

Электрохимия, 2020, T. 56, № 6, стр. 528-543
Комбинация электрохимических и адсорбционных методов разложения и удаления пестицида Padan 95SP (95% Cartap) из воды
Нгуен Тиен Хоанг a, Рудольф Хольц a, b, c, *
a Хемницкий технологический университет, Институт химии, Отделение электрохимии
D-09107 Хемниц, Германия
b Санкт-Петербургский государственный университет, Институт химии
199034 Санкт-Петербург, Россия
c Государственная главная лаборатория материаловедческого химического машиностроения,
Школа энергетических наук и технологии, Нанкинский технический университет
211816 провинция Цзянсу, Нанкин, Китай
* E-mail: rudolf.holze@chemie.tu-chemnitz.de
Поступила в редакцию 14.05.2019
После доработки 30.07.2019
Принята к публикации 08.10.2019
Аннотация
Комбинация электрохимических и адсорбционных методов показала эффективное снижение общего органического углерода (TOC) и удаление Padan 95SP (95% Cartap) из воды благодаря его окислению и адсорбции на гранулированном активированном угле. Влияние вспомогательных электролитов, скорости потока, высоты слоя, количества повторных циклов адсорбции, а также начальной концентрации было изучено, чтобы определить их влияние на удаление TOC и Cartap. Концентрацию Cartap определяли с помощью УФ и видимой спектроскопии в соответствии с процедурой для 5,5'-дитиобис(2-нитробензойной кислоты). Эта комбинация методов обеспечивает более 90% удаления Cartap и приблизительно 75% снижения TOC. Увеличение высоты слоя и повторная адсорбционная обработка раствора значительно не увеличивают удаление TOC. Высокоэффективная жидкостная хроматография была использована для характеристики образования электрохимических побочных продуктов. Гранулированный активированный уголь, используемый на стадиях адсорбции, исследовали до и после использования с помощью ИК-Фурье спектроскопии.
ВВЕДЕНИЕ
Роль Cartap в сельском хозяйстве
Инсектицид Cartap является предшественником нереистоксина, природного инсектицидного вещества, выделенного из морского кольчатого червя Lumbrineris. Инсектициды на основе гидрохлорида Cartap впервые были зарегистрированы в Японии в 1967 году и широко используются в Азии, Европе и Южной Америке. Гидрохлорид Cartap используется против широкого спектра насекомых, но особенно против рисового стебля, чешуекрылых вредителей овощей и колорадского жука. Коммерческие составы включают растворимые в воде порошки, пыль и гранулы. Примерно 70% мирового производства приходится на рис. В дельте реки Меконг (Вьетнам) около 20% использования инсектицидов на рисовых и рисовых рыбных фермах составляет Cartap [1]. В Китае, Японии и Корее он также является одним из наиболее часто используемых пестицидов для борьбы с насекомыми-вредителями [2, 3]. Гидрохлорид Cartap обычно используют из расчета 0.5–1.5 кг га–1 с интервалом до сбора урожая от 7 до 21 суток [4].
Несколько методов были разработаны для определения остатков Cartap. Газохроматографический (ГХ) метод представляется подходящим для регуляторного анализа. Этот метод основан на преобразовании исходного соединения и метаболитов в нереистоксин, который можно измерить с помощью пламенного фотометрического детектора, оборудованного серным фильтром. Предел определения составляет 0.005 мг кг–1 [4].
Учитывая широкое применение Cartap в качестве основного соединения в пестицидах, очень вероятно, что оно вызовет загрязнение окружающей среды и опасные аварии во время использования. Таким образом, необходимо провести исследования деградации настоящего Cartap, например в Padan 95 SP (95% Cartap).
Электрохимический процесс
В электрохимическом процессе (см. своевременный обзор [5]) органические вещества разрушаются по прямому или косвенному механизму окисления [6] (рис. 1). В процессе прямого окисления загрязненные органические вещества сначала адсорбируются на поверхности анода, а затем разрушаются в результате реакции анодного переноса электрона. В процессе косвенного окисления загрязненные органические вещества разрушаются сильными окислителями (радикалами ОН) или более слабыми окислителями (гипохлорит/хлор, озон, персульфат), которые генерируются электрохимическим методом [7]. В результате органические соединения распадаются на более мелкие молекулярные промежуточные продукты и/или конечные продукты (CO2, H2O и неорганические вещества).
Рис. 1.
Пути удаления загрязняющих веществ при электрохимическом окислении (косвенное и прямое окисление) [6].

Недавнее использование тонкопленочных электродов из легированного бором алмаза (BDD) в анодном окислении сильно повысило интерес к его применению для очистки воды. Этот электрохимический метод основан на окислении органических загрязнителей радикалами •ОН (уравнение (1)).
Сочетание перспективных процессов окисления и адсорбции активированным углем
Один только метод может быть недостаточным для обработки бионеразлагаемых органических соединений. Следовательно, многие исследователи пытаются объединить два или более методов обработки для успешного удаления [7]. Например, Guzzella и др. [8] сравнили новые инновационные методы обработки питьевой воды (адсорбция в колонке со смолой и усовершенствованные процессы окисления AOP) со старым фильтром и с гранулированным активированным углем (GAC) с точки зрения их эффективности удаления токсичных/мутагенных органических микрозагрязнителей. Rajkumar и др. [7] изучали комбинацию электрохимического разложения и адсорбционной обработки активированным углем (AC) для сточных вод, содержащих смешанные фенольные соединения. Точно так же Santhanam и др. [9] объединили несколько технологий (электрохимическое, вызванное солнечным светом окисление и биологические процессы) для обработки хлоридсодержащих текстильных стоков.
Активированный уголь является очень распространенным адсорбентом для адсорбционной технологии благодаря множеству преимуществ: адсорбционная способность, селективность, регенерируемость, кинетика, совместимость и низкая стоимость [10–12]. АС выпускается в гранулированной форме с размерами частиц в диапазоне от 0.5 до 4 мм или в виде порошка с размерами частиц <40 мкм. Этот метод эффективен при удалении нежелательного вкуса и запаха некоторых органических веществ, микроорганизмов, хлоридов, фторидов и радона из питьевой или сточной воды [13]. Однако он не эффективен для микробных загрязнений, ионов металлов, нитратов и других неорганических загрязнений [13]. Адсорбционная способность углеродных материалов не связана простым образом с их площадью поверхности и пористостью. Адсорбционная емкость зависит от доступа органических молекул к внутренней поверхности адсорбента, который зависит от их размера и других рабочих параметров. Таким образом, при соответствующих условиях эксперимента небольшие молекулы, такие как фенол, могут получить доступ к микропорам, природные органические вещества могут получить доступ к мезопорам, а бактерии могут иметь доступ к макропорам. Полярное вещество (вещество, которое хорошо растворяется в воде) не может быть адсорбировано (если только оно плохо удаляется), в то время как неполярное вещество может эффективно адсорбироваться на AC.
АС (в том числе порошкообразный и гранулированный) изготавливается из различных видов углеродистого сырья: например углей (антрацит, битум, лигнит), древесины, торфа и скорлупы кокосовых орехов и т.д. Использованный АС можно регенерировать или утилизировать. Активированный уголь очень неоднороден как по морфологии, так и по поверхностным характеристикам. Типичный элементный анализ показал, что основными элементами в каркасе из активированного угля являются углерод (92 мас. %) и кислород (7 мас. %) [14]. Для коммерческих активированных углей количество кислорода может варьироваться от <1 до 16%. Помимо углерода и кислорода в структуре могут присутствовать другие элементы – водород, азот, фосфор и сера. Структурированный АС построен из графеновых слоев, также называемых “базисными плоскостями” [15]. Различные функциональные группы, содержащие кислород, азот и серу, могут быть обнаружены на краях базисной плоскости, как показано на рис. 2 [14, 16].

Количество функциональных групп в АС, определяемое методом температурно-программируемой десорбции, может различаться в зависимости от сырья, из которого сделан АС. Краткое описание этого метода, а также содержание функциональных групп для некоторых типов GAC можно найти в литературе [14].
В этой статье мы сообщаем о подробном исследовании сочетания электрохимической обработки и адсорбционной технологии для удаления коммерческого пестицида Padan 95 SP при постоянной плотности тока 20 мА см–2 с использованием однокамерной ячейки. Чтобы прояснить влияние окислителей, более слабых, чем радикалы •ОН, такие как гипохлорит, хлор или персульфат, образующиеся из вспомогательных электролитов, на удаление Cartap, были измерены их соответствующие концентрации. Изменение концентрации вспомогательного электролита при электрохимической обработке также прослеживается. В случае последующих исследований адсорбции исследовали высоту слоя, скорость потока обработанного раствора в колонну, влияние числа повторных циклов адсорбции и насыщения адсорбента GAC. Выделение побочных продуктов в различных поддерживающих электролитах сопровождалось высокоэффективной жидкостной хроматографией (ВЭЖХ).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Химические реагенты
Товарный пестицид Padan 95SP (95% Cartap) был приобретен в филиале японской компании Sumitomo Chemical во Вьетнаме. Реагент Эллмана (5,5'-дитиобис(2-нитробензойная кислота), DTNB), приобретенный у компании Sigma-Aldrich, использовали для определения содержания Cartap в Padan 95SP после интервалов времени 5, 15, 30, 60, 90 и 120 мин в электрохимическом и послеадсорбционных процессах. Другие химические вещества (AgCl, HCl, H2SO4, BaCl2 · 2H2O, NaNO3, Na2CO3, Na2SO4, CH3OH, K2Cr2O7, H3PO4, K2CrO4, CH3COOH, C2H5OH, глицерин, борная кислота) были поставлены компаниями Sigma-Aldrich и Merck. Для процесса адсорбции гранулированный активированный уголь был получен от компании Sigma-Aldrich. Растворы готовили с использованием деионизованной сверхчистой воды (Seralpur Pro 90 C).
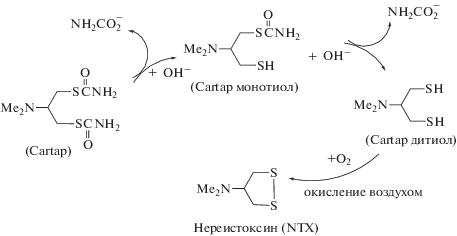
Определение содержания Cartap по методике для 5,5'-дитиобис(2-нитробензойной кислоты) (DTNB) [17–19]
Группа –SCO(NH2) Cartap реагирует с DTNB с образованием желтого аниона 3-карбокси-4-нитрофенилтиолата [20] (рис. 3), который детектируется на длине волны 412 нм с помощью УФ и видимой спектроскопии. Нереистоксин (NTX) (рис. 4), в котором отсутствует свободная тиоловая группа, не реагирует с DTNB. 0.2 мл испытуемого раствора и 0.8 мл раствора DTNB (1 г л–1) добавляли к 4 мл 0.1 М раствора фосфатного буфера (рН 9). После 1 ч времени реакции записывали УФ и видимый спектр. Калибровочная кривая для Cartap была построена в соответствии с процедурой, описанной выше [18].

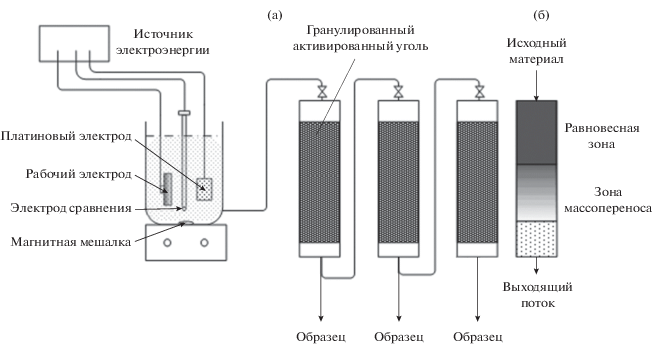
Электролитическая система и адсорбционная установка
Объемный электролиз проводили при комнатной температуре (22°С) в стеклянной ячейке объемом 400 мл с одним отсеком. Электрод BDD, изготовленный из BDD-диска (Neocoat, Швейцария), использовали в качестве рабочего электрода с открытой круглой поверхностью 3.8 см2; толщина алмазного слоя составляла 2.5–3 мкм в соответствии с информацией производителя. Платиновую фольгу и Ag/AgCl (3 М KCl) использовали в качестве противоэлектрода и электрода сравнения соответственно. В течение всего процесса 250 мл раствора электролита перемешивали с помощью магнитной планки. Перед началом экспериментов электрод BDD подвергали ультразвуковой обработке в течение 5 мин для удаления загрязнений, а затем промывали сверхчистой водой. Pt-электрод также промывали сверхчистой водой. Использовали растворы, содержащие Padan 95SP, с концентрациями 100, 300, 500 и 700 мг л–1. Значения рН раствора регулировали с помощью H2SO4 и NaOH, используя измеритель рН для контроля. Общее время процесса составило 120 мин. Приложенная плотность тока j = = 20 мА см–2 была определена в других работах как наиболее подходящая [21]. Грубая оценка показывает, что при этой плотности тока окисление Cartap ограничено массопереносом.
Активированный уголь упаковывали в стеклянные пробирки с внутренним диаметром 2.8 см. Затем раствор из электрохимической секции пропускали в колонку через клапан в верхней части для регулирования скорости потока притока. Вся установка изображена на рис. 5а (см. также [22, 23]).
Аналитические методы
Образцы раствора отбирали и сразу определяли оставшееся содержание Cartap (согласно процедуре, описанной в 2.2) с помощью спектрометра Getspec-2048-SPU. Интенсивность поглощения в ультрафиолетовом и видимом спектре 3‑карбокси-4-нитрофенилтиолат-аниона при длине волны максимума 412 нм была пропорциональна содержанию Cartap. Общий органический углерод (TOC) растворов определяли количественно в подкисленных растворах после продувки образцов кислородом стандартным методом NPOC (неотдуваемый органический углерод) с использованием анализатора TOC multi N/C 3100 (Analytik Jena). Поскольку в образцах нет другого источника углерода, содержание углерода (за исключением карбонатсодержащего электролита в одном образце) можно назвать TOC, как это делается в настоящей статье.
Продукты, образующиеся в процессе электрохимического окисления (объемного электролиза), анализировали с помощью ВЭЖХ (модель KNAUER Smartline). Хроматографическая колонка представляла собой колонку Eurospher 100 5 C8 (250 × 4.6 мм), а детектор представлял собой DAD 200-800 нм. Подвижная фаза состояла из 85 об. % воды и 15 об. % ацетонитрила со скоростью потока 1 мл мин–1 при 25°С. Объем впрыска составлял 20 мкл, а длина волны для количественного анализа составляла 210 нм.
Концентрацию ${\text{NO}}_{3}^{ - }$ определяли спектрофотометрическим методом [24], ${\text{CO}}_{3}^{{2 - }}$ количественно определяли как щелочность [25]. Концентрацию ${\text{C}}{{{\text{l}}}^{{\text{--}}}}$ измеряли методом титрования осаждением [26, 27], концентрацию ${\text{SO}}_{{\text{4}}}^{{{\text{2}} - }}$ измеряли методом мутности с использованием УФ-спектрофотометрии. Концентрации ${\text{Cl}}{{{\text{O}}}^{ - }}$ [25, 28] и в растворах измеряли йодометрическими методами.
Теоретическая реакция минерализации Cartap предлагается в уравнении (2):
(2)
$\begin{gathered} {{{\text{C}}}_{7}}{{{\text{H}}}_{{15}}}{{{\text{N}}}_{3}}{{{\text{O}}}_{2}}{{{\text{S}}}_{2}} + 20{{{\text{H}}}_{2}}{\text{O}} \to 2{\text{SO}}_{4}^{{2 - }} + \\ + \,\,3{\text{NH}}_{4}^{ + } + 7{\text{C}}{{{\text{O}}}_{2}} + 43{{{\text{H}}}^{ + }} + 42{\text{e}}. \\ \end{gathered} $Инфракрасные спектры с преобразованием Фурье (FTIR) регистрировали на спектрофотометре Bruker Vector 22 с использованием порошка GAC с платиновым блоком ATR. Все спектры были собраны между 7000 и 400 см–1, усредняя данные 64 последовательных сканирований.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Побочные продукты и их образование
Образование промежуточных продуктов во время разложения Cartap контролировали с помощью ВЭЖХ. Было отмечено, что большинство этих промежуточных продуктов являются начальными продуктами окисления, возникающими в результате первоначального окисления Cartap.
Чтобы получить общее представление о скорости высвобождения этих промежуточных соединений для различных поддерживающих электролитов (т.е. Na2SO4, NaCl, NaNO3 и Na2CO3), применяется метод сравнения относительной площади (RAR). Четыре пика наблюдаются на хроматограммах ВЭЖХ, как показано на рис. 6–9, их относительные соотношения площадей пиков в различные интервалы времени электролиза изображены на рис. 10.
Рис. 6.
Хроматограммы ВЭЖХ для Cartap и продуктов его разложения в электрохимическом процессе: [Padan 95SP]0 = 300 мг л–1, Vобраб. р-р = 250 мл, j = = 20 мА см–2, [NaCl] = 0.05 М, pH 3.

Рис. 7.
Хроматограммы ВЭЖХ для Cartap и продуктов его разложения после электрохимической обработки: [Padan 95SP]0 = 300 мг л–1, Vобраб. р-р = 250 мл, j = = 20 мА см–2, [Na2SO4] = 0.05 М, pH 3.

Рис. 8.
Хроматограммы ВЭЖХ для Cartap и продуктов его разложения после электрохимической обработки: [Padan 95SP]0 = 300 мг л–1, Vобраб. р-р = 250 мл, j = = 20 мА см–2, [NaNO3] = 0.05 М, pH 3.
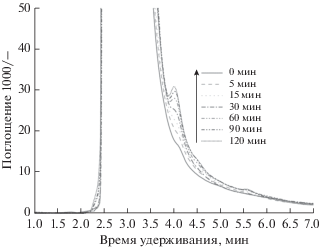
Рис. 9.
Хроматограммы ВЭЖХ для Cartap и продуктов его разложения после электрохимической обработки: [Padan 95SP]0 = 300 мг л–1, Vобраб. р-р = 250 мл, j = = 20 мА см–2, [Na2CO3] = 0.05 М, pH 3.

Рис. 10.
Относительные величины пиков Cartap и продуктов его разложения в зависимости от времени с различными фоновыми электролитами на основе хроматограмм ВЭЖХ, [Padan 95SP]0 = 300 мг л–1, Vобраб. р-р = 250 мл, j = 20 мА см–2, pH 3 (кроме Na2CO3, который был добавлен в электролитический раствор с начальным pH деионизованной воды).

На хроматограммах ВЭЖХ, полученных с образцами, приготовленными с NaCl и Na2SO4 в качестве электролита, пики появлялись при одинаковом времени удерживания. Пик 1 относится к Cartap (tr = 2.9 мин), а пик 3 к нереистоксину (tr = = 2.6 мин), причем они частично перекрывают друг друга.
Как видно на рис. 6 и 7, пик Cartap в случае NaCl в качестве электролита-носителя снижается быстрее, чем в случае Na2SO4, что приводит к более высокому RAR нереистоксина для NaCl, чем для Na2SO4. Пик 2 (${{t}_{{\text{r}}}}$ = 3.4 мин), по-видимому, достигает максимального значения через 30 мин, а затем уменьшается через 120 мин в случае NaCl, в то время как в случае Na2SO4 он медленно увеличивается (см. рис. 10). Тем не менее, пиком 4 можно пренебречь из-за его низкой интенсивности. Этот пик может быть вызван добавками в Padan 95 SP.
Хроматограммы ВЭЖХ для случая NaNO3 показывают выступ на главном пике (рис. 8), и больше нет никаких пиков, указывающих на промежуточные соединения. Возможно, присутствие ${\text{NO}}_{3}^{ - }$ в обработанных образцах вызывает некоторые помехи этому сигналу. Совсем другой результат можно увидеть и в случае Na2CO3 в качестве электролита: например, пик для Cartap сдвигается до tr = 3.4 мин, что приводит к исчезновению пика 2 (см. рис. 9). Нереистоксин постепенно накапливается во время процесса, что приводит к значительному увеличению его RAR.
Зависимость концентрации окисленных ионов от времени и влияние неорганических анионов фоновых электролитов
Во время электролиза на BDD-электроде при окислении Cl– образуются такие частицы, как СlO–, связанные с присутствием растворенного Cl2 и НClO, как показано в уравнениях (3)–(5), а при окислении ${\text{SO}}_{4}^{{2 - }}$ образуются ионы ${{{\text{S}}}_{2}}{\text{O}}_{8}^{{2 - }}$ (уравнение (6)):
(3)
${\text{BDD}}{{{\text{(}}}^{\centerdot }}{\text{OH) + C}}{{{\text{l}}}^{ - }} \to {\text{BDD + }}{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}{\text{C}}{{{\text{l}}}_{2}}{\text{ + O}}{{{\text{H}}}^{ - }},$(4)
${\text{C}}{{{\text{l}}}^{ - }} \to {\text{ }}{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}{\text{C}}{{{\text{l}}}_{2}}{\text{ + e}},$(5)
${\text{C}}{{{\text{l}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \to {\text{ C}}{{{\text{l}}}^{ - }}{\text{ + HClO + }}{{{\text{H}}}^{ + }},$Их накопление зарегистрировано на рис. 11. Непрерывное постепенное увеличение концентрации ClO– еще раз демонстрирует более высокую эффективность NaCl для разложения Cartap, чем Na2SO4. На рис. 11 накопление достигает 0.55 мМ через 120 мин, в то время как концентрация остается <0.1 мМ. Многие предыдущие исследователи [7, 9, 29] также обнаружили образование активного хлора (т.е., ClO–) в электрохимических процессах. Например, в исследовании Rajkumar и др. [7] в течение начального периода электролиза не было обнаружено хлора/гипохлорита, но после этого концентрация гипохлорита непрерывно увеличивалась. Santhanam и др. [9] обнаружили, что концентрация хлора и гипохлорита, соответственно, постепенно увеличивается после исчезновения окраски в растворе.
Рис. 11.
Концентрации гипохлорита и персульфата в случае 0.05 М NaCl и 0.05 М Na2SO4 во время электрохимического окисления, [Padan 95SP]0 = 300 мг л–1, Vобраб. р–р = 250 мл, j = 20 мА см–2, pH 3.

Чтобы оценить изменение концентрации анионов электролита, их концентрации показаны на рис. 12. Следует отметить, что деградация Cartap высвобождает неорганические ионы, такие как ${\text{SO}}_{4}^{{{\text{2}} - }}$ и ${\text{NO}}_{3}^{ - }$ [2, 30], которые также могут способствовать изменению их концентрации. как вполне видно с нитрат-ионами. В случае карбонат-ионов наблюдаемое снижение может быть связано с выделением CO2 из слабокислого раствора электролита. Несмотря на то, что при разложении Cartap высвобождается ${\text{SO}}_{4}^{{{\text{2}} - }}$, концентрация ${\text{SO}}_{4}^{{2 - }}$ значительно снижается в течение первых 30 мин и уменьшается в дальнейшем. Увеличение концентрации персульфата сравнительно невелико. Напротив, концентрация ${\text{NO}}_{{\text{3}}}^{ - }$ увеличивается примерно до 0.055 М через 120 мин, это связано с высвобождением ${\text{NO}}_{{\text{3}}}^{ - }$ в результате разложения Cartap.
Рис. 12.
Изменение концентрации поддерживающих электролитов при электрохимическом окислении при исходной концентрации электролитов 0.05 М, [Padan 95SP]0 = 300 мг л–1, Vобраб. р–р = 250 мл, j = = 20 мА см–2, pH 3 (за исключением Na2CO3, который был добавлен в электролитический раствор с типичным pH деионизованной воды около 6–6.5).

Сравнение затраченного количества электрического заряда и количества разложенного Cartap позволяет предположить очень низкий выход по току.
Удаление органических соединений в секции адсорбции после электрохимического процесса
Приток в секцию адсорбции представляет собой очищенный раствор от электрохимического (ЕС) процесса. Рабочие параметры в секции ЕС основаны на оптимизированных условиях (pH 3, [Na2SO4] = 0.05 М, плотность тока j = 20 мА см–2, время электролиза t = 120 мин). Схема комбинации показана на рис. 5. Были изучены как удаление Cartap, так и восстановление TOC путем адсорбции в GAC. Принимая во внимание, что измерения остающегося Cartap ограничены только одним соединением, исследование только в отношении ТОС дает лишь суммарный результат, который, возможно, содержит различные доли нескольких разлагающихся соединений, возможно даже изменяющихся как функция прогрессирующего процесса адсорбции.
Влияние скорости потока на удаление ТОС
Использование GAC является эффективным и экономичным способом удаления вкуса, запаха, цвета и нежелательных органических веществ, включая токсичные вещества, на постоянной основе без значительных капиталовложений [7]. Основываясь на этих преимуществах, GAC был выбран для дальнейшего сокращения TOC в этом исследовании. Обработанный раствор из процесса ЕС был передан в секцию адсорбции. Влияние скорости потока на удаление TOC было изучено. Как показано на рис. 13, содержание ТОС все еще присутствует после процесса ЕС на уровне 91.3% от его первоначального значения до электрохимической обработки, а затем снижается до 27–32% в секции адсорбции при различных скоростях потока притока. Как и ожидалось, электрохимическая обработка привела к довольно значительному разложению Cartap в продукты распада, но они, конечно, не были удалены из раствора электрохимическим процессом. Как правило, увеличение скорости потока приведет к уменьшению адсорбции органики на адсорбенте из-за уменьшения времени контакта между жидкостью и адсорбентом. Например, максимальное удаление адсорбируемых органических галогенов было достигнуто в другом месте, когда скорость потока поддерживалась на самом низком уровне (5 мл мин–1) [7]. Удивительно, что в диапазоне от 5 до 20 мл мин–1 самое высокое снижение TOC может быть достигнуто при относительно более высокой скорости потока (15 мл мин–1), тогда как обычно при более высокой скорости потока адсорбция ниже из-за сокращения времени контакта между адсорбатом и адсорбентом. Таким образом, управление скоростью потока является важным параметром для достижения высокой эффективности удаления, а также для сокращения рабочего времени процесса.

Влияние высоты слоя
Согласно конструкции установки колонны AC [31], оптимальное отношение высоты слоя AC к диаметру колонки составляет приблизительно 10 для практического применения. Однако в данной экспериментальной установке мы могли изменить высоту слоя с 8 до 14 см при внутреннем диаметре 2.8 см. Ясно, что увеличение высоты слоя приведет к более высокой адсорбционной способности из-за увеличенного количества частиц адсорбента и, следовательно, более доступных мест адсорбции. Однако отношение TOC/TOC0 уменьшается только на 5% (с 30.1 до 25.7%) при увеличении высоты слоя до 175% (с 8 до 14 см, см. рис. 14). Около 69.9% ТОС удаляется при высоте слоя 8 см. Можно сделать вывод, что при заданных рабочих параметрах зона массообмена не прошла через колонку, а некоторая активная зона осталась внизу (см. рис. 5б). Это также говорит о том, что прохождение элюента через дополнительные (свежие) колонки не приведет к дальнейшему снижению содержания ТОС. Из-за смешанного и по существу неизвестного состава растворенных продуктов разложения Cartap и их возможно различного адсорбционного поведения – это только предварительное заключение.
Рис. 14.
Удаление ТОС в зависимости от высоты слоя в процессе адсорбции после ЕС (рабочие параметры в процессе ЕС можно увидеть в описании рис. 13), диаметр частиц: 2.5 мм, Vобраб. р-р = 100 мл, внутренний диаметр колонки: 2.8 см, скорость потока: 10 мл мин–1).

Влияние анионов электролитов из процесса ЕС на процесс адсорбции
Как правило, хлорид, сульфат, нитрат и карбонат не только используются в качестве вспомогательных электролитов, они также могут быть найдены в сточных водах. Присутствие их в притоке может отрицательно сказаться на адсорбционном поведении органических соединений [32]. Вспомогательные электролиты оказывают аналогичное влияние на удаление ТОС в процессе ЕС, как можно видеть на рис. 15 количество оставшегося ТОС в присутствии этих анионов электролита очень схоже (от 24 до 29.5%). Если смотреть только на стадию адсорбции (см. вставку на рис. 15), то различия во влиянии анионов становятся видимыми: адсорбция карбонатов наиболее сильна, хлоридов слабее. По-видимому, влияние анионов зависит от природы ионов (то есть: одновалентных и двухвалентных ионов) и энергии гидратации целевых ионов. Значение энергии гидратации (молярные энтальпии) нитрата выше, чем у других анионов [33–36]. Таким образом, нитрат-ионы снижают адсорбционную способность по сравнению с другими анионами. Например, в исследовании Wilaimgam и др. [32] нитрат-ион оказал более сильное отрицательное влияние на адсорбционную способность перфторгексановой кислоты по GAC, чем хлорид. Об этом же явлении сообщили Galamos и др. [33] при использовании активированного угля для адсорбции пертехнетата.
Рис. 15.
Оставшийся TOC с различными неорганическими анионами в электрохимической обработке ([Padan 95SP]0 = 300 мг л–1, Vобраб. р-р = 250 мл, t = = 120 мин, j = 20 мА см–2, pH 3, (за исключением электролита Na2CO3 с неотрегулированным значением рН) и процессы адсорбции (Vобраб. р-р = 100 мл, диаметр частиц: 2.5 мм, высота слоя: 10 см, скорость потока: 10 мл мин–1). Вставка: удаление ТОС в адсорбционной колонке только после ЕС.

Влияние обработанного объема
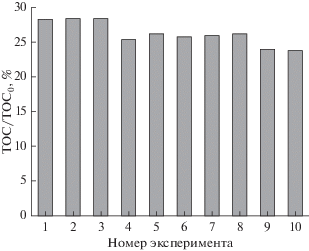
Чтобы оценить влияние обработанного объема на удаление TOC, было выполнено 10 последующих экспериментов с 50 мл. Ясно, что насыщенность абсорбента зависит от количества сорбата на поверхности/внутри адсорбента. Следовательно, адсорбционная емкость будет снижаться после нескольких раз использования. Удивительно, что TOC/TOC0 снижается лишь незначительно с 28.3 до 23.7% после 10 экспериментов в секции адсорбции (см. рис. 16), и, таким образом, среднее значение TOC остается на уровне 25% после 500 мл загружаемого объема притока. Объем обработанного раствора в 500 мл может быть слишком мал, чтобы указывать на любую потерю адсорбционной способности, возможно, вызванную накоплением адсорбата на поверхности/внутри GAC. Следовательно, мы не рассчитываем срок службы слоя. Тем не менее, это исследование предлагает долгосрочное использование GAC для удаления пестицидов из-за стабильности и высокой эффективности удаления GAC.
Влияние начальной концентрации Padan 95 SP
Для исследования влияния начальной концентрации Cartap и удаления TOC в обоих методах четыре разные начальные концентрации Padan 95SP (100, 300, 500 и 700 мг л–1) были проверены. Разложение Cartap в процессе EC уменьшается при увеличении начальной концентрации Padan 95 SP (рис. 17). Например, 18% Cartap оставалось в случае 100 мг л–1, где 60% оставалось в случае 700 мг л–1. Следует отметить, что увеличение начальной концентрации соединения увеличивает его градиент концентрации и массообмен через диффузионный слой и, таким образом, увеличивает его деградацию на электроде [37], что приводит к увеличению количества разлагаемых органических молекул. Однако эффективность разложения снижалась при более высокой концентрации Cartap. Влияние начальных концентраций было также исследовано предыдущими исследователями [37, 38] с использованием электрохимических методов разложения органических загрязнителей. В большинстве случаев увеличение концентрации органических загрязнителей снижает процент их удаления.
Рис. 17.
Разложение Cartap в электрохимическом процессе (Vобраб. р-р = 250 мл, j = 20 мА см–2, pH 3, [Na2SO4] = 0.05 М, t = 120 мин) и в процессе адсорбции после ЕС (высота слоя: 10 см, внутренний диаметр колонки: 2.8 см, диаметр частиц: 2.5 мм, Vобраб. р-р = 100 мл, скорость потока: 10 мл мин–1).

Промежуточные продукты разложения Cartap все еще оставались в стоке первой адсорбционной колонны (рис. 17, 18). Хотя изучение степени удаления в зависимости от высоты слоя уже показало, что даже 8 см высоты слоя дали максимальное удаление ТОС (ожидаемое удаление Cartap будет еще выше), обработанные растворы впоследствии пропускали через дополнительные колонки. После последующих стадий адсорбции с различными концентрациями Cartap, остающимися от EC, результаты показывают: 1) удаление Cartap немного выше при более высокой начальной концентрации, за исключением самой низкой начальной концентрации Cartap, 2) Cartap почти полностью удаляется в первой адсорбционной колонке с остаточным содержанием ниже 10% при небольших обработанных объемах и низких начальных концентрациях Cartap. Можно сделать вывод, что оставшийся Cartap (здесь ниже 10%) в стоках не зависит от начальной концентрации Padan 95 SP. После достижения максимального адсорбционного удаления уже в первой колонке оставшийся Cartap не может быть удален в последующих колонках.
Рис. 18.
Разложение TOC в электрохимическом процессе (Vобраб. р-р = 250 мл, j = 20 мА см–2, pH 3, [Na2SO4] = 0.05 М) и в процессе адсорбции после ЕС (высота слоя: 10 см, внутренний диаметр колонки: 2.8 см, диаметр частиц: 2.5 мм, Vобраб. р-р = 100 мл, скорость потока: 10 мл мин–1).

На рис. 18 показано удаление ТОС в обоих процессах. После процесса ЕС от 95 до 82% TOC все еще остаются в исходных концентрациях от 700 до 100 мг л–1, соответственно, потому что электрохимический процесс разрушает только Cartap, но не может удалять TOC. Это очевидно из сравнения удаления TOC (рис. 18) и удаления Cartap (рис. 17). Например, около 45, 35 и 20% ТОС остаются после первого эксперимента на стадии адсорбции для случаев 700, 500 и 100 мг л–1 соответственно. Дальнейшее удаление Cartap во второй колонке не может быть достигнуто, тогда как содержание TOC впоследствии снижается до достижения минимальных значений приблизительно 25% во 2-м и 4-м циклах для 500 и 700 мг л–1 соответственно.
Анализ GAC методом ИК-Фурье-спектроскопии
Удаление органических соединений путем обработки GAC происходит из-за физического сродства между органическими молекулами и GAC (например, ван-дер-ваальсовыми и электростатическими силами), а также из-за химических адсорбционных сил [39]. Инфракрасная спектроскопия широко используется для характеристики поверхностных групп различных оксидов, а также применяется к различным типам углерода и углеродистых материалов [40]. Таким образом, ИК-Фурье-спектроскопия использовалась для проверки различий между GAC до и после адсорбции (как показано на рис. 19). GAC после адсорбции показывает полосу около 1445 см–1, предварительно отнесенную к деформационным колебаниям C–H либо в –(CH3)2, либо в метиленовой группе (–CH2–). Кроме того, имеется слабый пик при 874 см–1, что позволяет предположить, что C–H, возможно, вызван колебательной модой в центре молекулы Cartap.

Таблица 1.
Отнесение полос в ИК-Фурье-спектре [41]
| Положение полосы, см–1 | Отнесение |
|---|---|
| 1612, 1631, 1656, 1657, 1695, 1732 | Валентные колебания C=O в кетоне |
| 1559, 1570, 1585 | Валентные колебания C=O в ароматическом соединении |
| 1467 | Деформационные колебания C–H в метиленовой группе |
| 1439 | Деформационные колебания C–H в –C(CH)3 |
| 1418 | Деформационные колебания C–H в алкане |
| 1161, 1206, 1218 | Валентные колебания C–O в спирте |
| <900 | Колебания C–H |
Механизм адсорбции Cartap
Хорошо известно, что поверхностные группы адсорбента могут взаимодействовать с органическим адсорбатом и влиять на адсорбцию последнего [42]. Mohammad и др. [43] предположили, что адсорбция пестицида Oxamyl, который имеет структуру, подобную структуре Cartap, происходит в основном за счет дисперсионных сил между электронами в структуре пестицида и электронами на поверхности активированного угля из фекалий шелкопряда. Кроме того, могут быть эффективными водородные связи между функциональными группами, такими как –COOH, –OH и –NH2 Cartap, а также продуктами его разложения и гидрофильными группами (например, –COOH-группами и –OH-группами) GAC [44, 45]. Эти варианты схематично изображены на рис. 20.

Согласно [45], ковалентные связи могут образовываться между органическими химическими веществами и углеродными нанотрубками (УНТ), если как адсорбируемые химические вещества, так и УНТ имеют функциональные группы, такие как –СООН, –ОН, –NH2, подходящие для образования ковалентных связей. Таким образом, на основании структурных формул GAC и Cartap, приведенных выше, также могут образовываться ковалентные связи между ними. Помимо адсорбционного удаления Cartap и продуктов его разложения, GAC может действовать как катализатор, усиливающий окислительное разложение различными окисляющими веществами, образующимися в процессе (персульфат, гипохлорит), как это предлагается в литературе [46, 47]. У авторов нет доказательств, подтверждающих или исключающих этот вариант.
ВЫВОДЫ
Отмечено влияние идентичности анионов вспомогательного электролита на электрохимическое образование продуктов распада Cartap. NaCl работает лучше, чем другие электролиты с точки зрения удаления ТОС. Это может быть связано с вкладом ClO– в разложение органического вещества. Удивительно, что уменьшение скорости потока притока и увеличение высоты слоя колонны не улучшают удаление TOC, как ожидалось. Авторы не обнаружили никаких потерь адсорбционной способности после 10 экспериментов. Стабильность с точки зрения удаления ТОС предполагает длительное использование гранулированного активированного угля в качестве адсорбента. При различных начальных концентрациях Cartap менее 1% его оставалось в сточных водах после 1‑го цикла, последующие повторные адсорбционные эксперименты не способствуют дальнейшему удалению Cartap, в то время как TOC постоянно снижается во время повторных адсорбционных экспериментов в случае более высоких начальных концентраций (500 и 700 мг л–1 Padan 95SP).
Список литературы
Berg, H., Pesticide use in rice and rice-fish farms in the Mekong Delta, Vietnam, Crop Protection, 2001, vol. 20, p. 897.
Tian, K., Ming, C., Dai, Y., and Ake, K.M.H., Fenton degradation of Cartap hydrochloride: identification of the main intermediates and the degradation pathway, Water Sci. Technol., 2015, vol. 72, p. 1198.
Choi, E., Cho, I.H., and Park, J., The Effect of Operational Parameters on the Photocatalytic Degradation of Pesticide, J. Environ. Sci. Health, 2004, vol. 39, p. 53.
Cartap (Pesticides residues in food: 1978 evaluation), http://www.inchem.org/documents/jmpr/jmpmono/ v076pr08.htm (accessed October 2, 2018).
Simon, R.G., Stöckl, M., Becker, D., Steinkamp, A.-D., Abt, C., Jungfer, C., Weidlich, C., Track, T., and Mangold, K.-M., Current to Clean Water - Electrochemical Solutions for Groundwater, Water, and Wastewater Treatment, Chem. Ing. Tech., 2018, vol. 90, p. 1832.
Chiang, L.C., Chang, J.E., and Wen, T.C., Indirect oxidation effect in electrochemical oxidation treatment of landfill leachate, Water Res., 1995, vol. 29, p. 671.
Rajkumar, D., Palanivelu, K., and Balasubramanian, N., Combined electrochemical degradation and activated carbon adsorption treatments for wastewater containing mixed phenolic compounds, J. Environ. Eng. Sci., 2005, vol. 4, p. 1.
Guzzella, L., Feretti, D., and Monarca, S., Advanced oxidation and adsorption technologies for organic micro-pollutant removal from lake water used as drinking-water supply, Water Res., 2002, vol. 36, p. 4307.
Santhanam, M., Selvaraj, R., Annamalai, S., and Sundaram, M., Combined electrochemical, sunlight-induced oxidation and biological process for the treatment of chloride containing textile effluent, Chemosphere, 2017, vol. 186, p. 1026.
Alafadehan, O.A., Junadu, O.W., Salami, L., and Popoola, O.T., Treatment of brewery wastewater effluent using activated carbon prepared from coconut shell, J. Appl. Sci. Technol., 2012, vol. 2, p. 178.
National Research Council (US) Safe Drinking Water Committee, Washington DC, Drinking Water and Health, (Volume 2), National Academies Press (US), 1980.
Ali, I., Water Treatment by Adsorption Columns: Evaluation at Ground Level, Sep. Purif. Rev., 2014, vol. 43, p. 175.
Mazille, F. and Spuhler, D., Adsorption (Activated Carbon), https://www.sswm.info/sswm-university-course/module-6-disaster-situations-planning-and-preparedness/further-resources-0/adsorption-%28activated-carbon%29 (accessed August 19, 2018).
de Ridder, D.J., Adsorption of organic micropollutants onto activated carbon and zeolites, Delft: Water Management Academic Press, (Doctoral Thesis) 2002.
Brennan, J.K., Bandosz, T.J., Thomson, K.T., and Gubbins, K.E., Review: Water in porous carbons, Physicochem. Eng. Aspects, 2001, vols. 187–188 p. 539.
Holze, R., Carbon as electrocatalyst in electrochemical energy conversion—An overview, 4th Int.Carbon Conf. Baden-Baden, 1986.
Lee, S.J., Caboni, P., Tomizawa, M., and Casida, J.E., Cartap Hydrolysis Relative to Its Action at the Insect Nicotinic Channel, J. Agric. Food Chem., 2004, vol. 52, p. 95.
Determination of cartap hydrochloride content (spectrophotometric method), Bureau of Indian Standards, IS 14159:1994.
Ellman, G.L., Tissue sulfhydryl groups, Arch. Biochem. Biophys., 1959, vol. 82, p. 70.
Riddles, P.W., Blakeley, R.L., and Zerner, B., Reassessment of Ellman’s reagent, Methods Enzymol., 1983, vol. 91, p. 49.
Hoang, N.T. and Holze, R., J. Appl. Electrochem., submitted.
Patel, H., Fixed-bed column adsorption study: a comprehensive review, Appl. Water Sci., 2019, vol. 9, p. 45.
Hawkins G.B., Fixed Bed Adsorber Design Guidelines, GBHEnterprises, 2013.
Analytical Method: Nitrate (direct measurement in the UV range) as per APHA 4500-${\text{NO}}_{{\text{3}}}^{ - }$ B, https://www.sigmaaldrich.com/technical-documents/articles/analytical-applications/photometry/nitrate-direct-measurement-in-the-uv-range-as-per-apha-4500-no3-b.html (accessed March 7, 2019).
Rice E.W., Standard methods for the examination of water and wastewater, Washington, DC: Amer. Public Health Association, 2012.
Strähle, J. and Schweda, E., Jander Blasius Einführung in das anorganisch-chemische Praktikum 15. edition, Stuttgart: S. Hirzel Verlag, 2005.
Kraemer, E.O. and Stamm, A.J., Mohr’s Method for the Determination of Silver and Halogens in other than Neutral Solutions, J. Am. Chem. Soc., 1924, vol. 46, p. 2707.
Machado, C.M.L., Braitt, A.H., Braitt, G.R., Rodrigues, E.A., and da Silveira Bueno C.E., Analysis of active chlorine releasing and pH of sodium hypochlorite solutions used in Endodontics, Revista Sul-Brasileira de Odontologia RSBO, 2014, vol. 11, p. 252.
Panizza, M. and Cerisola, G., Direct and mediated anodic oxidation of organic pollutants, Chem. Rev., 2009, vol. 109, p. 6541.
Choi, E., Cho, I.H., and Park, J., The Effect of Operational Parameters on the Photocatalytic Degradation of Pesticide, J. Environ. Sci. Health B, 2004, vol. 39, p. 53.
for some suggestions see: Anonymous, Chapter 3: activated carbon column plant design, http://bibing.us.es/ proyectos/abreproy/20087/fichero/CHAPTER+3.pdf (accessed 19.09.2018).
Wilaingam, K., Tanaka. S., Chularueanksorn, P., Suzuki, Y., Ono, R., and Fujii, S., Effect of Anions on Perfluorohexanoic Acid adsorption onto Anion Exchange Plolymers, Non-ion Exchange Polymers and Granular Activated Carbon, J. Jap. Soc. Civil Eng. G (Environ. Research), 2014, vol. 70, p. 65.
Galamos, M., Dano, M., Viglasova, E., Krivosudsky, L., Rosskopfova, O., Novak, I., Berek, D., and Rajec, P., Effect of competing anions on pertechnetate adsorption by activated carbon, J. Radioanal. Nucl. Chem., 2015, vol. 304, p. 1219.
Ohtaki, H. and Radnai, T., Structure and dynamics of hydrated ions, Chem. Rev., 1993, vol. 93, p. 1157.
Barret, J., Inorganic chemistry in aqueous solution, Royal Society of Chemistry, Cambridge: Education, 2003.
Kieland, J., Individual activity coefficients of ions in aqueous solutions, J. Am. Chem. Soc., 1937, vol. 59, p. 1675.
Chen, T.S., Tsai, R.W., Chen, Y.S., and Huang, K.L., Electrochemical Degradation of Tetracycline on BDD in Aqueous solutions, Int. J. Electrochem. Sci., 2014, vol. 9, p. 8422.
Zhang, C., Liu, L., Wang, J., Rong, F., and Fu, D., Electrochemical degradation of ethidium bromide using boron-doped diamond electrode, Sep. Purif. Technol., 2013, vol. 107, p. 91.
Gur-Reznik, S., Katz, I., and Dosoretz, C.G., Removal of dissolved organic matter by granular-activated carbon adsorption as a pretreatment to reverse osmosis of membrane bioreactor effluents, Water Res., 2008, vol. 42, p. 1595.
Lee, D., Hong, S.H., Paek, K.-H., and Ju, W.-T., Adsorbability enhancement of activated carbon by dielectric barrier discharge plasma treatment, Surf. Coat. Tech., 2005, vol. 200, p. 2277.
Socrates, G., Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd ed., West Sussex: John Wiley & Sons Ltd., 2001.
Liu, G., Li, X., and Campos, L.C., Role of the functional groups in the adsorption of bisphenol A onto activated carbon: thermal modification and mechanism, J. Water Supply Res. T., 2017, vol. 66, p. 105.
Mohammad, S.G. and Ahmed, S.M., Preparation of environmentally friendly activated carbon for removal of pesticide from aqueous media, Inter. J. Indust. Chem., 2017, vol. 8, p. 121.
Al-Qodah, Z. and Shawabkah, R., Production and characterization of granular activated carbon from activated sludge, Braz. J. Chem. Eng., 2009, vol. 26, p. 127.
Yang, K. and Xing, B., Adsorption of organic compounds by carbon nanomaterials in aqueous phase: Polanyi theory and its application, Chem. Rev., 2010, vol. 110, p. 5989.
Domínguez, C.M., Ocón, P., Quintanilla, A., Casas, J.A., and Rodriguez, J.J., Highly efficient application of activated carbon as catalyst for wet peroxide oxidation, Appl. Catal. B: Environm., 2013, vols. 140–141. p. 663.
Georgi, A. and Kopinke, F.-D., Interaction of adsorption and catalytic reactions in water decontamination processes: Part I. Oxidation of organic contaminants with hydrogen peroxide catalyzed by activated carbon, Appl. Catal. B: Environ., 2005, vol. 58, p. 9.
Дополнительные материалы отсутствуют.