭ), соответственно. Высокое перенапряжение выделения молекулярного кислорода в случае Pb/PbO2- и ДБА-электродов создает благоприятные условия для окисления органики непосредственно на аноде и/или с участием АФК.
Рис. 1.
Потенциодинамические поляризационные кривые ОРТА (1), Pb/PbO2 (2) и ДБА (3) в фоновом электролите 0.5 М Na2SO4 (рН 2). Скорость развертки: 50 мВ с–1.
Для получения большей информации о механизме (прямое или непрямое окисление) протекания процессов деструкции органических субстратов были получены ЦВА-характеристики исследуемых электродов. На рис. 2 представлены циклические вольтамперограммы для фонового электролита и электролита, содержащего модельный органический субстрат (200 мг л–1), на Pb/PbO2, ОРТА и ДБА при скорости развертки потенциала 50 мВ с–1.
Рис. 2.
а – Циклические вольтамперограммы Pb/PbO2-электрода в фоновом электролите (1) и при добавлении 200 мг л–1 ХТС (2), МО (3) и анилина (4). Скорость развертки: 50 мВ с‒1. На врезке – участки анодной ветви ЦВА-кривых (увеличено). б – Циклические вольтамперограммы ОРТА в фоновом электролите (1, 1 ') и при добавлении 200 мг л–1 ХТС (2) и анилина (3, 3 '). Скорость развертки: 50 мВ с–1. На врезке – участки анодной ветви ЦВА-кривых (увеличено). в – Циклические вольтамперограммы ДБА-электрода в фоновом электролите (1) и при добавлении 200 мг л–1 ХТС (2) и анилина (3). Скорость развертки: 50 мВ с–1. На врезке – участки анодной ветви ЦВА-кривых (увеличено).
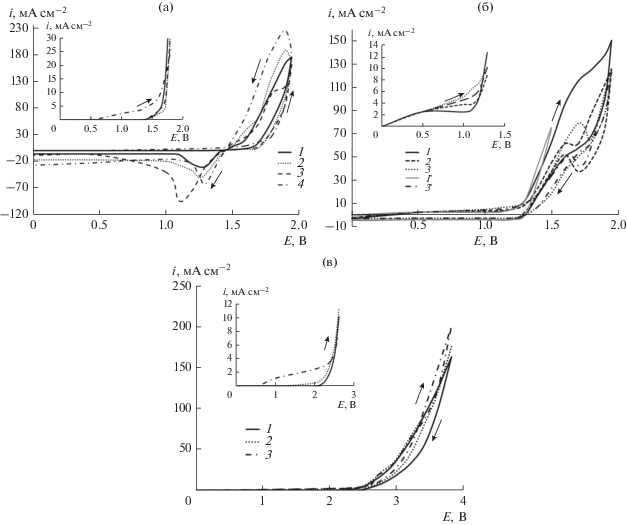
Вид фоновой ЦВА-кривой (рис. 2а, кривая 1) соответствует процессам, происходящим на поверхности (Pb/PbO2)-электрода в растворе электролита, содержащего серную кислоту [22, 23]. В ходе анодного сканирования потенциала наблюдается только пик выделения кислорода. Отсутствие пика в области потенциалов отрицательнее потенциала выделения кислорода, характеризующего образование β-PbO2 связано, по-видимому, с низкой активностью PbSO4. Вследствие этого анодный пик образования диоксида свинца смещен в сторону положительных значений и перекрывается с анодным пиком выделения кислорода [23]. Появление катодного пика при Е = 1.4–1.3 В указывает на обратный процесс восстановления β-PbO2 до PbSO4 [22–24]. При добавлении в электролит МО или ХТС никаких новых пиков на ЦВА-кривой не наблюдается. Однако в присутствии анилина отмечается рост плотности тока в области потенциалов стабильности воды, что свидетельствует о его прямом окислении на аноде (врезка, рис. 2а) [25]. Для всех исследуемых субстратов, тем не менее, наблюдается незначительное смещение потенциала выделения кислорода в анодную область. При сканировании потенциала в катодном направлении на анодной ветви кривой наблюдается увеличение плотности тока в присутствии анилина. В то же время для МО увеличения плотности тока на обратном ходе кривой не отмечалось, а для ХТС оно было незначительным. В присутствии органики на ЦВА-кривых наблюдается также рост плотности тока пика, относящегося к восстановлению диоксида свинца. При этом для красителей ХТС и МО наблюдается сдвиг потенциала пика в катодную область до 1.2 и 1.1 В соответственно.
Такое поведение, на наш взгляд, связано с тем, что в присутствии органики может изменяться морфология и размер кристаллов осадков PbSO4 и β-PbO2. Подобное явление наблюдали авторы в [26], исследовав влияние никотинамида на поведение PbO2-электрода в процессе заряда/разряда в сернокислых растворах. Смещение потенциала выделения кислорода в анодную область указывает на то, что часть активных центров на поверхности электрода блокирована адсорбированными молекулами субстрата и/или продуктами его окисления, т.е. происходит ингибирование процесса выделения О2 [27]. Другими словами, процесс прямого окисление органики конкурирует с процессом разряда электролита (образования АФК). В то же время наблюдаемый рост плотности тока при обратном ходе кривой, по-видимому, связан с увеличением эффективной площади поверхности электрода, в том числе и за счет образования PbO2. Поскольку PbO2 является катализатором реакции выделения кислорода, увеличение его количества приводит к росту числа адсорбированных молекул воды с последующим их разрядом и образованием •ОН/О2 [23]. С другой стороны, в случае анилина, образование полимерной пленки также может приводить к изменению скоростей этих реакций [12].
При исследовании электрохимической активности ОРТА было обнаружено интересное явление. Вольтамперограмма, полученная в растворе фонового электролита при сканировании потенциала от 0 до 1.5–1.6 В (рис. 2б, кривая 1 '), представляет типичное поведение для термально приготовленного оксидного слоя TiO2RuO2 с широким неявно выраженным пиком в области потенциалов стабильности воды. Форма пика отражает высокую неоднородность поверхности и отвечает редокс-процессу перехода низших оксидов рутения в высшие оксиды. Редокс-реакции на поверхности RuO2 связаны с переносом протона из раствора и переносом электрона и могут быть описаны следующим уравнением [28]:
(10)
$\begin{gathered} {\text{RuOx(OH}})y + \delta {{{\text{H}}}^{ + }} + \delta {{{\text{e}}}^{{}}} \leftrightarrow \\ \leftrightarrow {\text{RuOx}} - \delta ({\text{OH}})y + {\text{ }}\delta . \\ \end{gathered} $Однако сканирование потенциала в интервале 0–1.95 В приводит к изменению формы кривой фона (кривая 1) – изменение наклона кривой при Е > 1.8 В на прямом и Е < 1.8 В на обратном ходе ЦВА-зависимости. Вероятной причиной замедления процесса выделения молекулярного кислорода может быть изменение механизма (изменение лимитирующей стадии процесса разряда воды) его образования [28].
В ходе первого сканирования на ЦВА-кривых в растворах, содержащих анилин (кривая 3) и ХТС (кривая 2), в области потенциалов стабильности электролита наблюдается широкий пик, соответствующий прямому электроокислению на аноде, и уменьшение плотности тока при потенциалах выделения О2. Другим важным моментом является появление хорошо выраженного пика тока при Е = 1.6 В для ХТС в обоих направлениях сканирования потенциала и в прямом направлении – для анилина при Е = 1.7 В, и сдвиг потенциала выделения кислорода в сторону положительных значений. При последующем цикле снижается высота пика прямого электроокисления (см. врезка, кривая 3′ для анилина) с одновременным снижением пиков в области потенциалов разряда воды. Такое поведение, по-видимому, связано с дезактивацией электродной поверхности продуктами деструкции субстрата с образованием полимерных органических пленок, окисление которых при более высоких анодных потенциалах конкурирует с реакцией выделения кислорода [12]. Подобное поведение ранее наблюдали для электродов на основе смешанных оксидов металлов в присутствии ароматических соединений, таких как фенол [29], нафтол [30], метиловый красный [31]. Зависимость тока пика прямого окисления субстрата от скорости развертки потенциала (20–100 мВ/с), построенная в координатах Ia–v1/2, дает прямую линию, проходящую через начало координат, что указывает на диффузионно-контролируемый процесс. Следует отметить, что подобная зависимость сохраняется и для пиков, наблюдаемых в области потенциалов разложения электролита.
Циклические вольтамперограммы, записанные для ДБА-электрода в растворах, содержащих анилин или ХТС, представлены на рис. 2в в области потенциалов 0–3.8 В при скорости развертки 50 мВ с–1. Как видно из рисунка (см. врезку), в области потенциалов стабильности электролита наблюдается волна тока окисления анилина, что указывает на существование прямого электрохимического процесса на поверхности анода. Этот процесс конкурирует с реакцией выделения кислорода в области потенциалов разряда воды. Дальнейший анодный сдвиг потенциала приводит к росту плотности тока, что может свидетельствовать о дополнительном механизме окисления, включающем поверхностные реакции между адсорбированными/неадсорбированными (ионными или радикальными) частицами и $^{\centerdot }{\text{OH}}$-радикалами. В случае красителей незначительный отклик тока при потенциалах стабильности электролита может указывать на возможность существования прямого механизма окисления ХТС (аналогично и МО). Наряду с этим, сдвиг вольт-амперной кривой в область более низких значений потенциалов свидетельствует также об участии в процессе окисления красителя гидроксильных радикалов. Следует отметить, что увеличение количества циклов не приводит к изменению вольт-амперной кривой для ХТС, тогда как в случае анилина наблюдается незначительное снижение плотности тока. Такое поведение может означать, что ХТС практически не адсорбируется на поверхности ДБА в отличие от анилина, а промежуточные продукты подвергаются быстрой деструкции, без образования полимерной пленки, пассивирующей электрод. Продукты осмоления, образующиеся при деструкции анилина, легко разрушаются анодной поляризацией при потенциалах разряда воды. Увеличение скорости развертки потенциала в обоих случаях приводит к росту плотности тока, что свидетельствует о диффузионном характере процесса.
Таким образом, окисление выбранных ароматических соединений (анилин, МО, ХТС) может протекать как непосредственно на поверхности анода, так и с участием АФК, в зависимости от природы материала электрода и величины прикладываемого потенциала. Проведение процесса в области потенциалов разложения водного электролита позволяет избежать проблемы “отравления” электрода, однако при этом может существенно снижаться фарадеевская эффективность процесса [12, 15].
Скорость и селективность процесса деструкции, как известно, в значительной степени зависят от материала анода, его способности генерировать высокореакционные $^{\centerdot }{\text{OH}}$-радикалы [8, 12, 16]. Для оценки влияния параметров электролиза и материала анода на кинетику процесса окисления исследуемых органических субстратов были проведен ряд экспериментов в гальваностатическом режиме электролиза. На рис. 3 представлены кинетические кривые окисления 150 мг л–1 метилового оранжевого при плотности тока 50 мА см–2 в 0.5 М Na2SO4 (pH 2) на различных анодах. Полученные зависимости удовлетворительно описываются кинетическим уравнением скорости реакции псевдо-первого порядка (11) [31]:
(11)
$r = - {\text{d}}{{\left[ {{{c}_{{{\text{орг}}}}}} \right]} \mathord{\left/ {\vphantom {{\left[ {{{c}_{{{\text{орг}}}}}} \right]} {{\text{d}}\tau }}} \right. \kern-0em} {{\text{d}}\tau }} = k{{\left[ {^{\centerdot }{\text{OH}}} \right]}^{\alpha }}\left[ {{{c}_{{{\text{орг}}}}}} \right] = {{k}_{{{\text{каж}}}}}\left[ {{{c}_{{{\text{орг}}}}}} \right],$(12)
$\ln {{{{{\left[ {{{c}_{{{\text{орг}}}}}} \right]}}_{0}}} \mathord{\left/ {\vphantom {{{{{\left[ {{{c}_{{{\text{орг}}}}}} \right]}}_{0}}} {{{{\left[ {{{c}_{{{\text{орг}}}}}} \right]}}_{\tau }} = {{k}_{{{\text{каж}}}}}\tau }}} \right. \kern-0em} {{{{\left[ {{{c}_{{{\text{орг}}}}}} \right]}}_{\tau }} = {{k}_{{{\text{каж}}}}}\tau }},$Рис. 3.
Зависимость остаточной концентрации МО от времени и соответствующий кинетический анализ (врезка) для реакции псевдо-первого порядка на электродах: 1 – ОРТА; 2 – Pb/PbO2; 3 – ДБА. Исходная концентрация – 150 мг л–1, плотность тока – 50 мА см–2, рН 2.
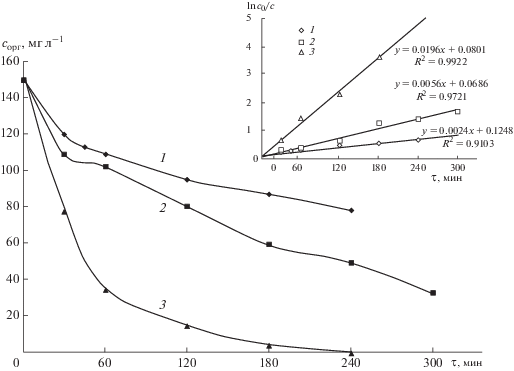
Рассчитанная в соответствии с уравнением (12) kкаж составила 2.4 × 10–3, 5.6 × 10–3 и 1.96 × 10–2 мин–1 для ОРТА, Pb/PbO2 и ДБА соответственно.
Анализ кинетических данных окисления МО на различных электродах показал, что, как и предполагалось, наиболее эффективным анодным материалом является ДБА. Можно видеть, что кажущаяся константа скорости для ДБА-электрода почти на порядок выше, чем при использовании ОРТА. Высокая окислительная способность ДБА обусловлена очень слабой адсорбцией гидроксил-радикалов на поверхности электрода, что благоприятствует их быстрой реакции с органическим субстратом. Вероятной причиной снижения скорости процесса окисления МО на электроде из диоксида свинца, как предполагает ряд авторов [10], может быть более сильная адсорбция $^{\centerdot }{\text{OH}}$ с гидратированным оксидным слоем (PbO(OH)2). Медленная деструкция красителя на ОРТА согласуется с поведением “активных” анодов, адсорбция •OH-радикалов на поверхности которых стабилизируется образованием высших оксидов МОx, что способствует выделению кислорода [15, 18].
С другой стороны, известно, что природа органического поллютанта, особенно наличие различных функциональных групп (азогруппа, карбоксильные, сульфогруппы), может существенно влиять на механизм окисления и эффективность электрохимического процесса [7, 10, 25].
На рис. 4 представлены зависимости относительной концентрации анилина, МО и ХТС от времени электролиза при плотности тока 50 мА см–2 в кислом электролите (рН 2) на ДБА-электроде.
Рис. 4.
Зависимость относительной концентрации анилина (1), МО (2) и ХТС (3) от времени окисления и соответствующий кинетический анализ (врезка) для реакции псевдо-первого порядка при i = 50 мА см–2 и рН 2 на ДБА-электроде. Кинетическая кривая окисления анилина (1') при плотности тока 100 мА см–2.
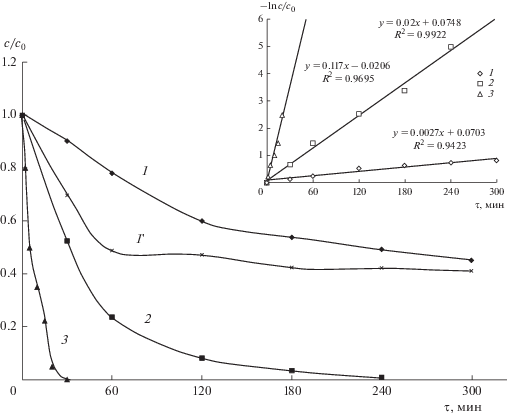
Как видно из полученных кинетических кривых, наблюдается существенное различие в скорости окисления органического субстрата, которая увеличивается в ряду: анилин < МО < ХТС. Определенные из наклона графиков ln[cорг]0/[cорг]τ–τ кажущиеся константы скорости окисления составили, соответственно, 2.7 × 10–3, 2.0 × 10–2 и 11.7 × × 10–2 мин–1. Достаточно низкая скорость окисления анилина по сравнению с азокрасителями, скорее всего, обусловлена его способностью к полимеризации в ходе прямого окисления на поверхности анода. Косвенным доказательством образования полимерной пленки может, по нашему мнению, служить тот факт, что при увеличении плотности тока (рис. 4, кривая 1 ') наблюдалось быстрое снижение остаточной концентрации анилина в течение первого часа электролиза, после чего она практически не менялась до конца эксперимента (5 ч). Следует отметить, что снижение концентрации МО и ХТС до нуля не означает, однако, их полной минерализации, а свидетельствует о быстрой реакции азокрасителя с ДБА(•OH), протекающей с разрывом хромофорной связи (–N=N–) в его молекуле и приводящей к образованию бесцветных или слабо окрашенных продуктов окисления. Разрыв хромофорной связи является первой стадией процесса деструкции азокрасителя (деколоризации раствора), после чего образующиеся интермедиаты подвергаются дальнейшему последовательному окислению до карбоксильных кислот [11]. Более низкая скорость окисления МО связана, по-видимому, с наличием в структуре красителя двух метильных групп (–СН3), окисление которых идет параллельно с разрушением азо-связи.
Одними из основных параметров, влияющих на эффективность процесса, являются плотность тока/потенциал и концентрация поллютанта. На рис. 5 представлена зависимость кажущейся константы скорости процесса окисления ХТС на ДБА при рН 2 от плотности тока и исходной концентрации красителя. Значения kкаж (R2 = 0.9573–0.9989) определены из полулогарифмической зависимости, в соответствии с уравнением реакции псевдо-первого порядка (12).
Рис. 5.
Зависимость kкаж от плотности тока и исходной концентрации ХТС при электролизе в 0.5 М Na2SO4 (рН 2).
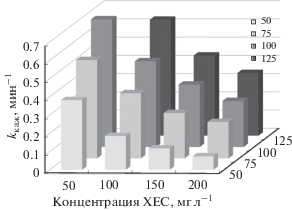
Как видно из диаграммы, наблюдается линейная зависимость kкаж–i, причем увеличение плотности тока в 2.5 раза приводит к более чем 3-кратному увеличению константы. Так, например, для исходной концентрации 100 мг л–1kкаж выросла в 3.2 раза (с 1.98 × 10–1 до 6.36 × 10–1 мин–1) при увеличении i от 50 до 125 мА см–2. Такая тенденция связана с увеличением скорости генерации $^{\centerdot }{\text{OH}}$ при повышении плотности тока, что способствует увеличению скорости разрыва хромофорной связи в молекуле ХТС и, соответственно, скорости его обесцвечивания [11, 15, 16]. Кроме того, при высоких плотностях тока в сульфатном растворе возможно образование других окислителей, таких как пероксидисульфат-анион (${{{\text{S}}}_{2}}{\text{O}}_{8}^{{2 - }}$) и/или сульфат-радикал (${\text{SO}}_{4}^{{ - \centerdot }}$), озон (О3), пероксид водорода (Н2О2), пергидроксил-радикал (HO2•) [19, 32].
С увеличением исходной концентрации ХТС наблюдается снижение кажущейся константы скорости. При повышении содержания красителя с 50 до 200 мг л−1 и i = 50 мА см–2kкаж уменьшилась более чем в пять раз и составила, соответственно, 3.82 × 10–1 и 7.36 × 10–2 мин–1, что указывает на недостаточное количество гидроксил-радикалов при данном токе. Однако, когда плотность тока возрастает, к примеру, до 100 мА см–2, наблюдается только 3-кратное уменьшение kкаж при увеличении концентрации ХТС. Фактически это означает, что процесс деградации красителя не соответствует псевдо-первому порядку, поскольку в этом случае величина константы не должна зависеть от начальной концентрации вещества. Такое поведение свидетельствует о сложности процесса окисления, в котором параллельно протекают реакции взаимодействия исходных молекул ХТС и промежуточных продуктов его деструкции с различными активными окисляющими частицами [8, 11].
Как показано многочисленными исследованиями [19, 33, 34], при высоких анодных потенциалах в сернокислых электролитах наблюдается образование ${\text{SO}}_{4}^{{ - \centerdot }}$-радикалов, которые наряду с $^{\centerdot }{\text{OH}}$-радикалами, принимают участие в окислении органического субстрата. В ряде работ [35‒37] образование ${\text{SO}}_{4}^{{ - \centerdot }}$ и $^{\centerdot }{\text{OH}}$ подтверждено спектроскопическим методом электронного парамагнитного резонанса (ЭПР). Также для идентификации ${\text{SO}}_{4}^{{ - \centerdot }}$/$^{\centerdot }{\text{OH}}$ достаточно широко применяется метод конкурирующих реакций (метод “ловушки” радикалов) как в системах с активированным персульфатом [36], так и в электрохимических системах [13, 34, 38].
Для того чтобы проверить образование радикальных частиц на исследуемых электродах и оценить их вклад в трансформацию ХТС, в данной работе использовали метод конкурирующих реакций. В качестве индикаторов ${{{\text{SO}}_{4}^{{ - \centerdot }}} \mathord{\left/ {\vphantom {{{\text{SO}}_{4}^{{ - \centerdot }}} {^{\centerdot }{\text{OH}}}}} \right. \kern-0em} {^{\centerdot }{\text{OH}}}}$ были выбраны этанол с α-водородом, который действует как поглотитель и ${\text{SO}}_{4}^{{ - \centerdot }}$, и $^{\centerdot }{\text{OH}}$, и трет-бутиловый спирт, являющийся эффективной “ловушкой” для $^{\centerdot }{\text{OH}}$ [35, 36]. В соответствии с [39], константа скорости реакции второго порядка для EtOH и ТБА с $^{\centerdot }{\text{OH}}$ составляет (1.2–2.8) × 109 и (3.8–7.6) × 108 М с–1 соответственно. Однако константа скорости между этанолом и ${\text{SO}}_{4}^{{ - \centerdot }}$ ((1.6– 7.7) × × 107 М с–1) в 18–193 раза выше, чем между ТБА и ${\text{SO}}_{4}^{{ - \centerdot }}$ ((4.0–9.1) × 105 М с–1). Таким образом, добавляя в избытке EtOH и ТБА и учитывая их различную реакционную способность по отношению к радикальным частицам, можно определить доминирующую роль $^{\centerdot }{\text{OH}}$ или ${\text{SO}}_{4}^{{ - \centerdot }}$ по изменениям скорости деградации поллютанта.
На рис. 6 представлены кинетические зависимости окисления 150 мг л–1 ХТС при плотности тока 50 мА см–2 в присутствии 0.6 М EtOH и 0.5 М ТБА на ДБА, Pb/PbO2 и ОРТА. Из рис. 6а видно, что 96% ХТС окисляется при этих условиях за 30 мин электролиза на ДБА-электроде, тогда как только 22.1 и 11.3% красителя деградирует в присутствии EtOH и ТБА соответственно. Существенное снижение скорости окисления ХТС в присутствии спиртов подтверждает, что $^{\centerdot }{\text{OH}}$- и ${\text{SO}}_{4}^{{ - \centerdot }}$-радикалы являются главными окисляющими частицами [34, 38]. Более сильное ингибирование процесса в присутствии ТБА, являющегося селективной “ловушкой” для $^{\centerdot }{\text{OH}}$, свидетельствует о преобладающей роли гидроксильных радикалов. Необходимо также отметить, что даже в присутствии ТБА, концентрация которого превышает более чем в 1700 раз концентрацию ХТС, около 15% красителя все еще окисляется, по-видимому, либо прямым окислением на аноде, либо с участием других АФК (${\text{O}}_{2}^{{ - {\kern 1pt} \centerdot }},{\text{HO}}_{2}^{\centerdot },$ О3 и др.).
Рис. 6.
Влияние EtOH и ТБА на скорость деградации ХТС на ДБА (а), Pb/PbO2 (б) и ОРТА (в): 1 – без добавления спирта; 2 – при добавлении 0.6 М EtOH; 3 – при добавлении 0.5 М ТБА.
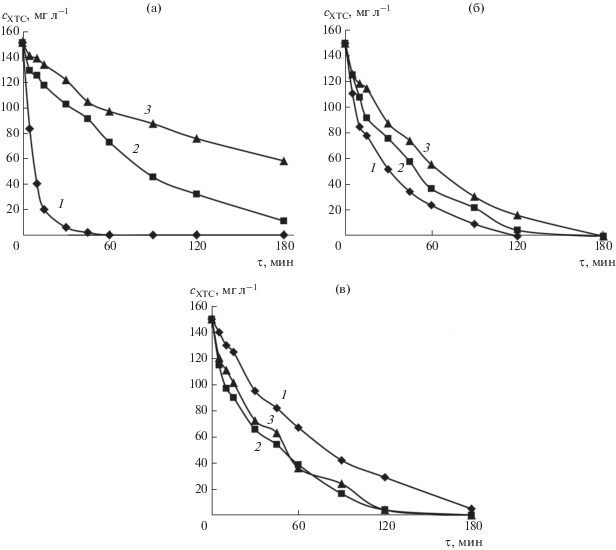
На аноде из диоксида свинца полная деструкция красителя (исчезновение окраски) наблюдалась через 120 мин электролиза. При этом эффективность окисления ХТС снижалась до 94.1% в присутствии EtOH и до 87.4% при добавлении ТБА. Полученные результаты указывают на образование $^{\centerdot }{\text{OH}}$- и ${\text{SO}}_{4}^{{ - \centerdot }}$-радикалов на Pb/PbO2-аноде, однако их вклад в процесс окисления ХТС незначительный. С другой стороны, считается, что окисление органики на Pb/PbO2, как и в случае ДБА, протекает с участием $^{\centerdot }{\text{OH}}$-радикалов [12, 15]. Возможной причиной различия в поведении двух анодов следует, по-видимому, считать различный характер взаимодействия $^{\centerdot }{\text{OH}}$-радикалов с поверхностью электрода: предполагается, что на поверхности ДБА радикалы более лабильны и, следовательно, более реакционноспособны по отношению к органике [10, 15, 31]. Кроме того, различная адсорбционная способность этих электродов может наблюдаться и в отношении органического субстрата [16–18]. Следует отметить, что и этанол, и ТБА являются гидрофильными “ловушками” радикалов, которые эффективно гасят их в гомогенной среде [40]. Поэтому возможно более гидрофобный краситель легче адсорбируется на поверхности Pb/PbO2, чем гидрофильные спирты, и взаимодействует с $^{\centerdot }{\text{OH}}$, следовательно, меньший ингибирующий эффект наблюдается. Другой причиной может быть изменение поверхностного слоя анода в ходе электролиза, поскольку наряду с разложением электролита, также может протекать реакция окисления свинцовой подложки (образование оксидного слоя PbO2).
Неожиданные и неоднозначные результаты были получены при использовании ОРТА. Как видно из рис. 6в, добавление и EtOH, и ТБА не только не тормозит процесс окисления, но приводит к росту скорости деструкции ХТС. Такой ход зависимости может служить подтверждением того, что на электродах типа ОРТА, которые относятся к “активным” анодам, не образуются (или практически не образуются) относительно свободные радикальные частицы ($^{\centerdot }{\text{OH}}$ и ${\text{SO}}_{4}^{{ - \centerdot }}$). Согласно [16, 17], адсорбция $^{\centerdot }{\text{OH}}$-радикалов на поверхности “активных” анодов сопровождается их внедрением в кристаллическую решетку оксида, что приводит к образованию высших оксидов (хемосорбированный “активный кислород”). По-видимому, окисление спиртов с участием “активного кислорода” протекает по иному механизму, отличному от механизма их взаимодействия со свободными радикальными частицами и может сопровождаться образованием алкоксильных радикалов [35]. В этом случае скорость деградации красителя, вероятно, может быть увеличена за счет его восстановительной реакции с органическими радикалами. Так в работе Zhu и соавторов [35] было изучено образование алкоксильных радикалов из соответствующих спиртов (EtOH и ТБА), используемых в качестве “ловушки” $^{\centerdot }{\text{OH}}$- и ${\text{SO}}_{4}^{{ - \centerdot }}$-радикалов в процессе активации пероксидисульфата и сделана оценка их вклада в деградацию органических загрязнителей. Полученные авторами результаты свидетельствуют о существенном влиянии алкоксильных радикалов не только на деградацию органики, но и на процесс разложения ${{{\text{S}}}_{2}}{\text{O}}_{8}^{{2 - }}$. Отметим в этой связи, что на идеально поляризуемых электродах, например, на Hg, при взаимодействии простейших алифатических спиртов (MeOH, EtOH) с $^{\centerdot }{\text{OH}}$-радикалами в основном образуются α-гидроксиалкильные радикалы:
(13)
${\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OH }} + {{\,}^{\centerdot }}{\text{OH }} \to {\text{ C}}{{{\text{H}}}_{3}}^{\centerdot }{\text{CHOH}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}},~$На основе данных УФ-спектроскопии было установлено, что процесс деструкции азокрасителей протекает с образованием ряда промежуточных продуктов. В спектрах растворов после электролиза в области более коротких длин волн наблюдается появление дополнительных полос поглощения, обусловленное образованием низкомолекулярных фрагментов окисления исходного субстрата и образовавшихся интермедиатов. В спектрах МО и ХТС были идентифицированы производные бензола, фенолсодержащие и хиноидные структуры. Анализ спектров анилина показал наличие интенсивного поглощения в области длин волн, относящихся к п-бензохинону и нитробензолу, а также, связанных c образованием отдельных звеньев при формировании полимерного продукта (смолистых веществ). Последним органическим продуктом деструкции являются малотоксичные карбоновые кислоты, которые подвергаются дальнейшему окислению до СО2 и Н2О.
ЗАКЛЮЧЕНИЕ
Исследовано электрокаталитическое окисление ароматических экополлютантов (анилин, метиловый оранжевый и хромовый темно-синий) на композиционных электродах (Pb/PbO2, ДБА и ОРТА).
С помощью циклической вольтамперометрии показано, что в зависимости от природы материала анода и прикладываемого потенциала электрохимическая конверсия органического субстрата может протекать как по прямому, так и непрямому механизмам. При этом окисление в области потенциалов стабильности электролита приводит к пассивации анодов в результате образования полиароматических продуктов деструкции.
Показано, что активность электродов в процессе электрохимической конверсии органического субстрата увеличивается в ряду ОРТА < < Pb/PbO2 < ДБА, при этом скорость деструкции поллютантов снижается в ряду ХТС > МО > анилин. Кинетические зависимости окисления азокрасителей удовлетворительно описываются уравнением скорости реакции псевдо-первого порядка. Рассчитанная кажущаяся константа скорости увеличивается с увеличением плотности тока и снижением исходной концентрации субстрата. Существенный рост скорости окисления при использовании ДБА-электрода может быть связан с генерацией дополнительных окисляющих частиц, таких как сульфат-радикал, гидропероксид-радикал, озон. Кинетика окисления анилина соответствует реакции псевдо-первого порядка только в первые 1–2 ч электролиза. Увеличение времени и тока окисления приводит к формированию пленки полимерных продуктов.
На примере красителя ХТС с использованием метода конкурирующих реакций подтверждено образование $^{\centerdot }{\text{OH}}$- и ${\text{SO}}_{2}^{{ - {\kern 1pt} \centerdot }}$-радикалов. Увеличение скорости окисления ХТС в присутствии спиртов (EtOH и ТБА) при использовании ОРТА может свидетельствовать о дополнительном вкладе алкоксильных радикалов в процесс деструкции красителя.
Список литературы
O’Neill, C., Hawkes, F.R., Hawkes, D.L., Lourenço, N.D., Pinheiro, H.M., and Delée, W., Colour in textile effluents–sources, measurement, discharge consents and simulation: a review, J. Chem. Technol. Biotechnol., 1999, vol. 74, p. 1009.
Dos Santos, A.B., Cervantes, F.J., and Van Lier, J.B., Review paper on current technologies for decolourisation of textile wastewaters: Perspectives for anaerobic biotechnology, Bioresour. Technol., 2007, vol. 98, p. 2369.
Sirés, I., Brillas, E., Oturan, M.A., Rodrigo, M.A., and Panizza, M., Electrochemical advanced oxidation processes: today and tomorrow. A review, Environ. Sci. Pollut. Res., 2014, vol. 21, p. 8336.
Харламова, Т.А., Алиев, З.М. Применение электролиза под давлением для деструктивного окисления фенола и азокрасителей. Электрохимия. 2016. Т. 52. С. 291. [Kharlamova, T.A. and Aliev, Z.M., Use of electrolysis under pressure for destructive oxidation of phenol and azo dyes, Russ. J. Electrochem., 2016, vol. 52, p. 251.]
Корниенко, В.Л., Чаенко, Н.В., Корниенко, Г.В. Непрямое электрохимическое деструктивное окисление ароматических соединений активными формами кислорода. Электрохимия. 2007. Т. 43. С. 4. [Kornienko, V.L., Chaenko, N.V., and Kornienko, G.V., Indirect electrochemical destructive oxidation of aromatic compounds with reactive oxygen species, Russ. J. Electrochem., 2007, vol. 43, p. 1243.]
Anglada, Á., Urtiaga, A., Ortiz, I., Mantzavinos, D., and Diamadopoulos, E., Boron-doped diamond anodic treatment of landfill leachate: evaluation of operating variables and formation of oxidation by-products, Water Res., 2011, vol. 45, p. 828.
Pacheco, M.J., Santos, V., Ciríaco, L., and Lopes, A., Electrochemical degradation of aromatic amines on BDD electrodes, J. Hazard. Mater., 2011, vol. 186, p. 1033.
Chaplin, B.P., Critical review of electrochemical advanced oxidation processes for water treatment applications, Environ. Sci.: Processes Impacts, 2014, vol. 16, p. 1182.
Forgacs, E., Cserháti, T., and Oros, G., Removal of synthetic dyes from wastewaters: a review, Environ. Int., 2004, vol. 30, p. 953.
Rodrigues, A.S., Nunes, M.J., Lopes, A., Silva, J.N., Ciríaco, L., and Pacheco, M.J., Electrodegradation of naphthalenic amines: Influence of the relative position of the substituent groups, anode material and electrolyte on the degradation products and kinetics, Chemosphere, 2018, vol. 205, p. 433.
Martınez-Huitle, C.A. and Brillas, E., Decontamination of wastewaters containing synthetic organic dyes by electrochemical methods: A general review, Appl. Catal. B: Environ., 2009, vol. 87, p. 105.
Panizza, M. and Cerisola, G., Direct and mediated anodic oxidation of organic pollutants, Chem. Rev., 2009, vol. 109, p. 6541.
Bu, L., Zhu, S., and Zhou, S., Degradation of atrazine by electrochemically activated persulfate using BDD anode: Role of radicals and influencing factors, Chemosphere, 2018, vol. 195, p. 236.
Oturan, M.A., An ecologically effective water treatment technique using electrochemically generate hydroxyl radicals for in situ destruction of organic pollutants: Application to herbicide 2,4-D, J. Appl. Electrochem., 2000, vol. 30, p. 475.
Martınez-Huitle, C.A. and Ferro, S., Electrochemical oxidation of organic pollutants for the wastewater treatment: direct and indirect processes, Chem. Soc. Rev. 2006, vol. 35, p. 1324.
Comninellis, C., Electrocatalysis in the electrochemical conversion/combustion of organic pollutants for wastewater treatment, Electrochim. Acta, 1994, vol. 39, p. 1857.
Marselli, B., Garcia-Gomez, J., Michaud, P.A., Rodrigo, M.A., and Comninellis, C., Electrogeneration of hydroxyl radicals on boron-doped diamond electrodes, J. Electrochem. Soc., 2003, vol. 150, p. 79.
Scialdone, O., Electrochemical oxidation of organic pollutants in water at metal oxide electrodes: a simple theoretical model including direct and indirect oxidation processes at the anodic surface, Electrochim. Acta, 2009, vol. 54, p. 6140.
Cañizares, P., Sáez, C., Sánchez-Carretero, A., and Rodrigo, M.A., Synthesis of novel oxidants by electrochemical technology, J. Appl. Electrochem., 2009, vol. 39, p. 2143.
Корниенко, Г.В., Чаенко, Н.В., Корниенко, В.Л. Непрямое электрохимическое окисление N-метил-n-аминофенола активными формами кислорода in situ генерированными из O2, H2O, and H2O2. Журн. прикл. Химии. 2008. Т. 81. С. 1285. [Kornienko, G.V., Chaenko, N.V., and Kornienko, V.L., Indirect electrochemical oxidation of N-methyl-n-aminophenol by active oxygen species generated in situ from O2, H2O, and H2O2, Russ. J. Appl. Chem, 2008, vol. 81, p. 1364.]
Фишер, Х. Практикум по общей химии: Вводный курс по экологически безопасной программе с экспериментами по регенерации химических реактивов. Ч. 2: Органическая и физическая химия. Новосибирск: Наука, 2002. 326 с. [Fisher, H., Praktikum in allgemeiner Chemie: Ein umweltschonendes Programm für Studienanfänger mit Versuchen zur Chemikalien-Rückgewinnung. Teil 2. Organische und Physikalische Chemie, Zürich, 1995.]
Abd El Aal, E.E., Cyclic voltammetric behavior of the lead electrode in sodium sulfate solutions, J. Power Source, 2001, vol. 102, p. 233.
Zhang, B., Zhong, J., Li, W., Dai, Z., Zhang, B., and Cheng, Z., Transformation of inert PbSO4 deposit on the negative electrode of a lead-acid battery into its active state, J. Power Source, 2010, vol. 195, p. 4338.
He, Z., Hayat, M.D., Huang, S., Wang, X., and Cao, P., PbO2 electrodes prepared by pulse reverse electrodeposition and their application in benzoic acid degradation, J. Electroanal. Chem., 2018, vol. 812, p. 74.
Santos, V., Diogo, J., Pacheco, M.J.A., Ciríaco, L., Morão, A., and Lopes, A., Electrochemical degradation of sulfonated amines on Si/BDD electrodes, Chemosphere, 2010, vol. 79, p. 637.
Sato, Y., Hishimoto, K., Togashi, K., Yanagawa, H., and Kobayakawa, K., The effect of nicotinamide on the charge/discharge behavior of PbO2 electrode in sulfuric acid solution, J. Power Source, 1992, vol. 39, p. 43.
Li, X., Xu, H., Yan, W., and Shao, D., Electrocatalytic degradation of aniline by Ti/Sb–SnO2, Ti/Sb–SnO2/Pb3O4 and Ti/Sb–SnO2/PbO2 anodes in different electrolytes, J. Electroanal. Chem., 2016, vol. 775, p. 43.
Over, H., Surface chemistry of ruthenium dioxide in heterogeneous catalysis and electrocatalysis: from fundamental to applied research, Chem. Rev., 2012, vol. 112, p. 3356.
Feng, Y.J. and Li, X.Y., Electro-catalytic oxidation of phenol on several metal-oxide electrodes in aqueous solution, Water Res., 2003, vol. 37, p. 2399.
Panizza, M. and Cerisola, G., Electrochemical oxidation of 2-naphthol with in situ electrogenerated active chlorine, Electrochim. Acta, 2003, vol. 48, p. 1515.
Panizza, M. and Cerisola, G., Electrochemical degradation of methyl red using BDD and PbO2 anodes, Ind. Eng. Chem. Res., 2008, vol. 47, p. 6816.
Hamza, M., Abdelhedi, R., Brillas, E., and Sirés, I., Comparative electrochemical degradation of the triphenylmethane dye methyl violet with boron-doped diamond and Pt anodes, J. Electroanal. Chem., 2009, vol. 627, p. 41.
Khamis, E., Mahé, D., Dardoize, F., and Devilliers, D., Peroxodisulfate generation on boron-doped diamond microelectrodes array and detection by scanning electrochemical microscopy, J. Appl. Electrochem., 2010, vol. 40, p. 1829.
Luo, H., Li, C., Sun, X., and Ding, B.B., Cathodic indirect oxidation of organic pollutant paired to anodic persulfate production, J. Electroanal. Chem., 2017, vol. 792, p. 110.
Zhu, C., Zhu, F., Dionysiou, D.D., Zhou, D., Fang, G., and Gao, J., Contribution of alcohol radicals to contaminant degradation in quenching studies of persulfate activation process, Water Res., 2018, vol. 139, p. 66.
Liang, C. and Su, H.-W., Identification of sulfate and hydroxyl radicals in thermally activated persulfate, Ind. Eng. Chem. Res., 2009, vol. 48, p. 5558.
Chen, L., Lei, C., Li, Z., Yang, B., Zhang, X., and Lei, L., Electrochemical activation of sulfate by BDD anode in basic medium for efficient removal of organic pollutants, Chemosphere, 2018, vol. 210, p. 516.
Bu, L., Zhou, S., Shi, Z., Deng, L., and Gao, N., Removal of 2-MIB and geosmin by electrogenerated persulfate: Performance, mechanism and pathways, Chemosphere, 2017, vol. 168, p. 1309.
Buxton, G.V., Greenstock, C.L., Helman, W.P., and Ross, A.B., Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (•OH/•O‒) in aqueous Solution, J. Phys. Chem. Ref. Data, 1988, vol.17, p.513.
Song, H., Yan, L., Jiang, J., Ma, J., Zhang, Z., Zhang, J., Liu, P., and Yang, T., Electrochemical activation of persulfates at BDD anode: Radical or nonradical oxidation?, Water Res., 2018, vol. 128, p. 393.
Бендерский, В.А., Кривенко, А.Г., Курмаз, В.А. Электродные реакции метанольного радикала на ртути. Докл. АН СССР. 1984. Т. 278. С. 896. [Benderskii, V.A., Krivenko, A.G., and Kurmaz, V.A., Electrode-reactions of a methanol radical on mercury (In Russian), Doklady Akademii Nauk SSSR, 1984, vol. 278, p. 896.]
Бендерский, В.А., Кривенко, А.Г., Курмаз, В.А. Электродные реакции метанольного и этанольного радикалов на ртути. Электрохимия. 1986. Т. 22. С. 644. [Benderskii, V.A., Krivenko, A.G., and Kurmaz, V.A., Electrode-reactions of methanol and ethanol radicals at mercury, Sov. Electrochem., 1986, vol. 22, p. 603.]
Rotenberg, Z.A. and Rufman, N.M., Photocurrents in nitrous-oxide solutions containing aliphatic-alcohols, J. Electroanal. Chem., 1984, vol. 175, p. 153.
Дополнительные материалы отсутствуют.