

Электрохимия, 2020, T. 56, № 8, стр. 675-686
Электрокаталитическое определение салициловой кислоты на стеклоуглеродном электроде, модифицированном сплавом Ni–Cr
Мерзак Дулаш a, *, Мохамед Трари a
a Лаборатория хранения и оценки возобновляемых источников энергии (LSVRE), химический факультет,
Научно-технологический университет им. Хурари Бумедьена (USTHB) BP 32 Эль-Алия
16111 Алжир, Алжир
* E-mail: doulache@yahoo.fr
Поступила в редакцию 24.07.2019
После доработки 02.11.2019
Принята к публикации 10.12.2019
Аннотация
Совместно нанесенный сплав никель–хром (Ni–Cr) на стеклоуглеродном электроде (GCE) успешно использован в качестве нового амперометрического датчика для определения салициловой кислоты (SA). SA обнаружена поверхностно-катализируемым окислением с участием оксигидроксидов никеля(III) в щелочном растворе. Характеристики биосенсора Ni–Cr/GCE охарактеризованы методами циклической вольтамперометрии, электрохимической импедансной спектроскопии (ЭИС) и хроноамперометрии. Электрохимическое поведение сплава Ni–Cr качественно аналогично поведению чистого никелевого электрода. Однако предполагается, что в верхней части сплава присутствует более высокая степень беспорядка структуры оксигидроксидного слоя. Электроактивность Ni–Cr/GCE изучается как функция молярной доли (Xf%) Cr3+ в ванне для осаждения. Результаты показывают, что Ni–Cr/GCE проявляет высокую электрокаталитическую активность в отношении окисления SA. Ni–Cr/GCE с 28Xf% Cr3+ демонстрирует наилучшую активность с высоким сигналом отклика, хорошей чувствительностью 71.22 мкА мМ–1, низким пределом обнаружения 0.1 мкМ (сигнал/шум = 3) и быстрым временем отклика (<3 с). Кроме того, удовлетворительно оцениваются воспроизводимость, селективность и применимость этого электрохимического датчика.
ВВЕДЕНИЕ
Салициловая кислота (SA) является биологическим веществом, широко используемым в косметических средствах, лосьонах и лекарствах благодаря своим антисептическим и жаропонижающим свойствам [1–3]. SA также представляет собой продукт первичного гидролиза ацетилсалициловой кислоты (ASA), которая обычно используется в качестве противовоспалительного средства. Поскольку ASA может быть немедленно гидролизована до SA в щелочном растворе, имеется много сообщений об ее выявлении с использованием косвенного метода определения [4–6]. Процесс выявления ASA основан на ее гидролизе до салициловой кислоты, которая затем обнаруживается.
В литературе сообщалось о многих аналитических методах определения SA, таких как спектрофотометрия в ультрафиолетовом и видимом диапазонах [7, 8], спектрофлуориметрия [9], комбинированная газовая хроматография–масс-спектрометрия [10] и высокоэффективная жидкостная хроматография (ВЭЖХ) [11, 12]. Однако такие методы требуют предварительной обработки образца, сложных операций и часто показывают низкую эффективность. Таким образом, существует необходимость в разработке простых и удобных методов титрования SA с использованием электрохимической технологии (потенциометрия, амперометрия и вольтамперометрия), которые доказали свою эффективность в аналитической химии. Электрохимическое обнаружение SA на открытых электродах (Pt, Au, углеродная паста и графит) не подходит для аналитических приложений из-за медленного переноса электронов и высоких перенапряжений [13, 14]. Кроме того, использование неизолированных электродов не всегда является удовлетворительным из-за загрязняющего характера адсорбции. В последнее время химические и электрохимические модификации электродов с посредниками для переноса электронов предлагают значительные преимущества для проектирования и разработки электрохимических сенсорных устройств. Поэтому существует большой интерес в области модифицированных электродов для преодоления этих проблем, и для этой цели были разработаны различные материалы [15–19].
В этом отношении оксидные и оксигидроксидные пленки переходных металлов были успешно использованы для технологических применений, включая биоэлектронные, каталитические, оптические, биомедицинские, а также химические и биохимические сенсоры благодаря их электрокаталитической активности по отношению к органическим молекулам и шероховатости границы чувствительности [20–25]. Среди 3d‑металлов для электродов использовались электроды на основе никеля, включая объемный металл [26], гидроксид [27], наночастицы [28], оксид [29], сплавы [30, 31] и комплексы [32–34], окисление как органических, так и биологических молекул. Их окисление включает образование валентностей с высоким содержанием никеля, действующих в качестве химического окислителя, а пара β-Ni(OH)2/β-NiOOH выступает в качестве эффективного электронного медиатора [29]. Механизм каталитической реакции обычно предлагается для объяснения результатов электроокисления нескольких органических молекул на оксигидроксид-модифицированных металлических электродах [27, 30]. Согласно этому механизму Ni(OH)2 сначала окисляется до NiOOH, который затем вступает в реакцию с органической молекулой для регенерации исходного материала. Предлагаемый общий механизм может быть представлен следующим образом:
(1)
${\text{Ni}}{{\left( {{\text{OH}}} \right)}_{2}} + {\text{O}}{{{\text{H}}}^{ - }} \leftrightarrow {\text{ NiOOH }} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} + {\text{e}},$(2)
$\begin{gathered} {\text{NiOOH}} + {\text{органическая молекула}} \to \\ \to {\text{Ni(OH}}{{{\text{)}}}_{2}} + {\text{продукты}}. \\ \end{gathered} $Стратегия, которая улучшает электрокаталитические характеристики, заключается в совместном нанесении переходных металлов на поверхность электрода. Повышенная активность была интерпретирована с точки зрения уменьшения отравления электродов, стабилизации окислительно-восстановительного посредника и синергетической активности. Модифицированные электроды из никелевых сплавов получают широкое распространение благодаря их синергетическому эффекту и большому разнообразию составов, структур и свойств. Перестраиваемые химические и физические свойства сплавов Ni, обусловленные зависимой от состава структурой поверхности и поведением атомной сегрегации, являются перспективными для разработки новых катализаторов с лучшей активностью и селективностью [35, 36]. Электрохимические исследования электродов, модифицированных никелевыми сплавами, показали, что различные виды электроактивных веществ могут быть определены с высокой скоростью переноса электрона, хорошей чувствительностью и воспроизводимостью [37–40].
Насколько известно авторам, не было проведено никаких работ по определению SA с электродом Ni–Cr/GCE. В этой статье представлен амперометрический датчик SA на основе Ni–Cr/GCE. Электрод получают электроосаждением никеля и хрома на поверхность GCE, которая проявляет высокую каталитическую активность в отношении окисления SA. По сравнению с другими модифицированными электродами (см. табл. 1) Ni–Cr/GCE обладает такими преимуществами, как высокая электроаналитическая эффективность, легко возобновляемая поверхность, низкая стоимость и простота модификации. Эти достоинства делают довольно привлекательным альтернативный метод определения SA в реальных образцах.
Таблица 1.
Сравнение предложенного модифицированного электрода с другими электродами для определения салициловой кислоты
| Электрод | Способ | Чувствительность, мкА мМ–1 | Линейный диапазон, мкМ | DL, мкМ | Литература |
|---|---|---|---|---|---|
| SPE | SWV* | 1.20 | 16–300 | 5.6 | [15] |
| CFE | DPV** | – | 2–3000 | 1.68 | [16] |
| PNP/Pt | Амперометрия | 0.219 | 20–500 | 6.4 | [17] |
| Co/Al HTLC/ Pt | DPV* | 12.01 | 10–500 | 6 | [18] |
| Ce/ZrO2–CPE | SWV | – | 5–1000 | 1.1 | [19] |
| Ni–Cr/GCE | Амперометрия | 71.22 | 2–300 | 0.1 | Настоящая работа |
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты и материалы
Все химические вещества имели аналитическое качество, а растворы готовили с использованием дважды дистиллированной воды. NiSO4, Cr(NO3)3, салициловая кислота (99.5%), H3BO3 и NaOH приобретены у компаний Fluka и Merck. Фосфатные буферные растворы (0.1 М) готовили из H3PO4, NaH2PO4 и Na2HPO4. Значение pH доводили до желаемого уровня с помощью HCl и NaOH и контролировали с помощью цифрового измерителя pH (Hanna pH 211).
Электрохимические приборы и измерения
Электрохимические измерения выполнены в трехэлектродной ячейке с использованием потенциостата/гальваностата PGZ301 (Radiometer Analytical). Температуру регулировали на уровне 25 ± 0.5°C с помощью термостатированной ванны. Использовали модифицированный и неизолированный GCE (∅ = 3 мм) в качестве рабочих электродов, лист Pt в качестве вспомогательного электрода и Ag/AgCl с насыщенным KCl в качестве электрода сравнения. Электрохимическая импедансная спектроскопия (EIS) проведена в диапазоне частот (100 кГц–2.5 мГц) при потенциале 0.55 В. Каждое испытание повторяли три раза, чтобы подтвердить воспроизводимость результатов. Для стационарных амперометрических экспериментов рабочий потенциал фиксировали на уровне 0.55 В при умеренном перемешивании.
Модифицированный электрод
Электрод GCE полировали суспензией Al2O3 (1 и 0.4 мкм) и тщательно промывали водой до получения зеркальной поверхности. Затем его очищали ультразвуком (5 мин) в смеси HNO3 и абсолютного этанола (1 : 1), промывали водой и сушили при комнатной температуре. После этого электрод циклировали от 0 до +1 В в H2SO4 (0.1 М) со скоростью развертки 50 мВ с–1. Пленки Ni и Ni–Cr сначала электрохимически осаждали на поверхности GCE путем катодного восстановления из смешанного раствора {NiSO4 (0.01 M) + + H3BO3 (0.05 M)} в течение 90 с в отсутствие и в присутствии Cr3+ при различной молярной доле (Xf% = 9, 16, 28, 37 и 50) путем приложения потенциала –1.2 В. После промывания дистиллированной водой модифицированный электрод погружали в раствор NaOH (0.1 М) и циклировали в течение 10 циклов в пределах диапазона потенциалов от 0.25 до 0.75 В со скоростью развертки 50 мВ с–1 до достижения стабильной циклической вольтамперограммы (CV). С другой стороны, эти параметры экспериментально оптимизированы для полного превращения Ni(II) в Ni(III) и максимальной активации поверхности электрода для электрокаталитического окисления SA. Кристаллическую структуру идентифицируют с помощью дифракции рентгеновских лучей (XRD, дифрактометр Philips PW 1710) с использованием излучения CuKα (λ = 0.15417 нм). Сканирующая электронная микроскопия (SEM) выполнена с помощью микроскопа JOEL JSM 6360-LV.
Приготовление реального образца
Фармацевтические таблетки аспирина (ASPIRINE UPSA), приобретенные у компании Propharmal (Algiers), исследовали для определения ASA. Скорость разложения ASA до SA и уксусной кислоты (AA) зависит как от температуры, так и от pH. В диапазоне pH (11–12) ASA быстро гидролизуется, в то время как в диапазоне (4–8) ее гидролиз медленный, и максимальная стабильность ASA достигается при pH 2–3 [41]. Обычно ASA определяется косвенно после ее превращения в SA и AA путем щелочного гидролиза. Таблетки измельчали в агатовой ступке, и точную навеску растворяли в растворе NaOH (0.1 М) в течение 30 мин при 80°C, чтобы подвергнуть гидролизу. Образцы разбавляли, чтобы соответствовать линейному диапазону концентраций. Концентрацию ASA определяли путем интерполяции аналитической кривой, ранее построенной из стандартных растворов. Все растворы готовили с использованием воды сверхвысокого качества (Millipore).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Исследование электрода Ni–Cr/GC
На рис. 1a и 1б показаны изображения SEM электроосажденного сплава Ni и Ni–Cr с 28Xf% Cr3+. Электрод Ni–Cr имеет много полостей на своей поверхности и является более грубым, чем электрод Ni. Такая морфология увеличивает активную площадь поверхности, и, следовательно, доступно больше активных центров по сравнению с Ni/GCE. Химический анализ методом энергодисперсионной рентгеновской спектроскопии (EDX) (рис. 1в) показал, что композиционный материал содержит 76 мас. % Ni и 24 мас. % Cr. Рентгенограмма осадка показана на рис. 1г. Пики с центром при 2θ = 44.5°, 51.4° и 7.6° относятся к электроосажденному никелю, в то время как пики при 34.9°, 40.9°, 45.9°, 62° и 67.9° обусловлены хромом. Осадок имеет гранецентрированную кубическую структуру, ориентированную преимущественно в плоскостях (111) и (200). Присутствие Cr3+ не меняет кристаллическую структуру электроосажденного никеля (пространственная группа $Fm\bar {3}m$, N° 225), но влияет на интенсивность пиков. Кроме того, рисунок демонстрирует широкие пики, что позволяет предположить, что пленка Ni–Cr менее кристаллизована, что неудивительно, поскольку пленка образуется при температуре окружающей среды. Размер кристаллитов осадка Ni–Cr, оцененный по полной ширине на половине максимума (FWHM) наиболее интенсивного пика рентгеновских лучей, составляет 9 нм.
Рис. 1.
Изображения SEM поверхности электрода после электроосаждения сплава (a) Ni и (б) Ni–Cr с 28Xf% Cr3+. (в) Cпектр EDX сплава Ni–Cr(28Xf% Cr3+). (г) Рентгенограммы электроосажденных сплавов Ni и Ni–Cr.

На рис. 2 представлены циклические вольтамперограммы (CV) Ni/GCE и Ni–Cr/CGE (с различными Xf% Cr3+) в растворе NaOH (0.1 М), снятые после 10 циклов. Потенциал рабочего электрода снижается от 0.25 до 0.75 В в течение 10 циклов, чтобы способствовать получению равных количеств NiOOH. Электрохимическое поведение Ni–Cr/GCE аналогично Ni/GCE, где анодно-катодные пики свидетельствуют о поверхностных окислительно-восстановительных парах NiIII/NiII [27].
Рис. 2.
CV после 10 циклов Ni/GCE и Ni–Cr/GCE, полученных с различными Xf% Cr3+ (9, 28, 37 и 50Xf%) в 0.1 М NaOH при скорости развертки 50 мВ с–1. На вставке показаны увеличенные пиковые токи.

Сообщалось, что существует четыре формы никель-гидроксид-оксигидроксидного электрода [30, 31], а именно α-Ni(OH)2, β-Ni(OH)2, β-NiOOH и γ-NiOOH. γ-NiOOH восстанавливается в гидратированную фазу α-Ni(OH)2, которая нестабильна в сильной щелочи и стареет до β-формы, а β‑Ni(OH)2 окисляется в β-NiOOH. С другой стороны, ожидается, что фаза β-NiOOH будет хорошим электроактивным материалом для высоких электрохимических характеристик в щелочном растворе из-за более низкого внутреннего сопротивления и, следовательно, лучшей эффективности электрода. Примечательно, что в настоящей работе при увеличении Xf% наблюдается существенное отрицательное смещение формального потенциала и резкое увеличение тока сигнала. Наибольший ток сигнала обнаружен для 28Xf%, выше которого наблюдается регрессия. Напротив, окислительно-восстановительные пики NiIII/NiII более четко определены, а пиковые токи на Ni/GCE ниже, чем на Ni–Cr/GCE. Более высокий пиковый ток, наблюдаемый на Ni–Cr (28Xf%) электроде, ранее был приписан [42] более высокой степени разупорядоченности оксигидроксидного слоя, образовавшегося на поверхности рабочего электрода. Эти результаты также могут быть объяснены с учетом того, что электрохимическая площадь, полученная на электродах из сплава Ni–Cr, увеличивается по сравнению с чистым никелевым электродом. Результаты CV также показывают, что ток выделения O2 увеличивается с увеличением содержания Cr на Ni–Cr/GCE, что указывает на меньший избыточный потенциал O2 и/или большую удельную площадь поверхности. Подобная форма CV уже наблюдалась для объемных сплавов Ni–Cr [38]. Следовательно, добавление хрома или других металлов стабилизирует никелевый электрод. Более того, Marioli et al. [43] сообщили, что никелевые сплавы сохраняют электрохимические характеристики никеля.
Также исследовано влияние скорости развертки (v) на электрохимическое поведение Ni/CGE и Ni–Cr/GCE с различными значениями Xf% в растворе NaOH (рис. 3). Результаты показывают, что пиковые токи пропорциональны скорости развертки (v) в диапазоне от 10 до 75 мВ/с, что указывает на электрохимическую активность поверхностной окислительно-восстановительной пары [44]. Поверхностное заполнение (Γ) частиц Ni(III) на Ni–Cr/GCE оценивается по уравнению Лавирона [41].
где А – площадь поверхности электрода, F – постоянная Фарадея (95 487 Кл моль–1), n – число обмененных электронов, T – абсолютная температура и R – универсальная газовая постоянная. На рис. 3 (вставка) показано, что поверхностное заполнение Γ частиц Ni(III) увеличивается с увеличением Xf%. В отличие от результата, приведенного на рис. 2, можно сделать вывод, что с увеличением Xf% количество электроактивного Ni(III), нанесенного на электроды, увеличивается, что приводит к более высокой электрокаталитической активности. Меньшее значение Γ для Ni/GCE может быть связано с его слабой электроосажденной эффективностью и различной морфологией, т.е. меньшей удельной площадью поверхности. Для сравнения, наибольшее значение Γ обнаружено для Ni–Cr/GCE, приготовленного с 28Xf% Cr3+. На основании приведенных выше результатов это оптимальное значение Xf% выбрано для дальнейших исследований.
Электрокаталитическое окисление SA на модифицированном электроде
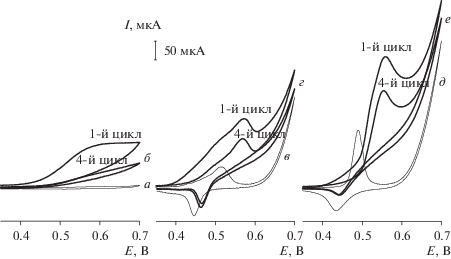
Электрокаталитическое поведение электродов Ni–Cr/GCE (28Xf%), Ni/GCE и GCE в отношении окисления SA изучали в растворе NaOH с использованием CV в диапазоне потенциалов от 0.25 до 0.70 В со скоростью развертки 50 мВ с–1. На рис. 4 показаны последовательные CV в отсутствие и в присутствии SA (10 мМ). Имеется необратимый пик окисления при 0.6 В для SA на GCE (кривая б), и пиковый ток значительно уменьшается с увеличением количества циклов. Такой результат ясно указывает на то, что SA обладает довольно высоким окислительным потенциалом и нестабильной реакцией на GCE. Напротив, на Ni/GCE (кривая в) и Ni–Cr/GCE (кривая д) наблюдается пара пиков, соответствующих обратимой окислительно-восстановительной реакции NiIII/NiII, в отсутствие SA. После добавления SA (кривые г и е) анодный пиковый ток значительно увеличивается, и отклик тока остается стабильным после четвертого цикла. Это окисление сопровождается уменьшением катодного пикового тока, что указывает на каталитический эффект пары NiIII/NiII в отношении окисления SA. Стабилизация пикового тока после четвертого цикла, вероятно, обусловлена стационарным состоянием контролируемых диффузией анодных процессов [18]. С другой стороны, Ni–Cr/GCE показывает самый отрицательный пиковый потенциал и самый высокий анодный пиковый ток, что указывает на то, что окисление SA происходит быстрее, чем на Ni/GCE. Этот усиленный отклик в основном обусловлен высокой шероховатостью поверхности Ni–Cr, что приводит к более высокой диффузии SA на пленке Ni(OH)2 и, таким образом, к лучшей электрокаталитической активности. Для сравнения, Ni–Cr/GCE больше подходит для обнаружения SA.
Рис. 4.
1-й и 4-й циклы чистого GCE (а и б), Ni/GCE (в и г) и Ni–Cr/GCE (д и е) в 0.1 М NaOH при отсутствии (а, б, д) и наличии (б, г, е) SA (10 мМ). Скорость развертки: 50 мВ с–1.
На рис. 5а приведены CV для анодного окисления SA (8 мМ) на Ni–Cr/GCE при различных скоростях развертки v от 5 до 300 мВ с–1. Ясно, что пиковый ток увеличивается с ростом v, в то время как пиковый потенциал смещается положительно. Ipa для окисления SA линейно возрастает с v1/2, как и ожидалось для реакции, контролируемой диффузией (вставка на рис. 5а). Наклон прямой линии (r = 0.996) зависимости Ipa от v1/2 составляет 29.40 мкА (мВ)–1/2 с1/2. С другой стороны, пиковый потенциал (Epa) смещается положительно при высоких скоростях развертки из-за уменьшения времени сканирования. Это показывает, что каталитическим током можно управлять с помощью кинетики каталитической реакции.
Рис. 5.
(а) CV на Ni–Cr/GCE в NaOH (0.1 М) в присутствии SA (8 мМ). Потенциальная скорость развертки: 5, 10, 15, 25, 50, 75, 100, 125, 150, 200, 250, 300 мВ с–1. Вставка: график зависимости Ipa от v1/2; (б) влияние pH на ток пика окисления и потенциал пика окисления SA. Скорость развертки: 50 мВ с–1.

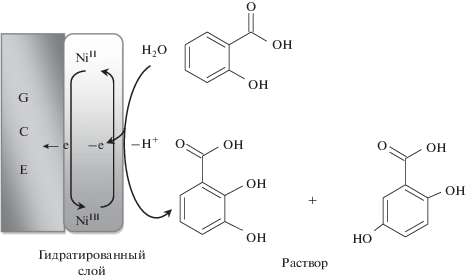
Влияние pH на окисление SA (10 мМ) на Ni–Cr/GCE исследовано с помощью циклической вольтамперометрии в диапазоне pH от 10 до 13.5 (рис. 5б). Более высокий анодный пик тока SA достигается при pH 13; поэтому это значение pH выбрано для дальнейших аналитических экспериментов. Кроме того, потенциал пика окисления (Epa) SA смещается отрицательно с увеличением pH от 10.0 до 13.5, как показывает следующее уравнение:
(5)
${{E}_{{\text{p}}}}\left( {{\text{мВ}}} \right) = 1.557 - 0.076{\text{pH}}\,\,\left( {{{r}^{2}} = 0.997} \right).$Согласно наклону уравнения Ep–pH, механизм окисления SA в водных средах включает в себя то же количество электронов и протонов, как описано в литературе [4, 6, 45]. Предложенный механизм электрохимической реакции SA показан на рис. 6.
Чтобы изучить воспроизводимость модификации электрода, эксперименты повторяют три раза. Профили CV модифицированного электрода регистрируют в растворе NaOH (50 мВ/с), и результаты почти одинаковы для всех испытаний. Измеряя пиковый ток анода, получают относительное стандартное отклонение (RSD), равное 4.5%, что указывает на хорошую повторяемость процесса модификации.
Хроноамперометрические исследования
Каталитическое окисление SA на Ni–Cr/GCE также изучено методом хроноамперометрии при 0.55 В в отсутствие и в присутствии SA в диапазоне концентраций от 0.2 до 1.4 мМ (рис. 7). Построение графика суммарного тока относительно времени в степени –1/2 показывает линейную зависимость, что указывает на преобладание диффузионно-контролируемого процесса. Коэффициент диффузии (D) SA может быть получен из наклона (рис. 7, вставка (а)) в соответствии с уравнением Коттрелла [44]:
(6)
$I = nFA{{D}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}{{c}_{{\text{o}}}}{{\left( {\pi t} \right)}^{{ - {1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}},$(7)
${{{{I}_{{{\text{cat}}}}}} \mathord{\left/ {\vphantom {{{{I}_{{{\text{cat}}}}}} {{{I}_{{\text{L}}}} = {{\gamma }^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}}}} \right. \kern-0em} {{{I}_{{\text{L}}}} = {{\gamma }^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}}}\left[ {{{\pi }^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}{\text{erf}}\left( {{{\gamma }^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}} \right) + \exp {{\left( { - \gamma } \right)} \mathord{\left/ {\vphantom {{\left( { - \gamma } \right)} {{{\gamma }^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}}}} \right. \kern-0em} {{{\gamma }^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}}}} \right],$(8)
${{{{I}_{{{\text{cat}}}}}} \mathord{\left/ {\vphantom {{{{I}_{{{\text{cat}}}}}} {{{I}_{{\text{L}}}} = {{\gamma }^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}{{\pi }^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}}}} \right. \kern-0em} {{{I}_{{\text{L}}}} = {{\gamma }^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}{{\pi }^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}}} = {{\pi }^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}{{\left( {k{{c}_{{\text{o}}}}t} \right)}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}.$Рис. 7.
Хроноамперограммы для Ni–Cr/GCE при отсутствии (кривая а) и наличии (кривые б–ж) SA в диапазоне концентраций (0.2–1.4 мМ). Вставка (а): график зависимости Icat от t–1/2; вставка (б): график зависимости Icat/IL от t 1/2.
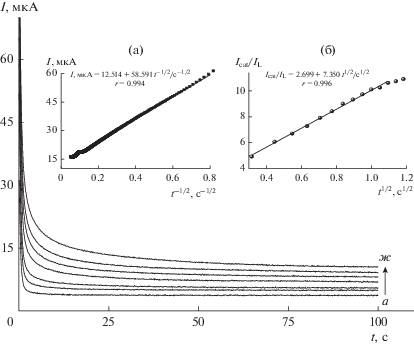
Из графика зависимости (Icat/IL) от t1/2 (рис. 7, вставка (б)), k составляет 1.43 × 104 М–1 с–1, что примерно в 3 раза больше, чем рассчитано для Ni/GCE (5 × 103 М–1 с–1).
Амперометрическое обнаружение SA на Ni–Cr/GCE
Чтобы оценить применимость Ni–Cr/GCE как селективной и чувствительной платформы, амперометрический отклик при последовательных добавлениях SA (с шагом 10 мкМ) регистрировали при 0.55 В в щелочном растворе (рис. 8). Отклик на Ni–Cr/GCE в 2.5 раза выше, чем на Ni/GCE, что указывает на то, что Ni–Cr/GCE обладает самой высокой электрокаталитической активностью в отношении окисления SA. Кроме того, отклик на Ni–Cr/GCE не уменьшается со временем. Ni–Cr/GCE показывает пропорциональное увеличение отклика тока на инжекцию SA, создавая устойчивые сигналы менее чем за 3 с. Кроме того, сигналы сопровождаются низким уровнем шума, что позволяет количественно определять SA со стабильной и эффективной каталитической способностью. Более интересно то, что отклик тока на Ni–Cr/GCE демонстрирует линейное поведение с концентрацией SA в широком диапазоне от 2 мкМ до 0.3 мМ с низким пределом обнаружения 0.1 мкМ (сигнал/шум = 3). Чувствительность Ni–Cr/GCE равна 71.22 мкА мМ–1 с коэффициентом корреляции 0.997 (вставка на рис. 8). Для более высоких концентраций (>0.3 мМ) отклик тока снижается из-за образования полимерной пленки на поверхности электрода. Это наблюдение соответствовало предыдущему сообщению [18, 19]. В начале окислительного потенциала SA молекулы SA окисляются и связаны друг с другом радикальными реакциями; образование малых молекул в результате радикальных реакций едва ли пассивирует поверхностную реакцию электрода. Дальнейшая цепная и разветвленная полимеризация димерных продуктов SA генерирует большие молекулы, которые прочно прилипают к поверхности электрода. Обычно известно, что эти пленки поли-SA связаны либо со связями, либо с углерод-углеродными связями и обладают плохой проводимостью, тем самым ингибируя процесс окисления SA [47].
Рис. 8.
Амперометрический отклик на GCE (кривая а), Ni/GCE (кривая б) и Ni–Cr/GCE (кривая в) при последовательных добавлениях SA (10 мкМ) в растворе NaOH (0.1 М). На вставке: калибровочный график SA для Ni–Cr/GCE, построенный при рабочем потенциале 0.55 В.

Таким образом, Ni–Cr/GCE проявляет превосходную электрокаталитическую активность в отношении электроокисления SA с повышенным током отклика по сравнению с Ni/GCE и GCE. Стабильный отклик без видимой потери чувствительности наблюдается при амперометрическом обнаружении в течение 25 мин. С другой стороны, получение Ni–Cr/GCE является простым и дешевым, и результаты, представленные в этой работе, являются благоприятными по сравнению с большинством описанных датчиков.
Исследование помех
Три потенциальных мешающих вещества со сходной структурой: бензойная кислота, аскорбиновая кислота и фенол, были использованы для оценки селективности Ni–Cr/GCE. Амперометрические отклики при Ni–Cr/GCE на добавление трех мешающих веществ в 0.1 М растворе NaOH (рН 13.0) при 0.55 В показаны на рис. 9. Значительного изменения токового отклика после добавления мешающих веществ не наблюдается. Напротив, ток заметно увеличивается после введения SA, что указывает на селективность реакции электрода в отношении SA.

Анализ реальных образцов
Чтобы проверить применимость предложенного электрохимического датчика, Ni–Cr/GCE использовали для титрования ASA в реальных фармацевтических образцах (ASPIRINE UPSA). Непрямое определение ASA основано на его гидролизе до SA, который впоследствии обнаруживается. SA имеет сильный пик поглощения при 294 нм, и УФ-спектрофотометрия используется в качестве контрольного теста для проверки способности Ni–Cr/GCE (табл. 2). Результаты анализа препарата с использованием предлагаемого способа находятся в хорошем соответствии с заявленными значениями и спектрофотометрическим анализом. Таким образом, эти результаты указывают на отсутствие помех от часто встречающихся фармацевтических наполнителей, используемых в выбранной композиции. Они демонстрируют, что Ni–Cr/GCE можно рекомендовать для чувствительного определения SA в таблетках, и открывают новые перспективы для других лекарств.
Таблица 2.
Определение ацетилсалициловой кислоты в промышленных фармацевтических образцах (ASIRINE UPSA с составом: ASA 0.330 г, аскорбиновая кислота 0.200 г)* методами амперометрии и спектрофотометрии
| Номер образца | Номинальное значение (г/таблетка) | Найдено ацетилсалициловой кислоты (г/таблетка) | |
|---|---|---|---|
| амперометрия | спектрофотометрия | ||
| 1 | 0.330 | 0.322 | 0.331 |
| 2 | 0.334 | 0.332 | |
| 3 | 0.343 | 0.332 | |
| 4 | 0.324 | 0.332 | |
| X ± SD | 0.332 ± 0.013 | 0.332 ± 0.005 | |
ВЫВОДЫ
В этой работе на стеклоуглеродном электроде была сформирована тонкая пленка из сплава никеля и хрома путем потенциостатического осаждения, которая была успешно исследована в отношении электроокисления салициловой кислоты в щелочных средах. Для сравнения, Ni–Cr/GCE показал более высокую каталитическую активность и лучшую устойчивость к электрохимическому окислению. Электрокаталитическая активность Ni–Cr/GCE была повышена за счет регулирования содержания Cr3+ в растворе для осаждения. Более высокая кинетика переноса заряда и лучшая константа скорости катализа были получены на сплаве Ni–Cr. Конечная поверхностная концентрация частиц гидроксида Ni зависит от присутствия хрома. Высокий беспорядок структуры слоя гидроксида Ni формируется на Ni–Cr по сравнению с Ni/GCE. Более конкретно, Ni–Cr/GCE (28Xf%) демонстрирует наилучшую производительность, с быстрым откликом, линейной зависимостью при оптимизированных условиях при широкой концентрации SA с низким пределом обнаружения 0.1 мкМ. Предложенная методология обеспечивает возможность подготовки электродов, высокую чувствительность, низкий предел обнаружения и короткое время измерения. Следовательно, метод может быть использован для создания датчика салициловой кислоты для практического применения.
Список литературы
Mikami, E., Goto, T., Ohno, T., Matsumoto, H., and Nishida, M., Simultaneous analysis of dehydroacetic acid, benzoic acid, sorbic acid and salicylic acid in cosmetic products by solid-phase extraction and high-performance liquid chromatography, J. Pharma. Biomed. Anal., 2002, vol. 28, p. 26.
Abdolmohammad-Zadeh, H., Kohansal, S., and Sadeghi, G.H., Nickel-aluminum layered double hydroxide as a nanosorbent for selective solid-phase extraction and spectrofluorometric determination of salicylic acid in pharmaceutical and biological samples, Talanta, 2011, vol. 84, p. 368.
Saha, U. and Baksi, K., Spectrophotometric determination of salicylic acid in pharmaceutical formulations using copper (II) acetate as a color developer, Int. J. Pharm., Analyst, 2001, vol. 23, p. 95.
Zhang, W.D., Xu, B., Hong, Y.-X., Yu, Y-X.,Ye, J.-S., and Zhang, J.-Q., Electrochemical oxidation of salicylic acid at well-aligned multiwalled carbon nanotube electrode and its detection, J. Solid State Electrochem., 2010, vol. 14, p. 1713.
Zhihua, W., Xiaole, L., Bowan, W., Fangping, W., and Xiaoquan, L., Voltammetric Determination of salicylic acid by molecularly imprinted film modified electrodes, Int. J. Polym. Anal. Ch., 2012, vol. 17, p. 122.
Supalkova, V., Petrek, J., Havel, L., Krizkova, S., Petrlova, J., Adam, V., Potesil, D., Babula, P., Beklova, M., Horna, A., and Kizek, R., Electrochemical sensors for detection of acetylsalicylic acid, Sensors, 2006, vol. 6, p. 1483.
Ruiz-Medina, A., Fernàndez-de Córdova, M. L., Ortega-Barrales, P., and Molina-Díaz, A., Flow-through UV spectrophotometric sensor for determination of (acetyl)salicylic acid in pharmaceutical preparations, Int. J. Pharm., 2001, vol. 216, p. 95.
Kokot, Z. and Burda, K., Simultaneous determination of salicylic acid and acetylsalicylic acid in aspirin delayed-release tablet formulations by second derivative UV spectropohotometry, J. Pharm. Biomed. Anal., 1998, vol. 18, p. 871.
Marcelo, M.S., Marcello, G.T., and Ronei, J.P., Combining standard addition method and second-order advantage for direct determination of salicylate in undiluted human plasma by spectrofluorimetry, Talanta, 2006, vol. 68, p. 1707.
Kakkar, T. and Mayersohn, M., Simultaneous quantitative analysis of methyl salicylate, ethyl salicylate and salicylic acid from biological fluids using gas chromatography–mass spectrometry, J. Chromatogr., B, 1998, vol. 718, p. 69.
Kees, F., Jehnich, D., and Grobecker, H., Simultaneous determination of acetylsalicylic acid and salicylic acid in human plasma by high-performance liquid chromatography, J. Chromatogr., B, 1996, vol. 677, p. 172.
Jen, J.F., Tsai, Y.Y., and Yang, T.C., Microdialysis of salicylic acid from viscous emulsion samples prior to high-performance liquid chromatographic determination, Chromatogr., A, 2001, vol. 912, p. 39.
Petrek, J., Havel, L., Petrlova, J., Adam, V., Potesil, D., Babula, P., and Kizek, R., Analysis of salicylic acid in willow barks and branches by an electrochemical method, Russ. J. Plant. Physiol., 2007, vol. 54, p. 553.
Torriero, A.A.J., Luco, J.M., Sereno, L., and Raba, J., Voltammetric determination of salicylic acid in pharmaceuticals formulations of acetylsalicylic acid, Talanta, 2004, vol. 62, p. 247.
Rawlinson, S., McLister, A., Kanyong, P., and Davis, J., Rapid determination of salicylic acid at screen printed electrodes, Microchemical J., 2018, vol. 137, p. 71.
Park, J. and Eun, C., Electrochemical Behavior and Determination of Salicylic Acid at Carbon-fiber Electrodes, Electrochim. Acta, 2016, vol. 194, p. 346.
Wang, Z., Ai, F., Xu, Q., Yang, Q., Yu, J. H., Huang, W.H., and Zhao, Y. D., Electrocatalytic activity of salicylic acid on the platinum nanoparticles modified electrode by electrochemical deposition, Colloids Surf., B, 2010, vol. 76, p. 370.
Gualandi, I., Scavetta, E., Zappoli, S., and Tonelli, D., Electrocatalytic oxidation of salicylic acid by a cobalt hydrotalcite-like compound modified Pt electrode, Biosens. Bioelectron., 2011, vol. 26, p. 3200.
Alizadeh, T. and Nayeri, S., Electrocatalytic oxidation of salicylic acid at a carbon paste electrode impregnated with cerium-doped zirconium oxide nanoparticles as a new sensing approach for salicylic acid determination, J. Solid State Electrochem., 2018, vol. 22, p. 1.
Doulache, M., Saidat, B., and Trari, M., Electrocatalytic performance of cobalt microparticles film-modified platinum disk electrode for amperometric detection of ascorbic acid, J. Anal. Chem., 2017, vol. 72, p. 333.
Wei, Y., Wanga, A., and Liu, Y., Development of a glassy carbon electrode modified with graphene/Au nanoparticles for determination of acetaminophen in pharmaceutical preparation, Russ. J. Electrochem., 2018, vol. 54, p. 1141.
Babu, K.J., Senthilkumar, N., Kim, A. R., and Kumar, G.G., Three-dimensional dendrite Cu–Co/reduced graphene oxide architectures on a disposable pencil graphite electrode as an electrochemical sensor for nonenzymatic glucose detection, Sens. Actuators B, 2017, vol. 241, p. 541.
Sanghavi, B.J., Kalambate, P.K., Karna, S.P., and Srivastava, A.K., Voltammetric determination of sumatriptan based on a graphene/gold nanoparticles/Nafion composite modified glassy carbon electrode, Talanta, 2014, vol. 120, p. 1.
Tadayon, F., Vahed, S., and Bagheri, H., Au–Pd/reduced graphene oxide composite as a new sensing layer for electrochemical determination of ascorbic acid, acetaminophen and tyrosine, Mater. Sci. Eng. C, 2016, vol. 68, p. 805.
Safaei, M., Beitollahib, H., and Shishehborea, M.R., Simultaneous Determination of Epinephrine and Folic Acid Using the Fe3O4@SiO2/GR Nanocomposite Modified Graphite, Russ. J. Electrochem., 2018, vol. 54, p. 851.
Zhao, C., Li, M., and Jiao, K., Determination of formaldehyde by staircase voltammetry based on its electrocatalytic oxidation at a nickel electrode, J. Anal. Chem., 2006, vol. 61, p. 1204.
Majdi, S., Jabbari, A., and Heli, H., Electrocatalytic oxidation of some amino acids on a nickel-curcumin complex modified glassy carbon electrode, J. Solid State Electrochem., 2007, vol. 11, p. 601.
Safavi, A., Maleki, N., and Farjami, E., Fabrication of a glucose sensor based on a novel nanocomposite electrode, Biosens. Bioelectron., 2009, vol. 24, p. 1655.
Roushani, M., Shamsipur, M., and Pourmortazavi, S.M., Amperometric detection of glycine, L-serine, and L‑alanine using glassy carbon electrode modified by NiO nanoparticles, J. Appl. Electrochem., 2012, vol. 42, p. 1005.
Jafarian, M., Forouzandeh, F., Danaee, I., and Gobal, F., Electrocatalytic oxidation of glucose on Ni and NiCu alloy modified glassy carbon electrode, J. Solid State Electrochem., 2009, vol. 13, p. 1171.
Danaee, I., Jafarian, M., Mirzapoor, A., Gobal, F., and Mahjani M.G., Electrooxidation of methanol on NiMn alloy modified graphite electrode, Electrochim. Acta, 2010, vol. 55, p. 2093.
Elahi, M. Y., Heli, H., Bathaie, S. Z., and Mousavi, M.F., Electrocatalytic oxidation of glucose at a Ni-curcumin modified glassy carbon electrode, J. Solid State Electrochem., 2007, vol. 11, p. 273.
Gholivanda, M.B., Pashabadi, A., Azadbakht, A., and Menati, S., A nano-structured Ni(II)–ACDA modified gold nanoparticle self-assembled electrode for electrocatalytic oxidation and determination of tryptophan, Electrochim. Acta, 2011, vol. 56, p. 4022.
Zheng, L. and Song, J.F., Nickel(II)-baicalein complex modified multiwall carbon nanotube paste electrode and its electrocatalytic oxidation toward glycine, Anal. Biochem., 2009, vol. 391, p. 56.
Zhong, D.C., Aranishi, K., Singh, A.K., Demirci, U.B., and Xu, Q., The synergistic effect of Rh–Ni catalysts on the highly-efficient dehydrogenation of aqueous hydrazine borne for chemical hydrogen, Chem. Commun., 2012, vol. 48, p. 1945.
Jiang, H.L. and Xu, Q., Porous metal-organic framewerks as platforms for function application, Mater. J. Chem., 2011, vol. 21, p. 3705.
Tao, B., Zhang, J., Hui, S., and Wan, L., An amperometric ethanol sensor based on a Pd–Ni/SiNWs electrode, Sens. Actuators, B, 2009, vol. 142, p. 298.
Mattos, I.L., Melo, D., and Zagatto, E.A.G., Nickel–chromium electrode as a detector in flow-injection amperometry: determination of glycerol, Anal. Sciences, 1999, vol. 15, p. 537.
Ojani, R., Raoof, J.B., and Norouzi B., Performance of glucose electrooxidation on Ni–Co composition dispersed on the poly (isonicotinic acid)(SDS) film, J. Solid State Electrochem., 2011, vol. 15, p. 1139.
Yeo, I.H. and Johnson, D.C., Electrochemical response of small organic molecules at nickel–copper alloy electrodes, J. Electroanal. Chem., 2001, vol. 495, p. 110.
Connors, K.A., Amidon, G.L., and Kennon, L., Chemical Stability of Pharmaceuticals, Wiley: New York, 1979, p. 151.
Marioli, J.M. and Sereno, L.E., The potentiodynamic behavior of nickel–chromium (80 : 20) alloy electrodes in 0.10 N sodium hydroxide, Electrochim. Acta, 1995, vol. 40, p. 983.
Marioli, J.M., Luo, P., and Kuwana, T., Nickel–chromium alloy electrode as a carbohydrate detector for liquid chromatography, Anal. Chim. Acta, 1993, vol. 282, p. 571.
Bard, A. J. and Faulkner, L.R., Electrochemical Methods: fundamentals and applications, John Wiley press, 2001, 2nd ed., p. 196.
Ghoreishi, S.M., Kashani, F.Z., Khoobi, A., and Enhessari, M., Fabrication of a nickel titanate nanoceramic modified electrode forelectrochemical studies and detection of salicylic acid, J. Mol. Liq., 2015, vol. 211, p. 970.
Kutner, W., Wang, J., Lher, M., and Buck, R.P., Analytical aspects of chemically modified electrodes: Classification, critical evaluation and recommendations, Pure Appl. Chem., 1998, vol. 70, p. 1301.
Park, J. and Eun, C., Electrochemical behavior and determination of salicylic acid at carbon-fiber electrodes, Electrochim. Acta, 2016, vol. 194, p. 346.
Дополнительные материалы отсутствуют.
Инструменты
Электрохимия