

Электрохимия, 2020, T. 56, № 4, стр. 356-364
Редокс-свойства и реакционная способность органических трисульфидов в реакциях с алкенами
Д. А. Бурмистрова a, *, И. В. Смолянинов a, b, **, Н. Т. Берберова a
a ФГБОУ ВО “Астраханский государственный технический университет”
414056 Астрахань, ул. Татищева, 16, Россия
b ФГБОУ Федеральный исследовательский центр Южный научный центр РАН
344006 Ростов-на-Дону, ул. Чехова, 41, Россия
* E-mail: burmistrova.da@gmail.com
** E-mail: ivsmolyaninov@gmail.com
Поступила в редакцию 22.01.2019
После доработки 19.03.2019
Принята к публикации 15.07.2019
Аннотация
Исследованы редокс-реакции органических трисульфидов, содержащих различные углеводородные группы, с алкенами в апротонных растворителях. Электроокисление трисульфидов протекает необратимо по механизму ЕСЕ с образованием сероцентрированных интермедиатов RS+ и RSS+. Генерированные катионы вступают в реакции электрофильного присоединения с алкенами с образованием несимметричных ди- и моносульфидов. Электрохимическое восстановление трисульфидов приводит к образованию анион-радикала, который фрагментируется на RSS-анион и RS-радикал. В присутствии уксусной кислоты катодная активация трисульфидов сопровождается образованием алкил-, фенилгидродисульфидов (RSSH).
ВВЕДЕНИЕ
В настоящее время значительное внимание уделяется синтезу и свойствам биологически активных полисульфидов [1–4]. Особый интерес вызывают органические трисульфиды, проявляющие противоопухолевую активность [5], антиоксидантное действие на живые организмы [6], а также являющиеся источниками эндогенного сероводорода [7]. Биологическая активность сераорганических соединений, содержащих полисульфидные фрагменты, обусловлена наличием нескольких редокс-состояний атомов серы, что делает привлекательным использование электрохимических методов при изучении свойств подобного рода соединений [8].
Существуют различные способы формирования связи C–S, которые представляют собой актуальную область исследований в органическом синтезе [9, 10]. Электрохимические подходы к формированию новых C–C-, C–S-, C–N-, C–O-связей в последние несколько лет являются особо востребованными [11]. Большой синтетический потенциал электрохимических методов связан с их эффективностью, атомной экономичностью [12], высокой селективностью активации той или иной функциональной группы (фрагмента), отсутствием опасных, токсичных реагентов и металлсодержащих катализаторов [13, 14]. Электросинтез соединений серы также относится к одному из активно развивающихся направлений органической химии [15, 16]. Наиболее изученными являются редокс-превращения ди- и моносульфидов [17, 18]. Исследовано влияние заместителей на величину потенциалов окисления и условий проведения электрохимических реакций для тиоэфиров различного строения [19, 20]. Электроокисление симметричных дисульфидов в смеси с тиолом или другим дисульфидом приводит к образованию несимметричного дисульфида [21]. В условиях электрохимической активации (CH3)2S2 происходит тиолирование антрацена и фенантрена [22]. Анодная активация арилдисульфидов способствует генерированию катионов ArS(ArSSAr)+, где Ar – aryl, участвующих в реакциях с алкенами, что ведет к получению продуктов присоединения [23]. Реакция электрохимического кросс-сочетания арил- и алкантиолов приводит к образованию несимметричных дисульфидов [24]. Во многих случаях состав продуктов электрохимического окисления дисульфидов зависит от природы заместителей, растворителя и условий синтеза [17].
Существует несколько основных подходов к получению трисульфидов: 1) взаимодействие тиолов с дихлоридом серы [25–27], 2) реакции галогеналканов с тио-производным 1,3,4-оксодиазола [28], 3) использование фосфорорганических тиопроизводных при взаимодействии с нуклеофильными агентами [29, 30]. Известен также электрохимический метод синтеза трисульфидов, основанный на взаимодействии электрогенерированных катионов серы с тиолами в дихлорметане [31]. Нашей группой разработаны как прямые методы электросинтеза R2Sn (n = 2–4) [32, 33], так и косвенные – с участием различных медиаторных редокс-систем и электрокатализаторов [34, 35].
Если электрохимические превращения ди- и моносульфидов являются хорошо изученной областью [17], то редокс-свойствам и реакционной способности трисульфидов уделено значительно меньше внимания. Актуальность применения электрохимической активации трисульфидов, рассматриваемых в настоящей работе, связана с возможностью генерирования короткоживущих интермедиатов, участвующих в различных реакциях. Подобного рода частицы могут потенциально формироваться в биологических системах в результате редокс-превращений полисульфидов. На основании полученных данных можно предложить механизм фрагментации интермедиатов и оценить дальнейшие пути их взаимодействия с различными субстратами. Процесс анодной активации трисульфидов в реакциях с алкенами изучен мало, хотя представляет практическую значимость для синтеза ценных сераорганических соединений.
Целью работы является изучение электрохимических свойств органических трисульфидов и реакционной способности их окисленных форм по отношению к алкенам. Данный подход позволяет обеспечить формирование C–S-связи, а также прямое введение S–S-фрагмента в молекулу углеводорода путем экологически чистой электрохимической активации R2S3 без использования металлсодержащих катализаторов при комнатной температуре.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали коммерчески доступные реактивы – дифенилдисульфид (99%, “Sigma-Aldrich”), дициклогексилдисульфид (95%, “Sigma-Aldrich”), дибутилдисульфид (97%, “Sigma-Aldrich”), циклогексен (99%, “Sigma-Aldrich”), циклопентен (97%, “Sigma-Aldrich”), гептен-1 (>96%, “Sigma-Aldrich”), уксусная кислота (99.5%, “AppliChem”), тиофенол (99%, “Sigma-Aldrich”), гидроксид тетрабутиламмония (99%, “Sigma-Aldrich”). Симметричные трисульфиды R2S3 получали по ранее опубликованной методике [36], несимметричный – метилпропилтрисульфид (50%, “Sigma-Aldrich”) использовали без дополнительной очистки. Очистку ацетонитрила и хлористого метилена (“х. ч.”) проводили известным способом [37].
Измерение редокс-потенциалов соединений (с = 5 мМ) проводили методом циклической вольтамперометрии (ЦВА) в трехэлектродной ячейке с помощью потенциостата “VersaSTAT 3” (США) в среде аргона. Рабочие электроды – стационарный платиновый электрод (Pt) диаметром 3 мм и стационарный стеклоуглеродный (СУ) электрод диаметром 2 мм, вспомогательный электрод – платиновая пластина (S = 32 мм2). Электрод сравнения (Ag/AgCl/KCl) с водонепроницаемой диафрагмой. Скорость развертки потенциала 0.2 В с–1. Фоновый электролит – 0.15 М n-Bu4NClO4 (99%, “Acros”) дважды перекристаллизованный из водного EtOH и высушенный в вакууме (48 ч) при 50°С. Пики окисления (восстановления) исследуемых трисульфидов имеют диффузионную природу, что определяется линейной зависимостью величины тока пика Iпa от v1/2 в диапазоне развертки потенциалов от 0.05 до 1.00 В с–1.
Микроэлектролиз трисульфидов (2 ч) проводили на потенциостате “VersaSTAT 3” при 25°С в бездиафрагменной трехэлектродной ячейке (V = = 2 мл) на Pt-электродах (S = 32 мм2) в потенциостатическом режиме (Е = 1.85 В). В качестве электрода сравнения использовали (Ag/AgCl/KCl) электрод с электропроводной водонепроницаемой диафрагмой. В предварительно деаэрированную электрохимическую ячейку, содержащую раствор фонового электролита (0.15 М n-Bu4NClO4) в ацетонитриле, добавляли трисульфид (c = 5 мМ). Микроэлектролиз трисульфида в присутствии алкена проводили в аналогичных условиях. Мольное соотношение трисульфид : алкен составило 1 : 15. Количество электричества в проведенных микроэлектролизах варьировалось от 2 до 4 Ф/моль.
Препаративный электролиз (4 ч) проводили на потенциостате ПИ-50-1.1 (Россия) в бездиафрагменной ячейке (40 мл) на платиновых электродах (S = 210 мм2) в CH2Cl2 в потенциостатическом режиме (1.85 В). Мольное соотношение трисульфид : : алкен составило 1 : 30. После окончания электролиза реакционную смесь концентрировали в вакууме. Фоновый электролит осаждали гексаном. Органические производные серы выделяли трехступенчатой экстракцией гексаном. Экстракт концентрировали в вакууме.
Для идентификации сераорганических соединений использовали методы хромато-масс-спектрометрии и рентгенофлуоресцентного анализа. Содержание элементной серы определяли на рентгенофлуоресцентном анализаторе АСЭ-1. Анализ смеси продуктов электролиза методом хромато-масс-спектрометрии проводили на приборе “Shimadzu GCMS-QP2010 Ultra” с масс-спектрометрическим детектором (метод ионизации – электронный удар, 70 эВ). Капиллярная колонка SPB-SULFUR (30 м × 0.32 мм), tmax = = 320°С. Газ-носитель – гелий. Температурный режим колонки программировали от 30 до 280°С. Результаты масс-спектрометрического анализа представлены в табл. 1.
Таблица 1.
Данные масс-спектрометрического анализа продуктов электролиза трисульфидов в присутствии алкенов
| Соединение | m/z (Iотн, %) |
|---|---|
| C6H11SC6H5 | 192 [M]+ (4), 190 (54), 177 (3), 147 (5), 109 (8), 82 (7), 81 (57) |
| C5H9SSC6H5 | 212 [M]+ (43), 210 (3), 142 (69), 109 (32), 77 (18), 69 (12) |
| (C6H11)2S2 | 230 [M]+ (14), 148 (38), 83 (100), 55 (40) |
| C6H11SC5H9 | 182[M]+ (4), 115 (13), 101 (8), 83 (73), 67 (100) |
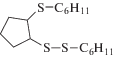 |
329 [M]+ (4), 247 (10), 132 (9), 113 (5), 83 (34), 67 (100) |
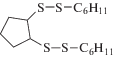 |
362 [M]+ (3), 309 (7), 279 (5), 198 (6), 164 (10), 115 (16), 100 (13), 83 (100), 67 (36) |
| C6H11SC7H15 | 213 [M]+ (6), 199 (5), 129 (10), 96 (58), 82 (100) |
| (C3H7)2S2 | 150 [M]+ (26), 108 (23), 74 (8), 66 (11), 47 (12) |
| CH3SSC3H7 | 122 [M]+ (84), 93 (9), 80 (100), 64 (24), 47 (32) |
| C5H9SSCH3 | 148[M]+ (5), 150 (17), 102 (8), 67 (100), 47 (6) |
| C5H9SSC3H7 | 176 [M]+ (5), 178 (10), 130 (12), 107 (10), 99 (12), 67 (100), 47 (9) |
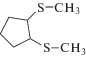 |
162 [M]+ (4), 160 (42), 145 (11), 131 (23), 99 (32), 67 (100), 47 (14) |
| CH3SC7H15 | 146 [M]+ (3), 148 (8), 131 (83), 100 (12), 68 (18), 47 (28) |
| C3H7SSC7H15 | 206 [M]+ (9), 174 (42), 131 (58), 107 (25), 99 (46), 47 (35) |
Квантово-химические расчеты проводили в программе Hyper Chem 8.0.8 методом функционала плотности (DFT) (базис: B3LYP/6-31++G(d,p)). Влияние растворителя (CH3CN) учитывали с помощью модели поляризуемого континиума (PCM).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
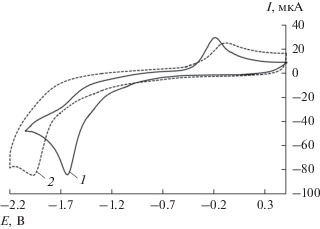
Исследованы редокс-превращения серии органических трисульфидов, содержащих ароматические, ациклические и алифатические углеводородные группы: (C6H5)2S3 (1), (C6H11)2S3 (2), (C4H9)2S3 (3) и (CH3)S3(C3H7) (4). Вольтамперограммы окисления трисульфидов 1–4, зарегистрированные в среде CH3СN−n-Bu4NClO4 (0.15 М), имеют одинаковую морфологию (рис. 1). Электроокисление трисульфидов представляет собой необратимый процесс в диапазоне скорости развертки потенциала до 1.0 В с–1. Ток пика Iпа соответствует переносу двух электронов в расчете на молекулу относительно стандарта – ферроцена и пропорционально возрастает с увеличением концентрации вещества в интервале 2.5 × 10–4–5 × 10–3 М. Анодный пик имеет диффузионную природу, что определяется линейной зависимостью величины тока пика Iпa от v1/2. Методом ЦВА определены значения потенциалов окисления исследуемых соединений, которые на платиновом аноде фиксируются в диапазоне от 1.68 до 1.80 В, а на СУ-аноде – от 1.57 до 1.75 В (табл. 2). Независимо от природы рабочего электрода (C6H11)2S3 окисляется при потенциале, смещенном в катодную область по сравнению с (C6H5)2S3 и (C4H9)2S3. Таким образом, не наблюдается корреляция между значениями Eпa и природой углеводородных групп в исследуемых трисульфидах.
Рис. 1.
ЦВА-кривая окисления (C6H5)2S3 (CH3CN, 0.15 М n-Bu4NClO4, c = 2.5 мМ, v = 0.2 В с–1, Pt-анод, Ag/AgCl/КСl).
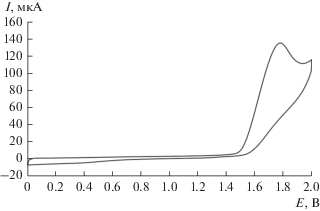
Таблица 2.
Редокс-потенциалы три- и дисульфидов по данным метода ЦВА (Pt- и СУ-электроды, CH3CN, 0.15 М n-Bu4NClO4, c = 3 мМ, v = 0.2 В с–1, Ar, отн. Ag/AgCl/KCl (нас.))
| № | Соединение | Pt-электрод | CУ-электрод | ||||
|---|---|---|---|---|---|---|---|
| Eпа, В | Iпа, мкА | Eпа, В | Iпа, мкА | Епк, В | Iпк, мкА | ||
| 1 | (С6Н5)2S3 | 1.77 | 165 | 1.64 | 176 | –1.62 | –83 |
| 2 | (C6H11)2S3 | 1.68 | 160 | 1.55 | 153 | –1.96 | –71 |
| 3 | (C4H9)2S3 | 1.80 | 178 | 1.75 | 180 | –1.96 | –85 |
| 4 | (CH3)S3(C3H7) | 1.77 | 253 | 1.57 | 191 | –1.94 | –90 |
| 5 | (С6Н5)2S2 | 1.67 | 200 | 1.64 | 175 | –1.62 | –82 |
| 6 | (C6H11)2S2 | 1.53 | 193 | 1.50 | 170 | –1.66 | –73 |
| 7 | (C4H9)2S2 | 1.47 | 217 | 1.41 | 198 | –1.65 | –93 |
Известно, что механизм электроокисления дисульфидов симметричного строения предполагает первоначальное образование катион-радикала с последующей его димеризацией в димерный дикатион [38]. Для электроокисления трисульфидов 1–3 можно предположить механизм ЕСЕ, учитывающий генерирование неустойчивого катион-радикала на первой стадии, диссоциация которого способствует образованию соответствующих катиона и радикала. Последующее окисление радикала до катиона приводит к суммарному двухэлектронному процессу окисления по схеме 1 :
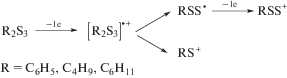
Схема 1 .
Подобное поведение R2S3 в условиях анодной активации позволяет рассматривать симметричные трисульфиды 1–3 в качестве источника двух видов электрофильных частиц (RS+ и RSS+). Электроокисление несимметричного трисульфида 4 в аналогичных условиях приводит к смеси электрофильных частиц (CH3S+, ${{{\text{C}}}_{3}}{{{\text{H}}}_{7}}{\text{S}}_{2}^{ + },$ ${\text{C}}{{{\text{H}}}_{3}}{\text{S}}_{2}^{ + },$ С3H7S+). На вольтамперограммах окисления продуктов микроэлектролиза трисульфидов (Е = 1.85 В, τ = 2 ч) фиксируются пики, характерные для ди- и тетрасульфидов: R2S2 (1.45–1.67 В) и R2S4 (1.96–2.05 В). Подобные превращения трисульфидов наблюдали ранее в процессе их электросинтеза [32].
Следует отметить, что метод ЦВА эффективен для разделения и идентификации ди-, три- и тетрасульфидов, поскольку с увеличением числа атомов серы в молекуле полисульфида значение потенциала пика окисления смещается в область положительных значений [39]. Сравнительный анализ значений потенциалов пиков окисления исследуемых трисульфидов и ряда соответствующих дисульфидов показал, что потенциалы пиков окисления трисульфидов 1–3 смещены в анодную область по сравнению с дисульфидами 5–7. Разность потенциалов ΔЕпа пиков окисления R2S3 и R2S2 для (C4H9)2S2 составила 0.35 В, (С6Н5)2S2 − 0.10 В, (C6H11)2S2 − 0.15 В, что согласуется с полученными ранее данными.
Известны примеры электрохимического восстановления серы, тиолов и дисульфидов [31], однако данных по катодной активации R2S3 недостаточно. Для трисульфидов 1–4 в катодной области на Pt-электроде не обнаружена какая-либо электрохимическая активность. Замена платинового электрода на стеклоуглеродный позволила зафиксировать на ЦВА-кривых выраженные пики восстановления для всех исследуемых соединений. Подобное поведение R2S3, возможно, связано с меньшей адсорбцией сераорганических соединений на поверхности СУ-электрода по сравнению с Pt-катодом. Ток пика Iпк соответствует переносу одного электрона в расчете на молекулу относительно стандарта – ферроцена. Электровосстановление трисульфидов представляет собой необратимый процесс в диапазоне скорости развертки потенциала до 1.0 В с–1. Пики восстановления имеют диффузионную природу, что определяется линейной зависимостью величины тока пика Iпк от v1/2. Величины потенциалов восстановления Eпк для (C6H11)2S3 и (C4H9)2S3 равны и составляют –1.96 В, для (CH3)S3(C3H7) значение Епк = –1.94 В. Для (С6Н5)2S3 потенциал восстановления смещен на 0.34 В в область положительных значений, что связано с электроноакцепторным характером фенильных групп (табл. 2, рис. 2).
Рис. 2.
ЦВА-кривая восстановления трисульфидов: 1 – (С6Н5)2S3, 2 – (C4H9)2S3 (CH3CN, 0.15 М n‑Bu4NClO4, c = 3 мМ, v = 0.2 В с–1, СУ-анод, Ag/AgCl/КСl).
Одноэлектронное восстановление трисульфидов приводит к образованию нестабильного анион-радикала, способного фрагментироваться по двум возможным направлениям, ведущим к генерированию: а) RS-радикала и RSS-аниона; б) пертиильного радикала (${\text{RS}}_{2}^{\centerdot }$) и RS-аниона (схема 2 , а, б). На обратной ветви ЦВА-кривой восстановления трисульфидов 1–4 фиксируется пик окисления $E_{{{\text{па}}}}^{'}$ в диапазоне значений ‒0.22…–0.11 В, отвечающий продукту распада анион-радикала. Помимо генерирования на катоде сероцентрированных анионов одновременно происходит процесс образования радикальных частиц (RS• или ${\text{RS}}_{2}^{\centerdot }$), способных к димеризации до R2S2 и R2S4.
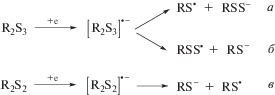
Схема 2 .
Для более полного понимания электрохимического поведения R2Sn (n = 2, 3) дополнительно были исследованы катодные превращения дисульфидов 5–7, потенциалы восстановления которых оказались смещены в область положительных значений относительно потенциалов восстановления трисульфидов 2–4 и находятся в диапазоне –1.66…–1.62 В (табл. 2). В отличие от установленной для процесса окисления зависимости величины потенциала от числа атомов серы в молекулах R2Sn (n = 2, 3), при восстановлении наблюдается обратная зависимость. При переходе от ди- к трисульфидам происходит смещение значения Епк в катодную область.
Электрохимическое восстановление (C6H5)2S2 приводит к фиксации на обратной ветви ЦВА-кривой пика окисления генерированного C6H5S-аниона при Eпа = –0.09 В (схема 2 , в). Аналогичный пик при Eпа = –0.1 В наблюдается в модельной реакции тиофенола с n-Bu4NOH (1 экв.), что подтверждает образование тиофенолят-аниона. Различие значений потенциалов окисления анионов, образующихся в ходе восстановления R2S2 и R2S3, позволяет сделать вывод, что анион-радикал [R2S3]•– расщепляется преимущественно с образованием RSS-аниона (схема 2 , а). Квантовохимические расчеты тепловых эффектов реакций фрагментации анион-радикала трисульфида, проведенные на примере (C6H5)2S3, также подтверждают распад по направлению 2а с формированием соответствующего аниона (ΔH = –9.42 кДж/моль). Фрагментация по направлению 2б термодинамически менее благоприятна (ΔH = 17.36 кДж/моль).
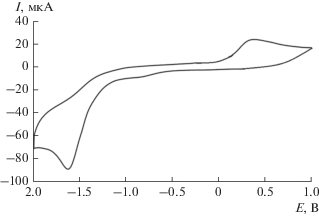
Электрохимическое восстановление R2S3 в присутствии уксусной кислоты (c = 0.01 М), используемой в качестве донора протона, приводит к исчезновению пиков реокисления RSS-анионов. При этом фиксируется новый анодный пик в диапазоне потенциалов от 0.26 до 0.32 В (рис. 3). Полученные значения потенциалов пиков смещены в катодную область по сравнению с данными для неорганических полисульфанов (0.4–1.5 В) [40]. Фиксируемые пики продуктов взаимодействия предположительно соответствуют образованию органических гидродисульфидов RSSH, биологическая ценность которых заключается в выполнении роли прекурсоров эндогенного сероводорода в живых организмах [41].
Рис. 3.
ЦВА-кривая восстановления (C6H5)2S3 в присутствии CH3COOH (CH3CN, 0.15 М n-Bu4NClO4, c = = 3 мМ, v = 0.2 В с–1, СУ-анод, Ag/AgCl/KCl).
Изучена реакционная способность электрогенерированных электрофильных частиц, образующихся при окислительной активации R2S3, в реакциях с алкенами. В результате проведения микроэлектролиза трисульфидов в присутствии избытка алкена получена смесь моно- и дисульфидов различного строения (схема 3 ).
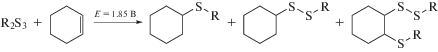
Схема 3 .
Известна реакция присоединения серосодержащих электрофильных частиц, образующихся при электроокислении диарилсульфида, к молекуле циклогексена, что позволяет получать наряду с моносульфидами, 1,2-бис-арил-тиопроизводные [35]. На ЦВА-кривой окисления продуктов микроэлектролиза трисульфидов с алкенами зарегистрированы пики окисления в диапазоне потенциалов от 1.45 до 1.90 В, отвечающие окислению смеси несимметричных ди- и моносульфидов. Однако ввиду образования близких по структуре серосодержащих соединений метод ЦВА не позволяет дифференцировать образующиеся продукты реакции. На основании этого идентификация серосодержащих продуктов, полученных в результате проведения препаративного электролиза исследуемых трисульфидов (1.85 В) с алкенами, проведена методом хромато-масс-спектрометрии (табл. 3).
Таблица 3.
Выходы и состав продуктов электролизов R2S3 в присутствии алкенов в условиях анодной активации R2S3 (CH2Cl2, 0.15 М n-Bu4NClO4, Pt-анод, Е = 1.85 В, Ag/AgCl)
| Трисульфид | Алкен | Выход продуктов реакции, % | |
|---|---|---|---|
| (C6H5)2S3 | C6H10 | C6H11SC6H5 | 6.6 |
| C5H8 | C5H9SSC6H5 | 7.8 | |
| (C6H11)2S3 | C5H8 | (C6H11)2S2 | 67.6 |
| C6H11SC5H9 | 12.4 | ||
| C6H11SSC5H9 | 4.5 | ||
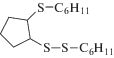 |
10.0 | ||
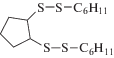 |
3.0 | ||
| C7H14 | Продукт полимеризации | 78.8 | |
| C6H11SC7H15 | 10.2 | ||
| (C6H11)2S2 | 8.0 | ||
| (CH3)S3(C3H7) | C5H8 | (C3H7)2S2 | 7.8 |
| CH3SSC3H7 | 3.8 | ||
| C5H9SSCH3 | 9.9 | ||
| C5H9SSC3H7 | 10.6 | ||
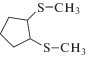 |
7.5 | ||
| C7H14 | (C3H7)2S2 | 23.5 | |
| CH3SSC3H7 | 19.2 | ||
| CH3SC7H15 | 5.1 | ||
| C3H7SSC7H15 | 3.3 | ||
Электролиз (C6H5)2S3 в присутствии избытка циклогексена (циклопентена) приводит к несимметричному моно- и дисульфиду: C6H11SC6H5 и C5H9SSC6H5 с выходом 6.6 и 7.8% соответственно. Невысокий выход продуктов присоединения по кратной связи обусловлен адсорбцией на поверхности рабочего электрода продуктов полимеризации.
Окислительная активация смеси (C6H11)2S3 с циклопентеном в аналогичных условиях способствует накоплению продуктов присоединения C6H11S+ и ${{{\text{C}}}_{6}}{{{\text{H}}}_{{11}}}{\text{S}}_{2}^{ + }$ к циклопентену. Выход несимметричного сульфида C6H11SC5H9 составил 12.4%, содержание дисульфида C6H11SSC5H9 не превышает 4.5%. Продукты присоединения двух видов сероцентрированных катионов по циклопентеновому кольцу представлены следующими соединениями: 1-циклогексил-2-(2-(циклогексилтио) циклопентил)дисульфаном (3%) и 1,2-бис(циклогексил-дисульфанил)циклопентаном (10%). Однако наибольший выход наблюдается для (C6H11)2S2 (67.6%), что обусловлено превращением R2S3 в R2S2 в условиях электролиза.
Образование C6H11SC7H15 (10.2%) происходит при анодной активации (C6H11)2S3 в присутствии гептена-1 в результате электрофильного присоединения катиона C6H11S+ к алкену. В то же время содержание (C6H11)2S2 не превышает 7–8%. Преобладающим продуктом электросинтеза является малорастворимый в CH2Cl2 серосодержащий продукт олигомеризации алкена (78.8%), инициируемой катионом C6H11SS+. Содержание серы в молекуле олигомера по данным рентгенофлуоресцентного анализа составляет 9.5%, что соответствует формуле (C6H11S2)2(С7Н14)n (n = 11).
Стоит отметить, что электролиз метилпропилтрисульфида в смеси с гептеном-1 не приводит к образованию полимерных продуктов. Основным направлением реакции является процесс превращения несимметричного (CH3)S3(C3H7) в дисульфиды: (С3Н7)2S2 (23.5%) и CH3SSC3H7 (19.2%). Также зафиксированы продукты присоединения катионов CH3S+ и C3H7SS+ к гептену-1: метилгептилсульфид (5.1%), пропилгептилдисульфид (3.3%). Электролиз (CH3)S3(C3H7) в присутствии избытка циклопентена способствует образованию смеси несимметричных дисульфидов: метилциклопентилдисульфида (9.9%), пропилциклопентилдисульфида (10.6%) и 1,2-бис(метилтио)циклопентана (7.5%). В данной реакции выход дисульфидов составил 7.8% (CH3SSC3H7) и 3.8% ((C3H7)2S2). Возможность восстановления RS+ до RS• на противоэлектроде в процессе препаративного электролиза в ячейке с неразделенным катодно-анодным пространством обуславливает возможность образования соответствующих дисульфидов.
ЗАКЛЮЧЕНИЕ
Таким образом, электрохимическое окисление органических трисульфидов ((С6H5)2S3, (C6H11)2S3, (C4H9)2S3, (CH3)S3(C3H7)) протекает необратимо по механизму ЕСЕ и приводит к генерированию катионов RS+ и RSS+. Электрохимическое восстановление R2S3 является одноэлектронным процессом, в результате которого преимущественно генерируются RS-радикал и RSS-анион, который в присутствии уксусной кислоты образует органические гидродисульфиды RSSH. Наличие в реакционной среде алкенов способствует протеканию реакции электрофильного присоединения с получением несимметричных моносульфидов, а также обеспечивает прямое введение дисульфидного мостика в молекулу углеводорода без участия металлсодержащих катализаторов при комнатной температуре.
Список литературы
Vo, C.D., Kilcher, G., and Tirelli, N., Polymers and sulfur: what are organic polysulfides good for? Preparative strategies and biological applications, Macromol. Rapid Commun., 2009, vol. 30, p. 299. https://doi.org/10.1002/marc.200800740
Wu, D., Hu, Q., and Zhu, Y., Therapeutic application of hydrogen sulfide donors: the potential and challenges, Front. Med., 2016, vol. 10, p. 18. https://doi.org/10.1007/s11684-015-0427-6
Saidu, N.E.B., Valente, S., Bana, E., Kirsch, G., Bagrel, D., and Montenarh, M., Coumarin polysulfides inhibit cell growth and induce apoptosis in HCT116 colon cancer cells, Bioorg. Med. Chem., 2012, vol. 20, p. 1584. https://doi.org/10.1016/j.bmc.2011.12.032
Putnik, P., Gabrić, D., Roohinejad, S., Barba, F.J., Granato., D., Mallikarjunan, K., Lorenzo, J.M., and Kovačevića, D.B., An overview of organosulfur compounds from Allium spp.: From processing and preservation to evaluation of their bioavailability, antimicrobial, and anti-inflammatory properties, Food Chem., 2019, vol. 276, p. 680. https://doi.org/10.1016/j.foodchem.2018.10.068
An, H., Zhu J., Wang, X., and Xu, X., Synthesis and anti-tumor evaluation of new trisulfide derivatives, Bioorg. Med. Chem. Lett., 2006, vol. 16, p. 4826. https://doi.org/10.1016/j.bmcl.2006.06.070
Fukao, T., Hosono, T., Misawa, S., Seki, T., and Ariga, T., The effects of allyl sulfides on the induction of phase II detoxification enzymes and liver injury by carbon tetrachloride, Food Chem. Toxicol., 2004, vol. 42, p. 743. https://doi.org/10.1016/j.fct.2003.12.010
Cerda, M.M., Zhao, Y., and Pluth, M.D., Thionoesters: a native chemical ligation-inspired approach to cysteine-triggered H2S donors, J. Am. Chem. Soc. 2018, vol. 140, p. 12574. https://doi.org/10.1021/jacs.8b07268
Sochor, J., Dobes, J., Krystofova, O., Ruttkay-Nedecky, B., Babula, P., Pohanka, M., Jurikova, T., Zitka, O., Adam, V., Klejdus, B., and Kizek, R., Electrochemistry as a tool for studying antioxidant properties, Int. J. Electrochem. Sci., 2013, vol. 8, p. 8464
Nguyen, T.B., Recent advances in organic reactions involving elemental sulfur, Adv. Synth. Catal., 2017, vol. 359, p. 1066. https://doi.org/10.1002/adsc.201601329
Yi, H., Zhang, G., Wang, H., Huang, Z., Wang, J., Singh, A.K., and Lei, A., Recent advances in radical C−H activation/radical cross-coupling, Chem. Rev., 2017, vol. 117, p. 9016. https://doi.org/10.1021/acs.chemrev.6b00620
Tang, S., Liu, Y., and Lei, A., Electrochemical oxidative cross-coupling with hydrogen evolution: a green and sustainable way for bond formation, Chem, 2018, vol. 4, p. 27. https://doi.org/10.1016/j.chempr.2017.10.001
Tang, S., Zeng, L., and Lei, A., Oxidative R1-H/R2-H cross-coupling with hydrogen evolution, J. Amer. Chem. Soc., 2018, vol. 140, p. 13128. https://doi.org/10.1021/jacs.8b07327
Jiang, Y., Xu, K., and Zeng, C., Use of electrochemistry in the synthesis of heterocyclic structures, Chem. Rev., 2018, vol. 118 (9), p. 4485. https://doi.org/10.1021/acs.chemrev.7b00271
Baker, L.A., A perspective and prospectus on single-entity electrochemistry, J. Amer. Chem. Soc., 2018, vol. 140 (46), p. 15549. https://doi.org/10.1021/jacs.8b09747
Yuan, Y., Yu, Y., Qiao, J., Liu, P., Yu, B., Zhang, W., Liu, H., He, M., Huang, Z., and Lei, A., Exogenous-oxidant-free electrochemical oxidative C–H sulfonylation of arenes/heteroarenes with hydrogen evolution, Chem. Comm., 2018, vol. 54, p. 11471. https://doi.org/10.1039/c8cc06451b
Wang, Y., Deng, L., Mei, H., Du, B., Han, J., and Pan, Y., Electrochemical oxidative radical oxysulfuration of styrene derivatives with thiols and nucleophilic oxygen sources, Green Chem., 2018, vol. 20, p. 3444. https://doi.org/10.1039/C8GC01337C
Lund, O. and Hammerich, O., Organic electrochemistry, Boca Raton: CRC Press, Taylor & Francis Group, 5th ed., 2016, p. 1736
Do, Q.T., Elothmani, D., Simonet, J., and Guillanton, G.L., The electrochemical oxidation of dimethyl disulfide – anodic methylsulfanylation of phenols and aromatic ethers Electrochim. Acta, 2005, vol. 50, p. 4792. https://doi.org/10.1016/j.electacta.2005.02.033
Manmode, S., Matsumoto, K., Nokami, T., and Itoh, T., Electrochemical methods as enabling tools for glycosylation, Asian J. Org. Chem., 2018, vol. 7, p. 1719. https://doi.org/10.1002/ajoc.201800302
Manmode, S., Kato, M., Ichiyanagi, T., Nokami, T., and Itoh, T., Automated electrochemical assembly of the β-(1,3)-β-(1,6)-glucan hexasaccharide using thioglucoside building blocks, Asian J. Org. Chem., 2018, vol. 7, p. 1802. https://doi.org/10.1002/ajoc.201800345
Mandal, B. and Basu, B., Recent advances in S–S bond formation, RSC Adv., 2014, vol. 4, p. 13854. https://doi.org/10.1039/c3ra45997g
Glass, R.S., Jouikov, V.V., and Bojkova, N.V., Electrochemical activation of dimethyl disulfide for electrophilic aromatic substitution, J. Org. Chem. 2001, vol. 66, p. 4440. https://doi.org/10.1021/jo010156x
Matsumoto, K., Suga, S., and Yoshida, J., Organic reactions mediated by electrochemically generated ArS+, J. Org. Biomol. Chem., 2011, vol. 9, p. 2586. https://doi.org/10.1039/c0ob01070g
Huang, P., Wang, P., Tang, S., Fu, Z., and Lei, A., Electro-oxidative cross S–H/S–H coupling with hydrogen evolution: a facile access to unsymmetrical disulfides, Angew. Chem. Int. Ed., 2018, vol. 57, p. 8115. https://doi.org/10.1002/anie.201803464
Banerji, A. and Kalena, G.P., A new synthesis of organic trisulfides, Tetrahedron Lett., 1980, vol. 21, p. 3003. https://doi.org/10.1016/0040-4039(80)88021-X
Derbesy, G. and Harpp., D.N., A simple method to prepare unsymmetrical di- tri- and tetrasulfides, Tetrahedron Lett., 1994, vol. 35, N. 30, p. 5381. https://doi.org/10.1016/S0040-4039(00)73505-2
Zysman-Colman, E. and Harpp., D.N., Optimization of the synthesis of symmetric aromatic tri- and tetrasulfides, J. Org. Chem., 2003, vol. 68, p. 2487. https://doi.org/10.1021/jo0265481
Soleiman-Beigi, M. and Mohammadi, F., Simple and green method for synthesis of symmetrical dialkyl disulfides and trisulfides from alkyl halides in water; PMOxT as a sulfur donor, J. Sulfur Chem., 2017, vol. 38, p. 134. https://doi.org/10.1080/17415993.2016.1253696
Kertmen, A., Lach, S., Rachon, J., and Witt, D., Novel and efficient methods for the synthesis of symmetrical trisulfides, Synthesis, 2009, N. 9, p. 1459. https://doi.org/10.1055/s-0028-1088161
Xu, S., Wang, Y., Radford, M.N., Ferrell, A.J., and Xian, M., Synthesis of unsymmetric trisulfides from 9‑fluorenylmethyl disulfides, Org. Lett., 2018, vol. 20, p. 465. https://doi.org/10.1021/acs.orglett.7b03846
Guillanton, G.L., Electrochemical activation of sulfur in organic solvents - new syntheses of thioorganic compounds with a sacrificial carbon-sulfur electrode, Sulfur Reports, 1992, vol. 12(2), p. 405. https://doi.org/10.1080/01961779208048949
Берберова, Н.Т., Смолянинов, И.В., Шинкарь, Е.В., Кузьмин, В.В., Седики, Д.Б., Швецова, А.В. Электросинтез биологически активных дициклоалкилди- и трисульфидов с участием редокс-системы H2S–S8. Изв. РАН, Сер. хим. 2018. № 1. С. 108. [Berberova, N.T., Smolyaninov, I.V., Shinkar, E.V., Kuzmin, V.V., Sediki, D.B., and Shvetsova, A.V., Electrosynthesis of biologically active dicycloalkyl di- and trisulfides involving an H2S–S8 redox system, Russ. Chem. Bull., 2018, vol. 67, no. 1, p. 108.] https://doi.org/10.1007/s11172-018-2044-4
Шинкарь, Е.В., Швецова, А.В., Cедики, Д.Б., Берберова, Н.Т. Редокс-активация сероводорода в реакции с циклогептаном. Электрохимия. 2015. Т. 51. № 11. С. 1182. [Shinkar, E.V., Shvetsova, A.V., Sediki, D.B., and Berberova, N.T., Redox activation of hydrogen sulfide in reaction with cycloheptane, Russ. J. Electrochem., 2015, vol. 51, p. 1182.] https://doi.org/10.1134/S1023193515110178
Берберова, Н.Т., Шинкарь, Е.В., Смолянинов, И.В., Пащенко, К.П. Редокс-медиаторы окисления сероводорода в реакциях с циклоалканами. Докл. РАН. 2015. Т. 465. № 6. С. 683. [Berberova, N.T., Shinkar, E.V., Smolyaninov, I.V., and Pashchenko, K.P., Redox mediators of hydrogen sulfide oxidation in reactions with cycloalkanes, Dokl. Chem., 2015, vol. 465, № 6, p. 683.] https://doi.org/10.1134/S0012500815120058
Berberova, N.T., Smolyaninov, I.V., Shinkar, E.V., Burmistrova, D.A. Andzhigaeva, V.V., and Sultanova, M.U., Electrosynthesis of polysulfides R2Sn (n = = 2–4) based on cycloalkanes and S8 via bromide-mediated oxidation of H2S, Int. J. Electrochem. Sci., 2019, vol. 14, p.531. https://doi.org/10.20964/2019.01.15
Vineyard, B. D., Mercaptan-sulfur reaction. Alkyl trisulfides, J. Org. Chem., 1966, vol. 31 (2), p. 601. https://doi.org/10.1021/jo01340a511
Гордон, А., Форд, Р. Спутник химика, Мир, Москва, 1976, 437 с. [Gordon, A. J. and Ford, R. A., The chemist’s companion, Wiley Intersci., N. Y.–London–Sidney–Toronto, 1972.]
Lam, K. and Geiger, W.E., Anodic oxidation of disulfides: detection and reactions of disulfide radical cations, J. Org. Chem., 2013, vol. 78 (16), p. 8020. https://doi.org/10.1021/jo401263z
Guillanton, G.L., Determination of mixtures of polysulfides by cyclic voltammetry, J. Electrochem. Soc., 1996, vol. 143 (10), p. 223. https://doi.org/10.1149/1.1837151
Берберова, Н.Т., Шинкарь, Е.В. Катион-радикал сероводорода и органические реакции с его участием. Изв. РАН, Сер. хим. 2000. № 7. С. 1182. [Berberova, N.T. and Shinkar, E.V., The radical cation of hydrogen sulfide and related organic reactions, Russ. Chem. Bull., 2000, vol. 49, p. 1178.] https://doi.org/10.1007/BF02495758
Bailey, T.S., Zakharov, L.N., and Pluth, M.D., Understanding hydrogen sulfide storage: probing conditions for sulfide release from hydrodisulfides, J. Amer. Chem. Soc., 2014, vol. 136, p. 10573. https://doi.org/10.1021/ja505371z
Дополнительные материалы отсутствуют.