

Электрохимия, 2022, T. 58, № 9, стр. 551-568
Реакция восстановления кислорода на углеродных материалах, полученных из карбида хрома
Х. К. В. Нгуен a, Я. Нэрут a, Х. Касук a, В. Грозовский a, Т. Томберг a, И. Талло a, Р. Палм a, М. Коппель a, Т. Романн a, Р. Хярмас a, Я. Арувяли b, М. Кюлавиир b, Э. Луст a, *
a Институт химии Тартуского университета
Тарту, Эстония
b Институт экологии и наук о земле Тартуского университета
Тарту, Эстония
* E-mail: enn.lust@ut.ee
Поступила в редакцию 11.11.2021
После доработки 04.01.2022
Принята к публикации 20.01.2022
- EDN: SWUZTJ
- DOI: 10.31857/S0424857022090146
Аннотация
Углеродные материалы, полученные из карбида хрома, были синтезированы при различных температурах для использования в качестве возможных носителей катализатора в полимер-электролитных мембранных топливных элементах. Данные физико-химических исследований показали, что используемая температура синтеза существенно влияет на кристаллографическую структуру углерода, включая коэффициент графитизации, а также значения объемов микропор и мезопор. Электрохимические измерения в растворах KOH и HClO4 выявили емкостные свойства и пригодность изученных углеродных материалов в качестве носителей катализатора.
ВВЕДЕНИЕ
Возобновляемые источники энергии составили самую высокую долю в производстве первичной энергии – 29.9% в ЕС-28 в 2017 г. [1] . Полимерные электролитические мембранные топливные элементы (PEMFCs) – это устройства доминирующего типа среди других видов топливных элементов, и в последние годы PEMFCs генерировали наибольшее количество энергии [2] . Однако основными препятствиями, затрудняющими широкое распространение PEMFCs, являются долговечность и цена электроэнергии, вырабатываемой этими топливными элементами (FCs). В данный момент трудности также связаны с совершенствованием будущих катализаторов и их носителей.
Различные формы углеродных материалов, такие как углеродные нанотрубки, сажа, углеродные материалы на основе карбидов, графен, углеродные волокна и т.д., широко применяются в качестве носителей катализаторов для PEMFCs [3–5] . Морфология поверхности углеродов вносит значительный вклад в мощностные характеристики и долговечность разработанных FCs [6‒10] . Материалы-носители с оптимальной структурой и пористостью обеспечивают поверхность носителя для хорошей дисперсии металлического катализатора и также оказывают влияние на размер кристаллов металлического катализатора.
Поскольку широко доступный технический углерод имеет много проблем [11] , ученые ищут надежный источник углерода с прогнозируемыми долговечными и стабильными свойствами [4] . Микропористая поверхность обычно обеспечивает большее количество активных центров для реакции восстановления кислорода (ORR) и, следовательно, повышает производительность FC [12] . Впрочем, многие мезопористые углероды с большим количеством кислородных функциональных групп на поверхности увеличивают дисперсность катализатора за счет лучшего связывания углеродной поверхности и металлического катализатора [13] .
Углеродные материалы, синтезированные из различных карбидов металлов [14–18] , так называемые углеродные материалы на основе карбидов (CDC), широко изучались, поскольку можно контролировать их пористость, морфологию и уровень графитизации [19–29]. Температура синтеза и параметры исходного карбида металла оказывают большое влияние на структуру и кристалличность синтезированных углеродных материалов [14–18, 30]. Карбид хрома(II) является хорошим претендентом для получения микро- и мезопористых углеродных материалов [30], поскольку хлорирование этого бинарного карбида приводит к равномерному распределению атомов углерода в решетке и высокому соотношению количества микропор к мезопорам, большой удельной площади поверхности и хорошо развитому объему пор в полученном материале [31]. Таким образом, углеродные материалы на основе карбидов хрома (Cr-CDCs), полученные при различных условиях синтеза, по-видимому, являются перспективными материалами как носители катализатора с контролируемым размером пор, высокой удельной площадью поверхности и высокой пористостью, что может способствовать приросту скорости электрохимической окислительно-восстановительной реакции.
Чтобы предсказать характеристики несущих материалов в FCs, работающих на водороде и воздухе, необходимо изучить временнýю стабильность материала-носителя и кинетику ORR, поскольку это может существенно повлиять на производительность FC [15, 32–35]. В водном электролите ORR может быть либо прямым четырехэлектронным процессом, либо двухэлектронным процессом [36]. ORR в щелочной среде можно описать следующей общей химической реакцией (стандартные значения потенциала приведены по сравнению со стандартным водородным электродом) [36].
(1)
${{{\text{O}}}_{2}} + 2{{{\text{H}}}_{{\text{2}}}}{\text{O}} + 4{\text{e}} \to 4{\text{O}}{{{\text{H}}}^{ - }}\,\,\,\,\,E^\circ = 0.401\,\,{\text{В}}.$Возможен и другой путь:
(2)
${{{\text{O}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} + 2{\text{e}} \to {\text{HO}}_{2}^{ - }\, + {\text{O}}{{{\text{H}}}^{ - }}\,\,\,\,E^\circ = - 0.065\,\,{\text{В,}}$(3)
${\text{HO}}_{2}^{ - } + {{{\text{H}}}_{{\text{2}}}}{\text{O}} + 2{\text{e}} \to 3{\text{O}}{{{\text{H}}}^{ - }}\,\,\,\,\,E^\circ = 0.867\,\,{\text{В,}}$ORR в кислом растворе может быть описана следующим общим уравнением химической реакции [36]:
(5)
${{{\text{O}}}_{2}} + 4{{{\text{H}}}^{ + }} + 4{\text{e}} \to 2{{{\text{H}}}_{{\text{2}}}}{\text{O}}\,\,\,\,\,E^\circ = 1.229\,\,{\text{В}}$(6)
${{{\text{O}}}_{2}} + 2{{{\text{H}}}^{ + }} + 2{\text{e}} \to {{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{2}}\,\,\,\,\,E^\circ = 0.67\,\,{\text{В}}{\text{.}}$Перекись водорода восстанавливается или разлагается до воды [36]:
(7)
${{{\text{H}}}_{2}}{{{\text{O}}}_{2}} + 2{{{\text{H}}}^{ + }} + 2{\text{e}} \to 2{{{\text{H}}}_{{\text{2}}}}{\text{O}}\,\,\,\,\,E^\circ = 1.77\,\,{\text{В,}}$(8)
${\text{2}}{{{\text{H}}}_{2}}{{{\text{O}}}_{2}} \to 2{{{\text{H}}}_{{\text{2}}}}{\text{O}} + {{{\text{O}}}_{2}}.$Cr-CDCs были синтезированы из Cr3C2 при фиксированных температурах хлорирования. Физико-химическая характеристика Cr-CDCs была проведена для получения характеристик пористости, морфологии, стабильности и химического состава. Электрохимическое поведение (ORR, емкостные свойства, стабильность и данные импеданса) Cr-CDCs было исследовано в двух электролитах (раствор 0.1 моль дм–3 HClO4 и раствор 0.1 моль дм–3 KOH) с использованием методов циклической вольтамперометрии (CV), вращающегося дискового электрода (RDE) и электрохимической импедансной спектроскопии (EIS).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез углеродных материалов, полученных из карбида хрома
Cr3C2 (99.5% чистота, порошок –325 меш, Sigma-Aldrich) использовался в качестве прекурсора CDC в этом исследовании. Детали синтеза были разработаны и обсуждены ранее Thomberg и др. [30]. Четыре различных образца Cr-CDC были получены при температурах хлорирования 800, 900, 1000 и 1100°C. Однако характеристики полученных материалов несколько отличались по сравнению с исследованием Thomberg и др., вероятно из-за того, что использовалась другая партия карбида [37]. Кроме того, процесс очистки смесью H2/Ar (1 : 4) при 800°C проводился на 0.5 ч дольше, чем в предыдущем исследовании [30]. Упрощенное уравнение реакции процесса хлорирования [30]:
(9)
$2{\text{C}}{{{\text{r}}}_{{\text{3}}}}{{{\text{C}}}_{2}} + 9{\text{C}}{{{\text{l}}}_{2}} \to 6{\text{CrC}}{{{\text{l}}}_{3}} + 4{\text{C}}.$Материалы обозначаются как Cr-CDC (температура синтеза).
Методы физико-химической характеризации материалов
Для исследования пористости приготовленного Cr-CDC был использован низкотемпературный (–195.8°C) метод сорбции азота [38] с помощью прибора 3FLEX (Micromeritics, США). Расчет значений удельной площади поверхности SBET был выполнен в соответствии с теорией многоточечной адсорбции Брунауэра–Эммета–Теллера (BET) [39] в диапазоне относительных давлений от 0.05 до 0.2. Данные о распределении пор по ширине были оценены программным обеспечением SAIEUS [40] (v2.02, Micromeritics, США) с использованием метода, называемого моделью нелокальной теории функционала плотности “Углерод-N2-77, неоднородная поверхность 2D-NLDFT” [41]. Общий объем пор Vtot определялся количеством адсорбированного газа в точке относительного давления перед стадией конденсации, которая находится вблизи точки давления насыщения. Для расчета объема микропор Vmicro был использован метод t-графика [42, 43]. Удельная площадь поверхности SDFT и объем пор VDFT также были получены из данных распределения ширины пор, рассчитанных с помощью модели 2D-NLDFT. Принимая во внимание, что метод BET является стандартным методом расчета удельной площади поверхности и широко используется для характеризации пористых материалов, нужно учитывать, что ему присущи проблемы с переоценкой удельной площади поверхности микро- и ультра микропор [44]. Таким образом, значения SDFT приводятся для сравнения, что дает вероятный диапазон для наиболее реалистичной удельной площади поверхности, доступной для N2.
Для измерения дифрактограмм использовался усовершенствованный дифрактометр Bruker D8 с CuKα-излучением, пропущенным через Ni-фильтр (параллельный луч шириной 0.6 мм, две щели Соллера под углом 2.5° и детектор LynxEye). Применялся шаг сканирования 0.01° для 2θ в промежутке от 10° до 90°. Общее время подсчета на каждый шаг составляло 166 с.
Измерения сканирующей электронной микроскопии (SEM) проводились с использованием системы Zeiss EVO MA15 с переменным давлением. Образцы были покрыты слоем платины толщиной 5–10 нм с использованием ионного распылителя Leica EM SCD500, работающего в высоком вакууме. Изображения SEM были получены в режиме обратного рассеяния электронов, и детальная химическая характеристика образцов была проведена с помощью энергодисперсионного детектора рентгеновской спектроскопии Oxford AZTEC-MAX (EDX), подключенного к устройству SEM. Спектры были получены в режиме высокого вакуума с использованием ускоряющего напряжения 20 кэВ и сфокусированного электронного пучка. Измеренные спектры были обработаны программным обеспечением Aztec.
Термогравиметрический анализ (TGA) материалов проводили с помощью прибора NETZSCH STA449F3 с использованием чаши из Al2O3. Применяемый диапазон температур варьировался от 25 до 1000°C. Использовалась скорость нагрева 10 К мин–1 и расход газа был 50 см3 мин–1. Масса образца составляла 10 мг. В этих экспериментах использовались азот (99.999%, Linde Gas) и кислород (99.999%, Linde Gas).
Рамановская спектроскопия проводилась с помощью аргонового лазера (λ = 514 нм) и Рамановского спектрометра Renishaw. Спектры возбуждения были получены в различных точках материалов. Все приведенные спектры являются усредненными спектрами наиболее аморфных пятен.
Электрохимические измерения
Подготовка электродов. Стеклоуглеродный дисковый электрод (GCDE), т.е. рабочий электрод носитель (d = 5 см), был отполирован 0.05 мкм суспензией оксида алюминия на водной основе (Buehler Inc., США). Отполированный электрод промывали водой Milli-Q (18.2 Мом см–1 при 25°C) до и после обработки ультразвуком в ультразвуковой ванне в течение 30 с.
Навеска углеродного материала составляла 1 мг см–2, а отношение иономера к углероду (вес/вес) составляло 0.07, использовался иономер Nafion® 117 (~5%-ный раствор, Sigma-Aldrich). Раствор изопропанола (>99%, Sigma-Aldrich) и воды Milli-Q (1 : 4, по весу) с углеродным материалом и иономером обрабатывали ультразвуком с помощью ультразвукового рупора (ультразвуковой процессор Vibra-Cell VCX 500 Sonics and Materials, Inc.) в течение 30 с при амплитуде мощности 35%, на ледяной бане. Капля суспензии объемом 7 мкл наносилась на GCDE.
Электрохимические измерения. Посуда и платиновые электроды были залиты раствором горячей (80°C) концентрированной серной кислоты (конц. 95–97%, Sigma-Aldrich, ч. д. а., ACS) с добавлением нескольких капель перекиси водорода (конц. 30 мас. %, Perhydrol®, Merck). После этого его промывали деминерализованной и Milli-Q водой.
Электрохимические измерения проводились при температуре 22°C в трехэлектродной системе. Все углеродные материалы были исследованы в двух растворах электролитов: 0.1 моль дм–3 HClO4, приготовленных из 70% HClO4 (99.999%, повторно дистиллированная, Sigma-Aldrich), и 0.1 моль дм–3 KOH, приготовленного из гранул KOH (полупроводниковый сорт, гранулы, 99.99% на базе рассеянных элементов, Sigma-Aldrich).
В качестве электрода сравнения использовался динамический водородный электрод [45]. Все потенциалы показаны относительно этого электрода, и он обозначен как RHE. Электрод сравнения был подключен к ячейке через капилляр Луггина. Вспомогательный электрод представлял собой платиновую сетку с высокой площадью поверхности, которая была изолирована от основного измерительного отсека ячейки перемычкой со стеклянной мембраной.
Все электрохимические измерения проводились с помощью потенциостата Autolab PGSTAT204 (Metrohm, Нидерланды) и ротатора фирмы PINE Instrument.
Перед электрохимическими измерениями слой катализатора на GCDE был стабилизирован путем его электрохимического циклирования в диапазоне измеряемых потенциалов при 800 об/мин. Циклы подготовки проводились поочередно в атмосфере Ar (99.9999%, Linde Gas) и в атмосфере O2 (99.999%, Linde Gas) до тех пор, пока поверхность рабочего электрода не была полностью смочена и стабилизирована.
EIS выполнялась при 0.8 В (ΔE = 10 мВRMS, 0.1 ≤ f ≤ 10 000 Гц, 10 точек на декаду). Во время измерений CV и RDE потенциал корректировался с учетом сопротивления электролита, измеренного с помощью EIS.
Испытание на стабильность проводили с Cr-CDC (1000°C) в растворе KOH 0.1 моль дм–3. В первый день были проведены измерения CV и RDE. Затем система циклировалась при 5 мВ с–1 в течение 24 ч (180 циклов) в условиях покоя, в атмосфере Ar, протекающего над раствором. Эта процедура повторялась в течение следующих 6 дней.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Физико-химическая характеризация
Измерения низкотемпературной сорбции N2. Результаты измерений низкотемпературной сорбции N2 приведены в табл. 1 и на рис. 1. Значения удельной площади поверхности и общего объема пор зависят от температуры синтеза материала. Наблюдается огромная разница в удельной площади поверхности, общем объеме пор и данных о распределении пор по размерам при повышении температуры синтеза с 800 до 900°C. Таким образом, материалы можно разделить на две группы: группа по типу микропористого Cr-CDC (800°C) и группа микро- и мезопористого углеродного материала, представленная остальными углеродными материалами. По сравнению с данными предыдущего исследования, где использовался тот же метод синтеза и такие же прекурсоры карбида металла [30], Cr-CDC (800°C) в данной работе имеет в 8.5 раз более высокое значение площади удельной поверхности. Даже если текущее значение SDFT синтезированных материалов сравнить с SBET Cr-CDC (800°C) в [30], то разница составит 6 раз. Следует подчеркнуть, что процесс хлорирования карбида (в настоящем исследовании) проводился очень медленно, чтобы обеспечить полноту реакции. Таким образом, было предоставлено достаточно времени для того, чтобы хлориды хрома диффундировали из структуры, и первоначальная структура карбида была сохранена и не разрушена в ходе интенсивного процесса синтеза углеродного материала, проведенного Thomberg и др. [30]. Повышение температуры синтеза Cr-CDC способствовало образованию микро- и мезопористых углеродов (рис. 1).
Таблица 1.
Результаты измерения низкотемпературной сорбции азота для углеродов, полученных из карбида хрома (Cr-CDCs)
| Материал/ параметр | SBET, м2 г−1 | SDFT, м2 г−1 | Smicro, м2 г−1 | Smeso, м2 г−1 | Smicro, SBET | Vtot, см3 г−1 | VDFT, см3 г−1 | Vmicro, см3 г−1 | Vmeso, см3 г−1 |
|---|---|---|---|---|---|---|---|---|---|
| Cr-CDC (800°C) | 2287 | 1632 | 2233 | 55 | 0.98 | 1.14 | 1.14 | 1.14 | 0 |
| Cr-CDC (900°C) | 238 | 206 | 42 | 196 | 0.18 | 0.39 | 0.35 | 0.02 | 0.37 |
| Cr-CDC (1000°C) | 311 | 273 | 78 | 233 | 0.25 | 0.65 | 0.66 | 0.03 | 0.62 |
| Cr-CDC (1100°C) | 252 | 227 | 77 | 175 | 0.30 | 0.72 | 0.72 | 0.03 | 0.69 |
Примечание. SBET, SDFT – удельная площадь поверхности в соответствии с методом BET и в соответствии с моделью NLDFT, Smicro – площадь микропор, Smeso – площадь мезопор, Vtot, VDFT – общий объем пор, рассчитанный на основе адсорбированного количества вблизи давления насыщения и с использованием модели NLDFT, Vmicro – объем микропор, Vmeso – объем мезопор.
Рис. 1.
Распределение углеродных материалов (Cr-CDCs), полученных из карбида хрома (Cr-CDCs), по ширине пор, рассчитанное по данным модели низкотемпературной сорбции азота “Гетерогенная поверхность 2D-NLDFT”.
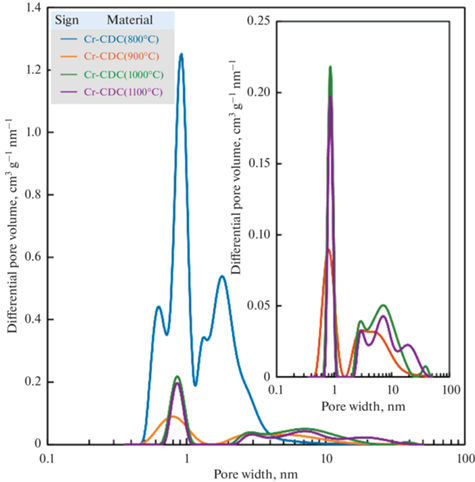
Размеры микропор для образцов Cr-CDCs распределяются вокруг значения 1 нм (рис. 1). Ширина мезопор для Cr-CDC (800°C) распределяется в диапазоне от 2 до 4 нм, в то время как для других Cr-CDCs ширина мезопор обширно распределена в пределах от 2 до 20 нм.
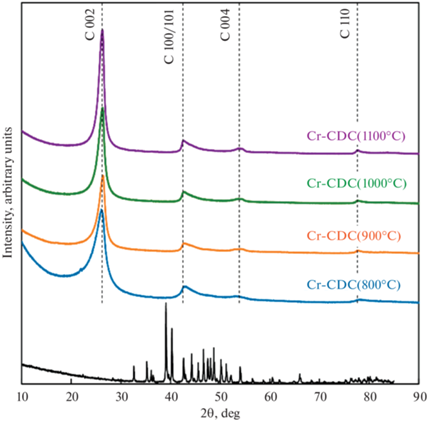
Дифракция рентгеновских лучей. Дифрактограмма для прекурсора Cr3C2, приведенная на рис. 2, представляет характерные пики карбида хрома [37, 46]. Появление этих острых пиков соответствует орторомбическим структурам решеток Cr3C2. Полученные Cr-CDC являются аморфными, и дифрактограммы для Cr-CDC, представленные пиками Брэгга, соответствующими плоскостям C(002), C(100/101), C(004) и C(110) при 2θ, наблюдаются примерно под углами 26°, 43°, 54° и 78°. Уровень графитизации немного увеличивается с повышением температуры синтеза, т.е. пик C(002) становится более острым, переходя от Cr-CDC (800°C) к Cr-CDC (1100°C). Вероятно, что большая часть прекурсора Cr3C2 прореагировала на стадии синтеза. Существует небольшой неопознанный пик примерно при 22° в случае Cr-CDC (800°C), который может быть обусловлен побочным продуктом синтеза, например, непрореагировавшими фазами CrO(OH) (брацевеллита) или/и оксида хрома(III).
Рис. 2.
Дифрактограмма прекурсора Cr3C2 и углеродных материалов, полученных из карбида хрома (Cr-CDCs).
Рамановская спектроскопия. По различию стадий графитизации на рис. 3а, как определено Schuepher и др. [47], Cr-CDC (800°C) может быть классифицирован как углеродный материал II стадии (по типу наночастиц). Материалы Cr-CDC (1000°C) и Cr-CDC (1100°C) уже относятся к материалам III стадии (неграфитовым) с узкими D- и G-полосами и присутствующей D'-полосой на 1620 см–1. Можно сказать, что углеродный материал Cr-CDC (900°C) находится между стадиями II и III, поскольку его спектр несколько напоминает спектры для Mo2C‑CDC (1100°C) [14, 48].
Рис. 3.
Рамановские спектры углеродных материалов, полученных из карбида хрома (Cr-CDCs) (а). Деконволюция спектров Cr-CDC (800°C) (б) и Cr-CDC (1000°C) (в).

Спектры материалов Cr-CDC (800°C) и Cr-CDC (900°C) были успешно разложены с помощью подхода, предложенного Sadezky и др. [49], который содержит пять функций аппроксимации (рис. 3б). Спектры углеродных материалов, полученные при 1000 и 1100°C, больше не содержали полос расположенных при ~1100 и 1500 см–1, и одним из физических объяснений было то, что эти полосы аналогичны D- и G-полосам, но происходят из сильно неупорядоченных областей [50]. Таким образом, спектры Cr-CDC (1000°C) и (1100°C) были разложены на три функции, соответствующие D-, G- и D'-полосам соответственно (рис. 3в).
Из параметров в табл. 2, которые описывают графитизацию, видно, что Cr-CDC (800°C) является наиболее неупорядоченным материалом. Это особенно заметно по ширине D-полосы, которая значительно шире для Cr-CDC (800°C) по сравнению с другими CDCs. Материалы Cr-CDC (1000°C) и Cr-CDC (1100°C) весьма схожи с точки зрения характеристик графитизации. Уменьшение отношения ID/IG можно интерпретировать как увеличение упорядоченности углеродного материала для этих образцов CDC, поскольку D- и G-полосы там относительно узкие.
Таблица 2.
Параметры, выведенные из рамановских спектров для углеродных материалов, полученных из карбида хрома (Cr-CDC)
| Tsyn, °C | ID/IG | FWHMD, см−1 | FWHMG, см−1 | SΣD/SΣG | SΣD/SΣG+D’ |
|---|---|---|---|---|---|
| 800 | 0.93 | 125.4 | 59.3 | 1.90 | 1.51 |
| 900 | 0.44 | 64.8 | 41.8 | 0.76 | 0.71 |
| 1000 | 0.16 | 52.2 | 26.7 | 0.33 | 0.30 |
| 1100 | 0.10 | 47.6 | 23.7 | 0.20 | 0.20 |
Хотя Cr-CDC (1000°C) и Cr-CDC (1100°C) являются умеренно графитными, теоретический минимум ширины G-полосы ~15 cм–1, характерный для высокоориентированного пиролитического графита, не был достигнут [51, 52].
Сканирующая электронная микроскопия с энергодисперсионной рентгеновской спектроскопией. Изображения SEM на рис. 4 свидетельствуют о том, что макроскопические и мезоскопические структуры углеродного материала были сохранены. Морфология полученных Cr-CDC не зависит от температуры синтеза. Изображения SEM для всех материалов показывают наличие крупных частиц углерода (площадью примерно 100 мкм2), а также мелких частиц углерода (площадью менее 1 мкм2).
Рис. 4.
Изображения со сканирующего электронного микроскопа и состав, полученный из данных энергодисперсионной рентгеновской спектроскопии (EDX) углеродных материалов, полученных из карбида хрома (Cr-CDCs).

Результаты EDX показывают ничтожные количества хрома, кремния и хлора во всех Cr-CDCs. Cr-CDC (800°C) содержит даже несколько процентов кислорода. Впрочем, некоторое количество кислорода, скорее всего, присутствует и в других углеродах. Этот качественный анализ согласуется с результатами рентгеновского анализа. Присутствие кислорода в материале Cr-CDC (800°C) наиболее очевидно, поскольку удельная площадь поверхности этого материала намного выше, и структура может содержать больше дефектов, что облегчает модификацию поверхности (хемосорбцию кислорода) атмосферным кислородом. Присутствие некоторого количества кремния во всех материалах может быть вызвано стационарным реактором с кварцевым слоем, используемым для синтеза углеродного материала [30].
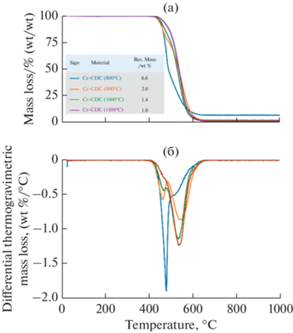
Термогравиметрия. Результаты TGA приведены на рис. 5. Все образцы содержат очень небольшое количество воды (около 0.3 мас. %), которая испаряется при температурах вплоть до 100°C. После этого термогравиметрические кривые не изменяются до тех пор, пока температура не достигнет 400°C. Двухступенчатый процесс окисления происходит в диапазоне от 400 до 650°C, что отображается двумя хорошо выраженными пиками на дифференциальных термогравиметрических кривых. Первый процесс, который начинается примерно при 470°C, – это окисление аморфных углеродных структур [53]. Второй пик, наблюдаемый примерно при 530°C, является результатом окисления более графитизированных областей углеродного материала [53]. Эти выводы подтверждаются результатами в дефрактограммах и в рамановской спектроскопии (рис. 2 и 3). Существует некоторая остаточная масса, которая может состоять из следового количества соединений кварца и хрома, в основном наблюдаемых у Cr-CDC (800°C). Количество остаточных соединений меньше для материалов, синтезированных при более высокой температуре, поскольку реакция хлорирования протекает быстрее и полнее. Количество кварца также можно было бы уменьшить, так как время хлорирования материалов, синтезированных при более высоких температурах, было короче. Наблюдаемое количество загрязняющих веществ согласуется с результатами EDX (рис. 4).
Результаты электрохимических измерений
Циклическая вольтамперометрия. Гравиметрические емкости (C, Ф г–1), приведенные на рис. 6, были рассчитаны как C = I/(mv), где m (г) – масса слоя материала (без Nafion), I (A) – измеренный ток, а v (В с–1) – скорость развертки потенциала.
Рис. 6.
Гравиметрические емкости для углеродных материалов, полученных из карбида хрома (Cr-CDCs), рассчитанные по результатам циклической вольтамперометрии в растворах 0.1 моль дм–3 KOH и 0.1 моль дм–3 HClO4, насыщенных аргоном, при скорости развертки потенциала 100 мВ с–1 (а), с увеличением на вставке. Зависимость емкости от скорости развертки потенциала продемонстрирована для Cr-CDC (800°C) в растворах 0.1 моль дм–3 KOH и 0.1 моль дм–3 HClO4 (б). Вставки показывают зависимость удельного тока от скорости развертки потенциала при различных потенциалах. Результаты испытаний на стабильность Cr-CDC (1000°C) в растворе 0.1 моль дм–3 KOH показаны на рисунке (в), на котором показана гравиметрическая емкость в течение 6 дней при скоростях развертки потенциала 100 и 10 мВ с–1.
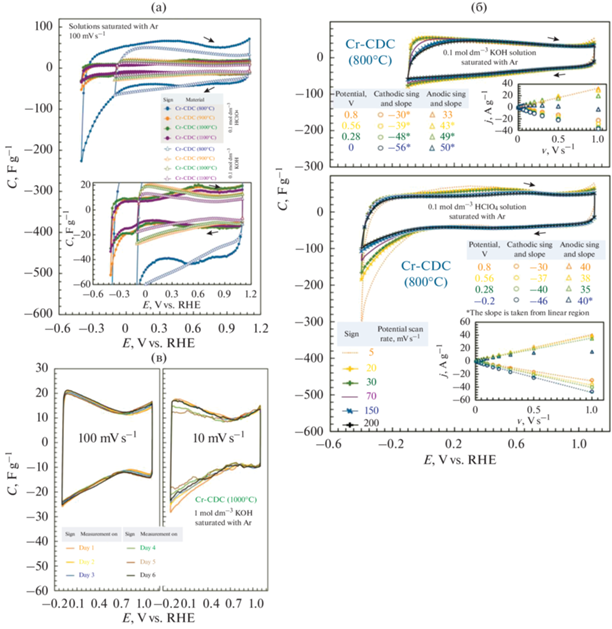
Кривые гравиметрической емкости, измеренные в растворе 0.1 моль дм–3 HClO4, имеют искаженную форму, т.е. C,E-кривые не являются прямоугольными. Поверхности электродов CDCs в кислой и щелочной среде начали окисляться при потенциалах более положительных, чем 0.9 В. Соответственно, плотность тока начинает увеличиваться при этих потенциалах. Окисление поверхности происходит очень медленно, и оно более выражено для наиболее аморфного Cr-CDC (800°C), который также имеет наибольшую удельную площадь поверхности.
В кислой среде происходит восстановление поверхностных >C=O-групп, и пики на C, E-кривых можно увидеть в области потенциалов от 0.55 до 0.6 В [54]:
(10)
$\rangle C = {\text{O}} + {{{\text{H}}}_{3}}{{{\text{O}}}^{ + }} + {\text{e}}\begin{array}{*{20}{c}} {\xrightarrow{{{\text{ Charge }}}}} \\ {\xleftarrow[{{\text{Discharge}}}]{}} \end{array}{\text{ }} \geqslant C{\text{--OH}}\left( {{{{\text{H}}}_{2}}{\text{O}}} \right),$В растворе 0.1 моль дм–3 KOH кривые зарядки/разрядки материалов CDC имеют искаженную прямоугольную форму [55]. Лимитирующее поведение медленной адсорбции обусловлено слабой специфической адсорбцией и образованием поверхностного комплекса [55, 56].
(11)
$\rangle C = {\text{O}} + {{\left[ {{\text{K}}{{{\left( {{{{\text{H}}}_{{\text{2}}}}{\text{O}}} \right)}}_{n}}} \right]}^{ + }} + {\text{e}}\begin{array}{*{20}{c}} {\xrightarrow{{{\text{ Charge }}}}} \\ {\xleftarrow[{{\text{Discharge}}}]{}} \end{array}{\text{ }} \geqslant C{\text{--OK}}{{\left( {{{{\text{H}}}_{2}}{\text{O}}} \right)}_{n}}.$Поскольку электрохимическая емкость углеродного материала зависит от специфической адсорбции ионов K+ или от накопленных зарядов в двойном электрическом слое (EDL), а также от псевдоемкостной реакции, модифицированные углеродные материалы могут достигать более высокой удельной емкости за счет псевдоемкостных эффектов [57]. Гравиметрическая емкость Cr-CDC (800°C) в области EDL при 0.8 В в 4–5 раз выше (53 и 35 Ф г–1 в 0.1 моль дм–3 HClO4 и 0.1 моль дм–3 KOH, соответственно). Эти высокие значения можно объяснить гораздо более высокой удельной площадью поверхности и более выраженными псевдоемкостными эффектами по сравнению с Cr-CDC, синтезированными при более высоких температурах. В области потенциала нулевого заряда C,E-кривые, измеренные в растворе 0.1 моль дм–3 HClO4, имеют более высокие значения емкости, чем в щелочных средах. По сравнению с измерениями в растворе 0.1 моль дм–3 HClO4, более низкие значения емкости в щелочных средах связаны с блокировкой поверхности из-за специфической адсорбции ионов K+ на электродах Cr-CDC.
Следует отметить, что было невозможно полностью смочить все поры в углеродном материале без выполнения циклов кондиционирования CV, достигая значительных отрицательных потенциалов, где происходило выделение водорода. Из-за этого циклы кондиционирования для всех материалов были расширены до области выделения водорода. После надлежащей подготовки значения емкости увеличились примерно в 2–3 раза. Выделение водорода в растворе 0.1 моль дм–3 HClO4 было наиболее выраженным для Cr-CDC (800°C), и наблюдалось уже при потенциалах меньших чем 0 В (рис. 6а). Для менее активных материалов выделение водорода начиналось при более отрицательных потенциалах (E < –0.2 В). Электрохимическая активность Cr-CDC в отношении выделения водорода, как правило, была выше в щелочной среде, чем в кислой (рис. 6а).
Электрод с нанесенным материалом Cr-CDC (800°C) был выбран для демонстрации зависимости значений емкости от скорости развертки потенциала (рис. 6б). В растворе 0.1 моль дм–3 HClO4, насыщенном аргоном, анодный пик смещается в сторону менее положительных потенциалов по мере увеличения скорости развертки потенциала. Однако значения емкости в области потенциала нулевого заряда не зависят от скорости развертки потенциала. Такое же явление наблюдается в растворах 0.1 моль дм–3 KOH, насыщенных аргоном. Значения емкости при катодной поляризации в щелочных средах более похожи на те, как при анодной поляризации, поскольку данные эксперименты проводятся при менее отрицательных потенциалах. Это явление указывает на влияние состояния поверхности электрода (морфологии) на адсорбцию/десорбцию реагентов при более высоких скоростях развертки потенциала.
Влияние поверхности электрода и реагентов более наглядно показано на вставках на рис. 6б. Зависимость (анодного и катодного) удельных токов от скорости развертки потенциала в обеих средах линейна при низких скоростях развертки потенциала (≤150 мВ с–1) и начинает отклоняться при более высоких скоростях развертки потенциала (≥200 мВ с–1). Наклон графика j, v при фиксированных применяемых удельных потенциалах подтверждает идею процессов, лимитированных стадией адсорбции/десорбции на поверхности электрода. В растворе 0.1 моль дм–3 KOH значения наклона для анодной и катодной поляризационной кривой при используемом значении потенциала одинаковы. Однако в растворе 0.1 моль дм–3 HClO4 значения наклона более различны. Это указывает на разницу в специфическом поведении адсорбции/десорбции реагентов двух растворов электролитов.
Для испытания на стабильность углеродный материал Cr-CDC (1000°C) исследовали в растворе 0.1 моль дм–3 KOH. Данные на рис. 6в показывают, что плотности тока не зависят от применяемого числа циклов, а кривые CV при скорости развертки потенциала 100 мВ с–1 остаются неизменными в течение 6-дневного цикла. Это указывает на отсутствие разрушения и отсутствие потери носителя катализатора с поверхности электрода в течение 6-дневного эксперимента. На рис. 6в кривые CV при скорости развертки потенциала 10 мВ с–1 изменяются очень незначительно в области менее положительных потенциалов (E < 0.7 В) по сравнению с кривой CV после однодневного цикла. Это явление может быть вызвано наличием толстого слоя оксида, накопленным после 6 дней на поверхности углеродного материала, который трудно восстановить.
Измерения CV проводились также в электролитах, насыщенных кислородом. Значения тока с поправкой на фоновую плотность тока (jc/A м–2) были рассчитаны для получения сведений об ORR в исследуемых материалах:
(12)
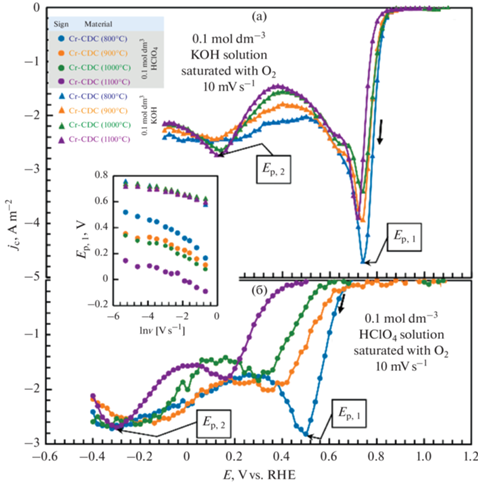
${{j}_{{\text{c}}}} = {{\left( {{{I}_{{{\text{oxygen}}}}} - {{I}_{{{\text{background}}}}}} \right)} \mathord{\left/ {\vphantom {{\left( {{{I}_{{{\text{oxygen}}}}} - {{I}_{{{\text{background}}}}}} \right)} S}} \right. \kern-0em} S},$В CV есть два необратимых восстановительных пика тока, характерных для ORR (рис. 7). Первый пик Ep, 1 при более положительных потенциалах соответствует ORR в процессе переноса двух электронов с образованием перекиси (уравнения (2), (6)). В качестве второй стадии (второй пик на jc,E-кривых) перекись водорода преобразуется в конечные продукты (уравнения (3), (4), (7), (8)) при менее положительном потенциале, чем Ep, 2. Почти линейная зависимость Ep, 1 от ln v, продемонстрированная на вставке рис. 7, указывает на процесс, лимитированный скоростью (стадией) адсорбции на поверхности электрода [58]. Перенапряжение ORR в растворе 0.1 моль дм–3 меньше по сравнению с таковым в растворе 0.1 моль дм–3 HClO4. Для материала Cr-CDC (800°C) разница в пиковых значениях потенциала высока (почти 0.24 В), и это значение еще выше для других материалов Cr-CDC. Последний аспект, вероятно, связан с различным состоянием функциональных групп кислорода на поверхности углерода в кислотной и щелочной средах. Интересно, что в кислых растворах состояние поверхности в большей степени зависит от температуры синтеза углеродного материала. В растворе 0.1 моль дм–3 KOH (рис. 7а) потенциал первого пика восстановления Ep, 1 почти не зависит от исследуемого материала. Однако в растворе 0.1 моль дм–3 HClO4 (рис. 7б), потенциал первого пика восстановления Ep, 1 сильно смещен в сторону более отрицательных значений (∆Ep, 1 = = 0.34 В, вставка на рис. 7) с увеличением температуры синтеза Cr-CDC-материалов, т.е. с увеличением кристалличности CDC активность ORR уменьшается. Второй пик Ep, 2 появляется примерно на 0.45 В при более отрицательных потенциалах в кислых средах по сравнению с щелочными средами, пики более плоские, а значение Ep, 1, а также Ep, 2, не зависит в значительной степени от материала в кислом растворе.
Рис. 7.
Плотности тока реакции восстановления кислорода с поправкой на фоновую плотность тока при 10 мВ с–1 для углеродных материалов, полученных из карбида хрома (Cr-CDCs), в растворах 0.1 моль дм–3 KOH (а) и 0.1 моль дм–3 HClO4 (б), насыщенных кислородом. Зависимость положения потенциала первого пика в ORR от скорости развертки показана на вставке.
Влияние скорости развертки потенциала на ORR продемонстрировано на рис. 8. На рис. 8а, первое пиковое значение потенциала Ep, 1 в растворе 0.1 моль дм–3 KOH изменилось на 110 мВ в сторону более отрицательного значения потенциала при увеличении скорости развертки потенциала с 5 до 200 мВ с–1. Однако изменение положения первого пика более выражено в растворе 0.1 моль дм–3 HClO4 (∆Ep, 1 = 0.22 V), поскольку константа скорости ORR намного ниже в кислых условиях, рис. 8б.
Рис. 8.
Влияние скоростей развертки потенциала на вольтамперограммы для углеродного материала, полученного из карбида хрома, Cr-CDC (800°C), в растворах 0.1 моль дм–3 KOH (а) и 0.1 моль дм–3 HClO4 (б), насыщенных кислородом. Вставки на рисунках показывают зависимость плотности тока первого пика от скорости развертки потенциала для исследуемых материалов.
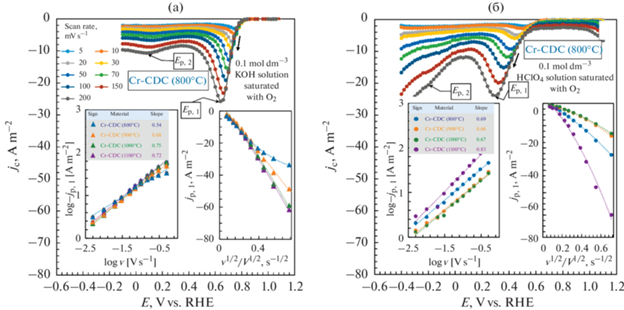
Зависимость первой пиковой плотности тока –jp, 1 от v1/2 на рис. 8а отклоняется от линейной зависимости для исследуемых материалов как в кислой, так и в щелочной среде. Плотности тока –jp, 1 при более высоких скоростях развертки выше по сравнению с ожидаемыми значениями, рассчитанными из уравнения для необратимого процесса переноса электронов [58]. Таким образом, происходит процесс со смешанной кинетикой, поскольку слой катализатора довольно толстый, можно ожидать отклонений от полубесконечной линейной диффузии, в связи с чем необходимо также учитывать процесс массопереноса в слое Nafion и внутри пор углеродного материала. Еще одной сложностью является геометрически шероховатая поверхность, которая вызывает отклонение плотности тока от однородного распределения на поверхности электрода. Плотности тока также были нанесены на логарифмические координаты как lg |–jp, 1| против lg v (вставка на рис. 8). Эти зависимости были линейными, а значения наклона были выше 0.5 (от 0.66 до 0.75), что указывало на отклонение системы в сторону процессов с лимитированной стадией адсорбции. Только для наиболее активного и аморфного материала Cr-CDC (800°C) значение наклона (0.54) в растворе 0.1 моль дм–3 KOH приближается к теоретическому значению, то есть, вероятно, что поверхность слоя катализатора была очень активной в отношении ORR, и скорость массопереноса в углеродной матрице была уже не столь важна.
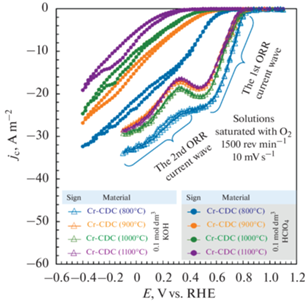
Измерения на вращающемся дисковом электроде. Результаты на рис. 9 явно подтверждают, что Cr-CDC были электрохимически более активны по отношению к ORR в щелочной среде. Активность ORR для Cr-CDCs в кислой среде возрастает вместе с развитием аморфной структуры (рис. 2) и увеличением удельной поверхности (табл. 1). Материалы Cr-CDC (900°C) и Cr-CDC (1100°C) обладают примерно одинаковыми значениями удельной площади поверхности и каталитической активностью ORR. Материал Cr-CDC (800°C), имеющий гораздо более высокое значение SBET, демонстрирует наибольшую активность по отношению к ORR в обеих изученных средах. В растворе 0.1 моль дм–3 HClO4 потенциалы начала реакции у исследуемых материалов уменьшаются вместе с повышением температуры синтеза исследуемых углеродных материалов. В растворе 0.1 моль дм–3 HClO4 нет плато тока, что указывает на невозможность достижения диффузионно-лимитирующего режима в используемом диапазоне потенциалов. Иначе происходит в 0.1 моль дм–3 растворе KOH, где потенциалы начала реакции гораздо более положительны, а влияние характеристик материалов менее выражено. В щелочных средах можно выделить двухэтапные процессы. В области около 0.5 В можно увидеть первый пик тока, где доминирует двухэлектронный процесс с образованием перекиси водорода (уравнение (2)). При менее положительных потенциалах перекись водорода дополнительно восстанавливается до OH–-ионов в области второй волны тока (E < < 0.3 В) (уравнения (3), (4)). На стеклоуглеродном электроде в 0.1 моль дм–3 растворе NaOH Taylor и Humffray [59] наблюдали при –0.3 и –0.6 В (относительно Ag/AgCl-электрода в насыщенном KCl) две волны при скорости развертки от 10 до 60 мВ с–1. Обе волны толковались как образование перекиси водорода (уравнение (2)), но в разных местах реакции. Таким образом, в разных кристаллографических областях (рис. 5) предполагалось, что восстановление/разложение перекиси до воды не происходило (уравнения (3), (4)) даже при умеренно отрицательных потенциалах, превышающих –1.3 В (относительно Ag/AgCl-электрода в насыщенном KCl). Все исследованные углеродные материалы являются очень пористыми по сравнению со стеклоуглеродом, а Cr-CDCs содержат незначительное количество примесей хрома. Следовательно, Cr-CDCs более активны, и, по-видимому, происходит совмещение (перекрытие) 2- и 4-электронных процессов.
Рис. 9.
Плотности тока реакции восстановления кислорода с поправкой на фон при 1500 об/мин для углеродных материалов, полученных из карбида хрома (Cr-CDCs), в растворах 0.1 моль дм–3 HClO4 и 0.1 моль дм–3 KOH при 10 мВ с–1.
В эксперименте RDE процесс ORR лимитирован переносом заряда, массопереносом и значением сопротивления пленки [9, 60]. Последний связан с диффузией реагентов в слое Nafion. Однако, по оценкам, толщина слоя Nafion очень мала или даже не влияет на лимитированные плотности тока ORR [61, 62]. Следовательно, в первом приближении может быть применено уравнение Коутецкого–Левича (K–L) [60, 63] в классической форме:
(13)
${1 \mathord{\left/ {\vphantom {1 {{{j}_{{\text{c}}}}}}} \right. \kern-0em} {{{j}_{{\text{c}}}}}} = {1 \mathord{\left/ {\vphantom {1 {{{j}_{{\text{k}}}}}}} \right. \kern-0em} {{{j}_{{\text{k}}}}}} + {1 \mathord{\left/ {\vphantom {1 {{{j}_{{\text{d}}}}}}} \right. \kern-0em} {{{j}_{{\text{d}}}}}},$(14)
$\begin{gathered} {1 \mathord{\left/ {\vphantom {1 {{{j}_{{\text{c}}}}}}} \right. \kern-0em} {{{j}_{{\text{c}}}}}} = \\ = {1 \mathord{\left/ {\vphantom {1 {\left( { - nF{{k}_{{{\text{het}}}}}{{c}_{{{{{\text{O}}}_{2}}}}}} \right)}}} \right. \kern-0em} {\left( { - nF{{k}_{{{\text{het}}}}}{{c}_{{{{{\text{O}}}_{2}}}}}} \right)}} + {1 \mathord{\left/ {\vphantom {1 {\left( { - 0.62nFD_{{{{{\text{O}}}_{2}}}}^{{{2 \mathord{\left/ {\vphantom {2 3}} \right. \kern-0em} 3}}}{{\vartheta }^{{ - {1 \mathord{\left/ {\vphantom {1 6}} \right. \kern-0em} 6}}}}{{c}_{{{{{\text{O}}}_{2}}}}}{{\omega }^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}} \right)}}} \right. \kern-0em} {\left( { - 0.62nFD_{{{{{\text{O}}}_{2}}}}^{{{2 \mathord{\left/ {\vphantom {2 3}} \right. \kern-0em} 3}}}{{\vartheta }^{{ - {1 \mathord{\left/ {\vphantom {1 6}} \right. \kern-0em} 6}}}}{{c}_{{{{{\text{O}}}_{2}}}}}{{\omega }^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}} \right)}}, \\ \end{gathered} $На графике Тафеля, в растворе 0.1 моль дм–3 KOH, имеются почти линейные области для различных Cr-CDCs, если значения плотности кинетического тока –jk малы (рис. 10a). Значения наклона графика Тафеля находятся в диапазоне от –120 до –129 мВ, что является приемлемыми значениями для толстых, пористых и шероховатых электродов [34]. Значения наклона графика Тафеля около –120 мВ в щелочном электролите соответствуют 2-электронному механизму. Аналогичное поведение обсуждалось для графитового электрода и объяснялось участием хиноновых групп, доступных на поверхности CDC. Основываясь на литературных данных, лимитирующая стадия O2(ads) + e → [O2(ads)]– была предположена и объяснена общим процессом 2-электронного восстановления кислорода с образованием ${\text{HO}}_{2}^{ - }$ [66]. Кроме того, наклоны Тафеля для GC-электрода отличаются и составляют около –60 мВ [66]. В 0.1 моль дм–3 KOH при потенциалах от 0.45 до 0.60 В значение n близко к двум (вставки на рис. 10). Это значение увеличивается до 2.5–3.0 при менее положительных потенциалах, т.е. перекись водорода частично переходит в гидроксид (так называемый механизм 2 + 2). Yeager и др. [67] утверждали, что вторая стадия (уравнения (3), (4)) может быть активирована/инициирована в пористом углеродном электроде достаточно активным катализатором разложения перекиси, как видно в нашем текущем случае (например, наличие соединений хрома в углеродной матрице и большое количество кромок на поверхности углерода). Количество электронов, переносимых в щелочных средах, сильно зависит от удельной площади поверхности, т.е. от характеристик поверхности катализатора. Таким образом, четко видно, что дальнейшее восстановление перекиси более вероятно в аморфном материале Cr-CDC (800°C) с высокой удельной площадью поверхности и с высокой концентрацией каталитически активных поверхностных дефектов.
Рис. 10.
Графики Тафеля для реакции восстановления кислорода на углеродных материалах, полученных из карбида хрома (Cr-CDCs), в 0.1 моль дм–3 KOH (а) и 0.1 моль дм–3 HClO4 (б). На вставке показано количество электронов, переносимых на одну молекулу кислорода, рассчитанное по графикам Коутецкого–Левича.
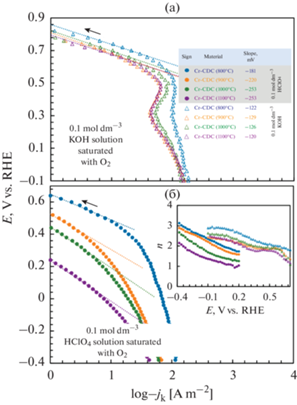
В растворе 0.1 моль дм–3 HClO4 в области низкой плотности кинетического тока –jk потенциал начала ORR смещается в сторону менее положительных значений, потому что количество графитизированной структуры в материалах увеличивается. Наиболее активный материал по отношению к ORR – это Cr-CDC (800°C), а другие материалы можно рассматривать как низко активные или неактивные материалы (рис. 9 и 10б). Увеличение активности ORR также коррелирует с увеличением удельной площади поверхности и количества побочных продуктов в Cr-CDC. Согласно результатам Taylor и Humffray [68], полученным для ORR на стеклоуглеродном электроде в насыщенном кислородом растворе, наклон линейной области графика Тафеля по расчетным данным (анодная поляризация) в 0.5 М уксусной кислоты и 0.5 М буфера ацетата натрия (рН 3.0–4.2) составляет приблизительно 140 мВ. Величина наклона, вызванная выделением водорода, составляла 220 мВ до того, как в раствор был введен газообразный кислород [68]. Наклоны участка на графике Тафеля для исследуемых материалов Cr-CDC довольно высоки, однако это не является чем-то необычным в кислых средах [35]. Эти наклоны на графике Тафеля, указывают на то, что энергия активации в кислом растворе значительно выше, чем в щелочном. Кривизна графиков Тафеля скорее всего вызвана отклонениями процессов от этапа лимитирования массопереноса в сторону процесса, лимитированного адсорбцией, в массивном электроде. На количество электронов, переносимых в кислых средах, по-видимому, существенно влияет количество аморфного углерода в материале электрода. Активность ORR для материала Cr-CDC (1100°C) в 0.1 моль дм–3 HClO4 и количество переносимых электронов достаточно низкое (всего 2 электрона, переносимых при –0.4 В). Этот материал содержит наибольшее количество графитизированных углеродных частиц, которые менее активны по отношению к ORR, чем частицы аморфного углерода с большим числом кромок [67, 69]. Число электронов, переносимых в случае материалов Cr-CDC (от 800 до 1000°C), составляет около 2 в области первого пика (рис. 7) и увеличивается до 3 при менее положительных потенциалах. Увеличение числа электронов, переносимых при менее положительных потенциалах, может быть вызвано образованием воды (уравнения (7), (8)). Количество электронов, переносимых в обоих электролитах, типично для немодифицированных, т.е. неактивированных углеродных материалов [15, 36, 69].
Электрохимическая импедансная спектроскопия. Результаты EIS отображаются в виде графиков фазового угла Боде (–φ относительно lg f) и графиков модуля импеданса (графики lg |Z| относительно lg f) (рис. 11), где f – частота (Гц), tg φ = = Z "/Z ' и модуль |Z| = (Z '2 + Z "2)1/2 (Z ' – действительная и Z " – мнимая часть импеданса). Значения сопротивлений электролита, Rel, (f → ∞) определялись по реальной части полного импеданса в области высоких частот. Были рассчитаны сопротивления электролита примерно 5.2 и 10.5 Ом см2 в растворах 0.1 моль дм–3 HClO4 и 0.1 моль дм–3 KOH соответственно.
Рис. 11.
Графики фазового угла и модуля Боде для углеродных материалов, полученных из карбида хрома (Cr-CDCs), при 0.8 В в растворах 0.1 моль дм–3 KOH (а) и 0.1 моль дм–3 HClO4 (б), насыщенных аргоном.
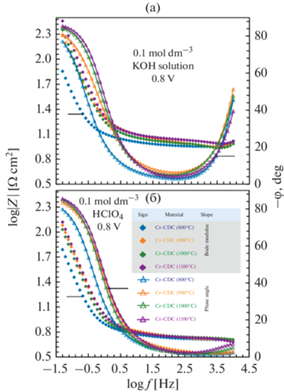
На рис. 11 фазовый угол, –φ, приближается к 83° в обеих средах – такое поведение типично для процесса с ограниченной передачей заряда [70]. Постоянная времени (τ = RelCEDL, где CEDL емкость EDL) для системы увеличивается по порядку: Cr‑CDC (1100°C) < Cr‑CDC (900°C) < < Cr‑CDC (1000°C) < Cr‑CDC (800°C) в растворе 0.1 M KOH и Cr‑CDC (1100°C) < Cr‑CDC (1000°C) < < Cr‑CDC (900°C) < Cr‑CDC (800°C) в растворе 0.1 моль дм–3 HClO4. Этот порядок согласуется с удельной площадью поверхности (табл. 1) и значениями емкости, рассчитанными при 0.8 В, используя кривые CV (рис. 6а).
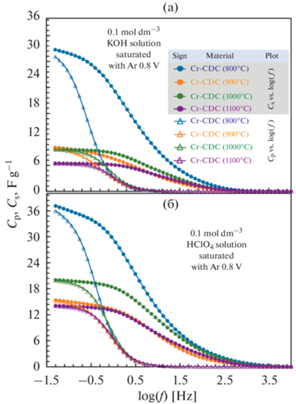
Зависимости емкостей последовательной цепи Cs и параллельной цепи Cp от lg f были рассчитаны с использованием формул Cs = –1/(2πfZ ") и Cp = –Z "/(2πf|Z|) и показаны на рис. 12 [70]. Характерные для емкостных материалов значения Cs и Cp совпадают на очень низких частотах как в кислых, так и в щелочных растворах. Значения емкостей при 0.8 В соответствовали значениям емкости, рассчитанным по данным CV, и емкость увеличилась в том же порядке (рис. 6а). Значения емкости, рассчитанные по данным EIS для Cr-CDC (800°C) в 0.1 моль дм–3 HClO4, немного выше по сравнению со значениями емкости, рассчитанными по данным CV. Вероятно, это связано с тем, что равновесие адсорбции не установилось, если метод CV применялся при v ≥ 10 мВ с–1.
ЗАКЛЮЧЕНИЕ
Различные углеродные материалы, полученные из карбида хрома (Cr-CDCs), были синтезированы и систематически изучены с использованием физических и электрохимических методов. Было установлено, что повышение температуры синтеза создает более мезопористую структуру и увеличивает количество графитированного углерода в CDC, но уменьшает удельную площадь поверхности Cr-CDC. Согласно изображениям SEM, применяемая температура синтеза очень незначительно повлияла на макроскопическую и мезоскопическую морфологию поверхности углерода.
Удельная площадь поверхности, количество аморфного углерода и следовое количество соединений хрома в синтезированных углеродах Cr-CDC влияют на электрохимическое поведение материалов CDC. Активность по отношению к ORR для материалов Cr‑CDCs в растворах 0.1 моль дм–3 KOH и 0.1 моль дм–3 HClO4, в основном протекает с помощью 2 + 2-электронного процесса. Кинетика ORR намного быстрее в растворе 0.1 моль дм–3 KOH.
Материал Cr-CDC (800°C) обладал наибольшей удельной площадью поверхности, SBET = = 2287 м2 г–1 (вероятно, немного преувеличено) и SDFT = 1632 м2 г–1. Он имеет наиболее аморфную структуру и в основном представляет собой микропористый углерод CDC. Электрохимическая активность этого Cr-CDC (800°C) как в кислой, так и в щелочной среде является самой высокой среди изученных материалов. Его гравиметрическая емкость, измеренная EIS в области EDL (53 и 35 Ф г–1 в кислой и щелочной средах соответственно), была сопоставима со значениями емкости для других материалов CDC [17, 24, 33, 71]. Другим интересным материалом является Cr-CDC (1100°C), который имеет микро- и мезопористую структуру, он слегка графитизирован и показывает наибольшее количество мезопор. В растворе 0.1 моль дм–3 HClO4, этот CDC проявлял умеренную активность к ORR и относительно низкую емкость. Все исследованные углеродные материалы обладают хорошей электрохимической стабильностью и могут быть использованы в качестве материалов-носителей катализатора для FC в обеих средах.
Список литературы
Energy Statistics – An Overview, Eurostat, 2019. https://ec.europa.eu/eurostat/statistics-explained/index.php?title=Energy_statistics_-_an_overview (accessed July 1, 2020).
The Fuel Cell Industry Review, E4tech, 2019. https://www.e4tech.com/news/2018-fuel-cell-industry- review-2019-the-year-of-the-gigawatt.php (accessed July 1, 2020).
Antolini, E., Carbon supports for low-temperature fuel cell catalysts, Appl. Catal. B-Environ., 2009, vol. 88, p. 1.
Samad, S., Loh, K.S., Wong, W.Y., Lee, T.K., Sunarso, J., Chong, S.T., and Wan Daud, W.R., Carbon and non-carbon support materials for platinum-based catalysts in fuel cells, Int. J. Hydrogen Energ., 2018, vol. 43, p. 7823.
Corti, H., Direct alcohol fuel cells: materials, performance, durability and applications. N.Y.: Springer, 2013. 370 p.
Sepp, S., Vaarmets, K., Nerut, J., Tallo, I., Tee, E., Kurig, H., Aruväli, J., Kanarbik, R., and Lust, E., Enhanced stability of symmetrical polymer electrolyte membrane fuel cell single cells based on novel hierarchical microporous-mesoporous carbon supports, J. Solid State Electr., 2016, p. 1.
Sepp, S., Vaarmets, K., Nerut, J., Tallo, I., Tee, E., Kurig, H., Aruväli, J., Kanarbik, R., and Lust, E., Performance of Polymer Electrolyte Membrane Fuel Cell Single Cells Prepared Using Hierarchical Microporous-Mesoporous Carbon Supported Pt Nanoparticles Activated Catalysts, Electrochim. Acta, 2016, vol. 203, p. 221.
Vaarmets, K., Nerut, J., Sepp, S., Kanarbik, R., and Lust, E., Accelerated Durability Tests of Molybdenum Carbide Derived Carbon Based Pt Catalysts for PEMFC, J. Electrochem. Soc., 2017, vol. 164, p. F338.
Lust, E., Vaarmets, K., Nerut, J., Tallo, I., Valk, P., Sepp, S., and Härk, E., Influence of specific surface area and microporosity-mesoporosity of pristine and Pt-nanoclusters modified carbide derived carbon electrodes on the oxygen electroreduction, Electrochim. Acta, 2014, vol. 140, p. 294.
Tae Hwang, J. and Shik Chung, J., The morphological and surface properties and their relationship with oxygen reduction activity for platinum-iron electrocatalysts, Electrochim. Acta, 1993, vol. 38, p. 2715.
Sharma, S. and Pollet, B.G., Support materials for PEMFC and DMFC electrocatalysts – A review, J. Power Sources, 2012, vol. 208, p. 96.
Kim, M., Soo Kim, H., Jong Yoo, S., Cheol Yoo, W., and Sung, Y.-E., The role of pre-defined microporosity in catalytic site formation for the oxygen reduction reaction in iron- and nitrogen-doped carbon materials, J. Mater. Chem. A, 2017, vol. 5, p. 4199.
Liang, C., Li, Z., and Dai, S., Mesoporous Carbon Materials: Synthesis and Modification, Angew. Chem. Int. Edit, 2008, vol. 47, p. 3696.
Jänes, A., Thomberg, T., Kurig, H., and Lust, E., Nanoscale fine-tuning of porosity of carbide-derived carbon prepared from molybdenum carbide, Carbon, 2009, vol. 47, p. 23.
Härk, E., Nerut, J., Vaarmets, K., Tallo, I., Kurig, H., Eskusson, J., Kontturi, K., and Lust, E., Electrochemical impedance characteristics and electroreduction of oxygen at tungsten carbide derived micromesoporous carbon electrodes, J. Electroanal. Chem., 2013, vol. 689, p. 176.
Jänes, A., Thomberg, T., and Lust, E., Synthesis and characterisation of nanoporous carbide-derived carbon by chlorination of vanadium carbide, Carbon, 2007, vol. 45, p. 2717.
Tallo, I., Thomberg, T., Kurig, H., Kontturi, K., Jänes, A., and Lust, E., Novel micromesoporous carbon materials synthesized from tantalum hafnium carbide and tungsten titanium carbide, Carbon, 2014, vol. 67, p. 607.
Schlange, A., dos Santos, A.R., Hasse, B., Etzold, B.J.M., Kunz, U., and Turek, T., Titanium carbide-derived carbon as a novel support for platinum catalysts in direct methanol fuel cell application, J. Power Sources, 2012, vol. 199, p. 22.
Gogotsi, Y., Nikitin, A., Ye, H., Zhou, W., Fischer, J.E., Yi, B., Foley, H.C., and Barsoum, M.W., Nanoporous carbide-derived carbon with tunable pore size, Nat. Mater., 2003, vol. 2, p. 591.
Gogotsi, Y., Portet, C., Osswald, S., Simmons, J.M., Yildirim, T., Laudisio, G., and Fischer, J.E., Importance of pore size in high-pressure hydrogen storage by porous carbons, Int. J. Hydrogen Energ., 2009, vol. 34, p. 6314.
Chmiola, J., Yushin, G., Gogotsi, Y., Portet, C., Simon, P., and Taberna, P.L., Anomalous Increase in Carbon Capacitance at Pore Sizes Less Than 1 Nanometer, Science, 2006, vol. 313, p. 1760.
Yushin, G., Hoffman, E.N., Barsoum, M.W., Gogotsi, Y., Howell, C.A., Sandeman, S.R., Phillips, G.J., Lloyd, A.W., and Mikhalovsky, S.V., Mesoporous carbide-derived carbon with porosity tuned for efficient adsorption of cytokines, Biomaterials, 2006, vol. 27, p. 5755.
Becker, P., Glenk, F., Kormann, M., Popovska, N., and Etzold, B.J.M., Chlorination of titanium carbide for the processing of nanoporous carbon: A kinetic study, Chem. Eng. J., 2010, vol. 159, p. 236.
Tallo, I., Thomberg, T., Kurig, H., Jänes, A., Kontturi, K., and Lust, E., Supercapacitors based on carbide-derived carbons synthesised using HCl and Cl2 as reactants, J. Solid State Electr., 2013, vol. 17, p. 19.
Batisse, N., Guérin, K., Dubois, M., Hamwi, A., Spinelle, L., and Tomasella, E., Fluorination of silicon carbide thin films using pure F2 gas or XeF2, Thin Solid Films, 2010, vol. 518, p. 6746.
Portet, C., Kazachkin, D., Osswald, S., Gogotsi, Y., and Borguet, E., Impact of synthesis conditions on surface chemistry and structure of carbide-derived carbons, Thermochim. Acta, 2010, vol. 497, p. 137.
Xu, J., Zhang, R., Wang, J., Ge, S., and Wen, F., Hollow carbon onions with larger lattice spacing obtained by chlorination of the ball-milled SiC, Mater. Lett., 2012, vol. 88, p. 168.
Xu, J., Zhang, R., Wang, J., Ge, S., Zhou, H., Liu, Y., and Chen, P., Effective control of the microstructure of carbide-derived carbon by ball-milling the carbide precursor, Carbon, 2013, vol. 52, p. 499.
Kormann, M. and Popovska, N., Processing of carbide-derived carbons with enhanced porosity by activation with carbon dioxide, Micropor. Mesopor. Mat., 2010, vol. 130, p. 167.
Thomberg, T., Kurig, H., Jänes, A., and Lust, E., Mesoporous carbide-derived carbons prepared from different chromium carbides, Micropor. Mesopor. Mat., 2011, vol. 141, p. 88.
Hoffman, E.N., Yushin, G., El-Raghy, T., Gogotsi, Y., and Barsoum, M.W., Micro and mesoporosity of carbon derived from ternary and binary metal carbides, Micropor. Mesopor. Mat., 2008, vol. 112, p. 526.
Vaarmets, K., Valk, P., Nerut, J., Tallo, I., Aruväli, J., Sepp, S., and Lust, E., Rotating Disk Electrode Study of Carbon Supported Pt-Nanoparticles Synthesized Using Microwave-Assisted Method, ECS Trans., 2017, vol. 80, p. 743.
Valk, P., Nerut, J., Tallo, I., Tee, E., Romann, T., and Lust, E., Influence of Molybdenum Carbide Additive on the Oxygen Reduction Reaction Kinetics at Molybdenum Carbide Derived Carbon Electrode, ECS Meet. Abst., vol. MA2014-01, 2014, p. 1156.
Taleb, M., Nerut, J., Tooming, T., Thomberg, T., and Lust, E., Oxygen Electroreduction on Platinum Nanoparticles Activated Electrodes Deposited onto D‑Glucose Derived Carbon Support in 0.1 M KOH, J. Electrochem. Soc., 2016, vol. 163, p. F1251.
Taleb, M., Nerut, J., Tooming, T., Thomberg, T., Jänes, A., and Lust, E., Oxygen Electroreduction on Platinum Nanoparticles Deposited onto D-Glucose Derived Carbon, J. Electrochem. Soc., 2015, vol. 162, p. F651.
Kinoshita, D.K., Electrochemical Oxygen Technology, N.Y.: John Wiley & Sons, 1992. 462 p.
Hirota, K., Mitani, K., Yoshinaka, M., and Yamaguchi, O., Simultaneous synthesis and consolidation of chromium carbides (Cr3C2, Cr7C3 and Cr23C6) by pulsed electric-current pressure sintering, Mater. Sci. Eng. A, 2005, vol. 399, p. 154.
Ravikovitch, P.I. and Neimark, A.V., Characterization of nanoporous materials from adsorption and desorption isotherms, Colloids Surf. A, 2001, vol. 187–188, p. 11.
Brunauer, S., Emmett, P.H., and Teller, E., Adsorption of Gases in Multimolecular Layers, J. Am. Chem. Soc., 1938, vol. 60, p. 309.
Jagiello, J. and Olivier, J.P., Carbon slit pore model incorporating surface energetical heterogeneity and geometrical corrugation, Adsorption, 2013, vol. 19, p. 777.
Jagiello, J. and Olivier, J.P., 2D-NLDFT adsorption models for carbon slit-shaped pores with surface energetical heterogeneity and geometrical corrugation, Carbon, 2013, vol. 55, p. 70.
de Boer, J.H., Lippens, B.C., Linsen, B.G., Broekhoff, J.C.P., van den Heuvel, A., and Osinga, Th.J., Thet-curve of multimolecular N2-adsorption, J. Colloid Interf. Sci., 1966, vol. 21, p. 405.
Lowell, S., Shields, J.E., Thomas, M.A., and Thommes, M., Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density, Dordrecht: Springer Science & Business Media, 2006. 370 p.
Rouquerol, J., Llewellyn, P., and Rouquerol, F., Is the BET Equation Applicable to Microporous Adsorbents?, Stud. Surf. Sci. Catal., 2007, vol. 160, p. 49.
Garsany, Y., Baturina, O.A., Swider-Lyons, K.E., and Kocha, S.S., Experimental Methods for Quantifying the Activity of Platinum Electrocatalysts for the Oxygen Reduction Reaction, Anal. Chem., 2010, vol. 82, p. 6321.
Zhao, Z., Zheng, H., Wang, Y., Mao, S., Niu, J., Chen, Y., and Shang, M., Synthesis of chromium carbide (Cr3C2) nanopowders by the carbonization of the precursor, Int. J. Refract. Met. Hard Mater., 2011, vol. 29, p. 614.
Schuepfer, D.B., Badaczewski, F., Guerra-Castro, J.M., Hofmann, D.M., Heiliger, C., Smarsly, B., and Klar, P.J., Assessing the structural properties of graphitic and non-graphitic carbons by Raman spectroscopy, Carbon, 2020, vol. 161, p. 359.
González-García, P., Navarro-Suárez, A.M., Carretero-González, J., Urones-Garrote, E., Ávila-Brande, D., and Otero-Díaz, L.C., Nanostructure, porosity and electrochemical performance of chromium carbide derived carbons, Carbon, 2015, vol. 85, p. 38.
Sadezky, A., Muckenhuber, H., Grothe, H., Niessner, R., and Pöschl, U., Raman microspectroscopy of soot and related carbonaceous materials: Spectral analysis and structural information, Carbon, 2005, vol. 43, p. 1731.
Ribeiro-Soares, J., Oliveros, M.E., Garin, C., David, M.V., Martins, L.G.P., Almeida, C.A., Martins-Ferreira, E.H., Takai, K., Enoki, T., Magalhães-Paniago, R., Malachias, A., Jorio, A., Archanjo, B.S., Achete, C.A., and Cançado, L.G., Structural analysis of polycrystalline graphene systems by Raman spectroscopy, Carbon, 2015, vol. 95, p. 646.
Ferrari, A. and Robertson, J., Interpretation of Raman spectra of disordered and amorphous carbon, Physical Review B – Condensed Matter and Materials Physics, 2000, vol. 61, p. 14095.
McCreery, R.L., Advanced Carbon Electrode Materials for Molecular Electrochemistry, Chem. Rev., 2008, vol. 108, p. 2646.
Bannov, A.G., Popov, M.V., and Kurmashov, P.B., Thermal analysis of carbon nanomaterials: advantages and problems of interpretation, J. Therm. Anal. Calorim., 2020, vol. 142, p. 349.
Nian, Y.-R. and Teng, H., Influence of surface oxides on the impedance behavior of carbon-based electrochemical capacitors, J. Electroanal. Chem., 2003, vol. 540, p. 119.
Frackowiak, E. and Béguin, F., Carbon materials for the electrochemical storage of energy in capacitors, Carbon, 2001, vol. 39, p. 937.
Marković, N.M., Gasteiger, H.A., and Ross, P.N., Oxygen Reduction on Platinum Low-Index Single-Crystal Surfaces in Alkaline Solution: Rotating Ring DiskPt (hkl) Studies, J. Phys. Chem., 1996, vol. 100, p. 6715.
Conway, B.E., Birss, V., and Wojtowicz, J., The role and utilization of pseudocapacitance for energy storage by supercapacitors, J. Power Sources, 1997, vol. 66, p. 1.
Bard, A.J. and Faulkner, L.R., Electrochemical Methods: Fundamentals and Applications, N.Y.: Wiley, 2001. 833 p.
Taylor, R.J. and Humffray, A.A., Electrochemical studies on glassy carbon electrodes: II. Oxygen reduction in solutions of high pH (pH > 10), J. Electroanal. Chem. Interfacial Electrochem., 1975, vol. 64, p. 63.
Lipkowski, J. and Ross, P.N., Electrocatalysis., N.Y.: John Wiley & Sons, 1998. 396 p.
Paulus, U.A., Schmidt, T.J., Gasteiger, H.A., and Behm, R.J., Oxygen reduction on a high-surface area Pt/Vulcan carbon catalyst: a thin-film rotating ring-disk electrode study, J. Electroanal. Chem., 2001, vol. 495, p. 134.
Higuchi, E., Uchida, H., and Watanabe, M., Effect of loading level in platinum-dispersed carbon black electrocatalysts on oxygen reduction activity evaluated by rotating disk electrode, J. Electroanal. Chem., 2005, vol. 583, p. 69.
Denuault, G., Sosna, M., and Williams, K.-J., Classical Experiments, in Handbook of Electrochemistry, Zoski, C.G., Ed., Amsterdam: Elsevier, 2007, p. 431.
Marković, N.M., Gasteiger, H.A., Grgur, B.N., and Ross, P.N., Oxygen reduction reaction on Pt(111): effects of bromide, J. Electroanal. Chem., 1999, vol. 467, p. 157.
Gubbins, K.E. and Walker, R.D., The Solubility and Diffusivity of Oxygen in Electrolytic Solutions, J. Electrochem. Soc., 1965, vol. 112, p. 469.
Yeager, E., Electrocatalysts for O2 reduction, Electrochim. Acta, 1984, vol. 29, p. 1527.
Yeager, E., Dioxygen electrocatalysis: mechanisms in relation to catalyst structure, J. Mol. Catal., 1986, vol. 38, p. 5.
Taylor, R.J. and Humffray, A.A., Electrochemical studies on glassy carbon electrodes: III. Oxygen reduction in solutions of low pH (pH < 10), J. Electroanal. Chem. Interfacial Electrochem., 1975, vol. 64, p. 85.
Shen, A., Zou, Y., Wang, Q., Dryfe, R.A.W., Huang, X., Dou, S., Dai, L., and Wang, S., Oxygen Reduction Reaction in a Droplet on Graphite: Direct Evidence that the Edge Is More Active than the Basal Plane, Angew. Chem. Int. Edit, 2014, vol. 53, p. 10804.
Retter, U. and Lohse, H., Electrochemical Impedance Spectroscopy, in: Electroanalytical Methods: Guide to Experiments and Applications, Scholz, F., Bond, A.M., Compton, R.G., Fiedler, D.A., Inzelt, G., Kahlert, H., Komorsky-Lovrić, Š., Lohse, H., Lovrić, M., Marken, F., Neudeck, A., Retter, U., Scholz, F., and Stojek, Z., Eds., Berlin: Springer, 2010, p. 159.
Zhao, Q., Wang, X., Liu, J., Wang, H., Zhang, Y., Gao, J., Liu, J., and Lu, Q., Surface Modification and Performance Enhancement of Carbon Derived from Chromium Carbide for Supercapacitor Applications, J. Electrochem. Soc., 2015, vol. 162, p. A845
Дополнительные материалы отсутствуют.