

Журнал эволюционной биохимии и физиологии, 2021, T. 57, № 4, стр. 273-288
ОСЬ КИШЕЧНИК–МОЗГ И РЕЦЕПТОРЫ, АКТИВИРУЕМЫЕ ПЕРОКСИСОМНЫМИ ПРОЛИФЕРАТОРАМИ КАК ФАКТОРЫ РЕГУЛЯЦИИ ЭПИЛЕПТОГЕНЕЗА
О. Е. Зубарева 1, *, Т. Б. Мелик-Касумов 2
1 Институт эволюционной физиологии и биохимии им. И.М. Сеченова РАН
Санкт-Петербург, Россия
2 Институт физиологии Национальной академии наук Беларуси
Минск, Беларусь
* E-mail: ZubarevaOE@mail.ru
Поступила в редакцию 11.03.2021
После доработки 03.04.2021
Принята к публикации 06.04.2021
Аннотация
Несмотря на активно проводимые исследования, значительная часть больных эпилепсией страдает фармакорезистентными формами заболевания. Это делает актуальным поиск новых методов лечения. В последние годы активно обсуждается возможная роль кишечно-мозговых взаимодействий в патогенезе эпилепсии. Последние экспериментальные и клинические исследования показывают корреляцию баланса кишечной микробиоты и выраженности эпилептогенеза. При этом различные методы модификации состава микробиоты показывают существенное влияние на течение эпилепсии. Тем не менее, остается открытым вопрос основного рецепторного звена оси кишечник–мозг, выступающего в роли интерфейса между кишечными микроорганизмами и регуляторными системами организма.
Целью настоящего обзора является анализ путей и степени вовлеченности кишечной микробиоты в патогенез и саногенез эпилепсии. Среди таких путей выделены нервно-проводниковый, метаболитный, иммунный и эндокринный. Анализ полученных на сегодняшний день данных показывает существенную роль в этих процессах рецепторов, активируемых пролифератором пероксисом (PPARs). Экспрессия этих рецепторов в основных структурах оси кишечник–мозг, наличие их лигандов среди метаболитов представителей микробиоты, а также описанные для агонистов некоторых PPARs противосудорожная и/или нейропротекторная активность позволяют выдвинуть гипотезу о роли PPARs в организме в качестве упомянутого выше сигнального интерфейса в оси кишечник–мозг. Отдельно в работе рассматривается терапевтический потенциал агонистов PPARs при лечении эпилепсии.
ВВЕДЕНИЕ
Височная эпилепсия – одно из наиболее тяжелых и сложно поддающихся лечению неврологических заболеваний. Распространенность эпилепсии в развитых странах составляет примерно 50 случаев на 100 000 населения, в менее развитых – 100 и более случаев на 100 000 человек [1].
Несмотря на активно проводимые исследования, около 30% пациентов остаются нечувствительными к применяемой терапии, что делает чрезвычайно актуальным поиск новых методов лечения. В последние годы активно обсуждается роль питания и пищеварительной системы в патогенезе различных видов нервно-психической патологии [2, 4], включая эпилепсию [5–7].
Значительная часть исследований по данным вопросам сосредоточена на изучении роли кишечной микрофлоры в патогенезе нервных расстройств [2]. Было показано, что микробиота кишечника может влиять на когнитивные функции, настроение, уровень тревожности и депрессивности. Микробный дисбаланс может коррелировать с различными нейродегенеративными расстройствами – болезнью Альцгеймера [8, 9], болезнью Паркинсона [10–12], аутизмом [13], депрессией [14], рассеянным склерозом [15], а также предрасположенностью к припадкам при эпилепсии [16]. Выявлено, что частота судорожных припадков повышена у пациентов с сочетанной патологией – эпилепсией и синдромом раздраженного кишечника [17]. При этом установлено, что патогенез последнего практически всегда связан с негативными изменениями в балансе микробиоты кишечника (клинический метаанализ – [18]). С другой стороны, А. Peng и соавт. [19] показали, что у пациентов с фармакорезистентной эпилепсией значительно изменяется состав кишечной микрофлоры, у них cущественно увеличиваются показатели видового разнообразия микробиоты (α-разнообразие), и отмечается увеличение содержания редких представителей. Содержание бифидобактерий и лактобацилл коррелирует с частотой приступов – их содержание выше у пациентов с четырьмя и менее судорогами в год по сравнению с больными, имеющими более частые приступы [19]. Изменения структуры микробиоты кишечника были выявлены также G. Xie и соавт. [20], обследовавшими 14 пациентов с рефрактерной эпилепсией и 30 здоровых испытуемых. Кроме того значительное улучшение качества жизни и снижение частоты судорог на 50% и более отмечалось у 28.9% пациентов с фармакорезистентной эпилепсией, получавших смесь из восьми видов бактерий: Lactobacillus acidophilus, Lactobacillus plantarum, Lactobacillus casei, Lactobacillus helveticus, Lactobacillus brevis, Bifidobacterium lactis (два штамма), Streptococcus salivarius subsp. Thermophiles [21].
Механизмы лечебных и побочных эффектов другого немедикаментозного метода лечения эпилепсии – кетогенной диеты – в последнее время также подвергаются значительному пересмотру в контексте кишечно-мозговых взаимодействий [22]. Предполагается, в частности, что существенное снижение поступления в организм перевариваемых углеводов при сохранении доли пищевых волокон при кетогенной диете может приводить к перестройке микробиома кишечника, в частности, к уменьшению доли дрожжеподобных и плесневых грибов. Умеренное ограничение потребления белков при такой диете в свою очередь может снижать долю гнилостных и условно-патогенных бактерий. Тем не менее отмечены и случаи негативных изменений микробиоты кишечника в случае кетогенной диеты [6, 23], выражающиеся в снижении доли полезных для здоровья бактерий, потребляющих клетчатку. Эти нарушения объясняются, вероятно, существенными для микробиома различиями в рационе, прежде всего пониженным содержанием в нем пищевых волокон и высоким содержанием насыщенных жирных кислот [24]. Изменение баланса микробиоты кишечника при кетогенной диете может существенно сказываться на ее эффективности [25]. Совместное использование курсового введения Lactobacillus fermentum MSK 408 кетогенной диеты усиливает защитные свойства последней в мышиной модели пентилентетразоловых судорог [26].
Данный обзор посвящен описанию основных механизмов, которые могут опосредовать влияние кишечной микробиоты и других компонентов пищеварительной системы на развитие эпилептических изменений в мозге. При этом особое внимание будет уделено рецепторам, активируемым пероксисомными пролифераторами, в связи с широкой вовлеченностью этих рецепторов в механизмы реализации кишечно-мозговых взаимодействий и перспективностью использования агонистов данных рецепторов для лечения фармакорезистентных форм эпилепсии.
I. ОСЬ КИШЕЧНИК–МОЗГ В ПАТОГЕНЕЗЕ ЭПИЛЕПСИИ: МЕТОДИЧЕСКИЕ АСПЕКТЫ ИССЛЕДОВАНИЯ ПРОБЛЕМЫ
Одним из методических подходов при изучении роли кишечной микробиоты в формировании нервно-психической патологии и, в частности, эпилепсии, является использование “стерильных” (свободных от микробов, англ. germ-free, GF) мышей. С помощью данной модели было показано влияние послеродовой микробной колонизации на формирование уровня тревожности [27, 28], мозговых механизмов стресс-реакции [29] и нейропластичности [28]. R. Konno и соавт. [30] показали, что у GF-мышей снижается уровень циркулирующего d-аланина, являющегося коагонистом NMDA-глутаматных рецепторов, после инокуляции бактерий его уровень восстанавливается. Роль NMDA-рецепторов в развитии эпилептических состояний была показана неоднократно [31, 32]. Было также показано, что состав микробиоты кишечника влияет на формирование и функциональную активность клеток миндалины [33–35] и гиппокампа [35] – как известно, это структуры мозга непосредственно вовлечены в патогенез эпилепсии. Нарушения развития нервной системы у GF-мышей отмечаются не только в центральном, но и периферическом (энтеральном) отделе: в возрасте Р3 у них выявляются морфологические изменения в ауэрбаховом сплетении тощей и подвздошной кишки, характеризующиеся снижением плотности нервных волокон, уменьшением количества нейронов в ганглиях и увеличением доли нитрергических нейронов [36], что может влиять на активность вегетативной нервной системы и, как следствие, на развитие эпилептических процессов в мозге.
Другой методический подход – трансплантация фекальной микробиоты – используется как в доклинических, так и в клинических исследованиях. Было показано, что трансплантация фекальной микробиоты от здоровых к психически больным людям приводила к уменьшению у последних депрессивных и тревожных симптомов. Также обнаружено и обратное – усиление депрессивных и тревожных симптомов в результате трансплантации микробиоты от психически больных доноров здоровым реципиентам [обзор – [37]]. Z. He и соавт. сообщают об успешном применении данного метода при лечении пациентки с болезнью Крона и эпилепсией. Трансплантация фекальной микрофлоры привела не только к ремиссии кишечных нарушений, но и позволила предотвратить рецидивы приступов после отмены противоэпилептических препаратов [38].
Для изменения состава кишечной микрофлоры применяют также антибиотики. Bercik и соавт. [39] вводили мышам смесь из антибиотиков неомицина, бацитрацина и пимарицина, что приводило к существенному изменению видового состава микробиоты. У экспериментальных мышей наблюдались нарушения исследовательского поведения в темно-светлой камере и изменения продукции в гиппокампе и миндалине нейротрофического фактора мозга (BDNF), вовлеченность которого в патогенез эпилепсии широко обсуждается в литературе [40].
Еще один методический подход в исследовании кишечно-мозговых взаимодействий заключается в хроническом введении экспериментальным животным пробиотиков (потенциально полезных представителей кишечной микробиоты) и пребиотиков (не перевариваемых пищевых волокон, стимулирующих рост полезных кишечных бактерий). Этот подход был использован при изучении действия микробиоты кишечника на нейромедиаторные системы мозга, вовлеченные в патогенез эпилепсии. Было показано, что Bifidobacterium infantis может влиять на метаболизм серотонина в мозге крыс [41]. При этом известно, что серотонин играет важную роль в патогенезе эпилепсии [42].
Интересны также исследования влияния про- и пребиотиков на состояние глутаматергической и ГАМК-ергической систем мозга, поскольку считается, что баланс этих нейромедиаторов играет ключевую роль в индукции судорожных состояний [43]. Исследование, проведенное с помощью магнитно-резонансной спектроскопии, показало, что хроническое введение Lactobacillus rhamnosus (JB-1) здоровым самцам мышей BALB приводит к увеличению уровня глутамата и глутамина (Glx), общего N-ацетиласпартата и N-ацетиласпартилглутаминовой кислоты (tNAA), а также ГАМК в головном мозге [44]. При этом ранее было показано, что уровень tNAA снижен у пациентов с височной эпилепсией [45]. Savignac и соавт. [46] исследовали эффекты хронического введения крысам фруктоолигосахаридов и галактоолигосахаридов – питательных веществ, являющихся субстратом для пробиотической микрофлоры кишечника – лактобацилл и бифидобактерий. Взрослые крысы, получавшие галактоолигосахариды, имели более высокие уровни белка субъединицы GluN1 NMDA-рецепторов в лобной коре и GluN2A-субъединицы в гиппокампе по сравнению с контрольными животными. Введение фруктоолигосахаридов увеличивало уровень GluN1 в гиппокампе. Williams и соавт. осуществляли ежедневное кормление новорожденных (P3-P21) крыс пребиотиком галактоолигосахаридом BGOS. В возрасте P22 они выявили увеличение экспрессии генов субъединицы GluN2A NMDA-глутаматных рецепторов и мозгового нейротрофического фактора BDNF в гиппокампе. В отношении BDNF эффект наблюдался также на Р56 (26 дней после прекращения кормления) [47]. С другой стороны, J. Bravo и соавт. [48] показали, что хроническое введение мышам Lactobacillus rhamnosus (JB-1) приводило к изменению экспрессии генов ГАМК-, B1b- и Aα2-рецепторов в гиппокампе, коре и миндалине. Ключевая роль рецепторов ГАМК в патогенезе эпилепсии была описана неоднократно [обзор – [49]].
II. МЕХАНИЗМЫ, ОПОСРЕДУЮЩИЕ ВЛИЯНИЕ КИШЕЧНОЙ МИКРОБИОТЫ НА МОЗГ
Среди возможных механизмов, опосредующих влияние кишечной микробиоты на мозг, можно выделить следующие основные пути: 1) нервнопроводниковый; 2) иммунный; 3) нейроэндокринный; 4) прямое влияние микробных метаболитов на нейроны мозга.
1) Нервнопроводниковый путь кишечно-мозговых взаимодействий
Hервнопроводниковый путь анатомически включает кишечную нервную систему (в частности, Мейсснерово и Ауэрбахово сплетение), превертебральные ганглии, симпатические и парасимпатические нервы, вегетативные центры головного мозга.
Ключевую роль в данном пути играет блуждающий нерв (nervus vagus, вагус), его ядра (в первую очередь, ядро солитарного тракта) и его проекции в лимбические структуры мозга. Доказательства роли вагуса в кишечно-мозговых взаимодействиях были получены в следующих исследованиях. Было показано, что увеличение в слепой кишке концентрации микробного метаболита индола приводит к активации блуждающего нерва [50]. Ваготомия подавляет действие пребиотического олигосахарида грудного молока 2-фукозиллактозы на эффективность ассоциативного обучения и показатели долговременной потенциации в нейронах гиппокампа [51], а также нивелирует поведенческие эффекты введений Lactobacillus rhamnosus (JB-1) [48]. Введение данной бактерии в просвет тощей кишки увеличивает частоту спонтанного возбуждения афферентных волокон брыжеечных нервов [52]. Было показано также, что вагус опосредует положительные эффекты Lactobacillus reuteri на социальное поведение в генетической модели расстройства аутистического спектра (мыши Shank3B –/–) [53].
С другой стороны, еще в конце 19 века было доказано, что стимуляция вагуса (VNS-терапия) положительно влияет на больных эпилепсией, снижая вероятность возникновения эпилептических приступов и улучшая психоэмоциональное состояние пациентов [54]. В настоящее время использование данного метода в комплексном лечении эпилепсии и депрессивных состояний разрешено в Европейском союзе и Канаде [55]. Этот метод лечения применяется также в России и Беларуси. Механизмы положительных эффектов VNS-терапии до конца не известны, но предполагается, что они опосредуются изменениями, происходящими в корковых проекциях ядра солитарного тракта посредством повышения синаптической ативности в клетках таламуса и таламокортикальных проекций, а также снижением синаптической активности в нейронах миндалины и гиппокампа [56].
Основным нейротрансмиттером вагусной афферентной сигнализации к ядру солитарного тракта является глутамат [57], однако множество других нейромедиаторов и нейропептидов также опосредуют кишечно-мозговые взаимодействия на уровне кишечной и вегетативной нервной систем. В частности, это предшественники и метаболиты триптофана, серотонин, ГАМК и катехоламины [33, 58], а также пептиды (холецистокинин, субстанция P, пептид YY, глюкагоноподобный пептид-1, грелин, лептин) [59–61].
2) Роль иммунной системы в кишечно-мозговых взаимодействиях
В желудочно-кишечном тракте сосредоточено до 70% иммунных клеток организма, постоянно взаимодействующих с триллионами микробов, населяющих кишечник. В основном иммунные клетки сосредоточены в лимфоидной ткани кишечника. Они представлены Т- и В-лимфоцитами, макрофагами, тучными и дендритными клетками.
Исследования, проведенные на GF-мышах, показали, что микробиота влияет на созревание иммунной системы хозяина, при этом введение Bacteroides fragilis нивелирует нарушения, наблюдаемые у GF-животных [63]. Специфическая микробиота направляет дифференцировку Т-хелперов, продуцирующих интерлейкин-17 (IL-17), в слизистой оболочке тонкой кишки [58]. Микробная колонизация кишечника GF-мышей приводит к индукции большого числа генов, участвующих в Т-клеточном иммунном ответе [64]. Дифференцировка регуляторных Т-лимфоцитов модулируется бутиратом – продуктом метаболизма пробиотической флоры кишечника, относящегося к короткоцепочечным жирным кислотам (англ. Short chain fatty acids, SCFA) [65].
Молекулярные механизмы, ответственные за взаимодействие микробиоты с клетками организма хозяина, включают в себя множество рецепторов распознавания образов, в том числе, Toll-подобные рецепторы (TLR), расположенные на мембранах иммунных клеток и клеток кишечника [66, 67].
Активация TLR четвертого типа (TLR4) приводит к индукции в иммунных клетках синтеза провоспалительных цитокинов, в частности, интерлейкинов 1 и 6 (IL-1, IL-6) и фактора некроза опухоли α (TNFα). Причем микробиота может влиять на изменение уровня провоспалительных цитокинов не только локально (в клетках кишечника), но и в крови, по крайней мере, при некоторых патологических состояниях. Исследование детей с неинфекционной диареей показало существенное увеличение в сыворотке их крови содержания IL-1, IL-6, TNFα и интерлейкина 17 (IL-17). При этом уровни IL-1, IL-6, IL-17 и TNF-α в сыворотке положительно коррелировали с численностью E. coli и Enterococcus sp. и отрицательно коррелировали с численностью Bifidobacterium sp. и молочнокислых бактерий в образцах фекалий [68]. Повышение уровня IL-1, IL-6 и TNF-α в крови приводит к усилению экспрессии аналогичных белков в глиальных клетках ЦНС, что в свою очередь может способствовать развитию эпилептических процессов [69, 70]. При этом показано, что с повышением уровня провоспалительных цитокинов в крови и мозге могут быть связаны не только развитие судорожной активности, но и появление характерных для эпилепсии нарушений социального и эмоционального поведения [71, 72].
3) Влияние микробиоты на гормональные системы организма
Можно выделить два основных уровня таких взаимодействий: первый связан с действием микробиоты на эндокринные клетки эпителиальной выстилки желудочно-кишечного тракта (энтероэндокринные клетки), производящие серотонин и кишечные пептиды, второй – с регуляцией активности гипоталамо-гипофизно-надпочечниковой оси.
Серотонин. Наиболее распространенные в желудочно-кишечном тракте энтероэндокринные клетки – энтерохромаффинные клетки. Они располагаются рядом с эпителием, выстилающим просвет пищеварительного тракта. Энтерохромаффинные клетки модулируют передачу сигналов нейронов в кишечной нервной системе посредством секреции серотонина и пептидов. Порядка 90% серотонина в организме производится в кишечнике [73]. Поскольку афферентные и эфферентные нервные волокна не выступают в просвет кишечника, энтерохромаффинные клетки необходимы для трансдукции сенсорного сигнала. Кишечный серотонин, действуя в синергии с другими пищеварительными гормонами, регулирует сенсорные и моторные желудочно-кишечные рефлексы. Кроме того, он обладает провоспалительным действием, воздействуя на рецепторы, расположенные на мембранах T- и B-лимфоцитов и дендритных клеток (5-HT7Rs) [74–76]. Показано также влияние периферического серотонина на углеводный и липидный обмен [74, 77].
Кишечная продукция серотонина регулируется микробиотой, эффект может опосредоваться SCFA-ацетатом и бутиратом [73, 78]. Действие микробиоты может носить не только локальный, но и системный характер. Существенное изменение в крови GF-мышей уровня индолсодержащих метаболитов, таких как индоксилсульфат и антиоксидант индол-3-пропионовая кислота (IPA), выявлено в исследовании, проведенном методом масс-спектрометрии. Интересно отметить, что производство IPA зависело от микрофлоры и могло быть скорректировано путем колонизации кишечника бактериями Clostridium porogenes [79].
Ряд данных указывает на возможное участие периферического и центрального пула серотонина в эпилептогенезе. Уровень серотонина в крови увеличивается после генерализованных судорог, что позволяет предположить усиление его синтеза энтерохромаффинными клетками кишечника [80]. Предполагается, что циркулирующий в крови серотонин может попадать в мозг лишь в ограниченном количестве [81], однако во время интенсивных приступов, таких как эпилептический статус, проницаемость гематоэнцефалического барьера увеличивается [82], потенциально облегчая обмен серотонина между периферическим кровообращением и центральной нервной системой. Вовлеченность мозгового серотонина в регуляцию эпилептических процессов в ЦНС показана во многих исследованиях (обзор – [42]).
Кишечные пептиды. Помимо доминирующих энтерохромаффинных клеток в диффузной энтероэндокринной системе обнаружено множество других клеток, обладающих секреторной активностью. В контексте рассматриваемой проблемы наибольший интерес представляют некоторые пептиды, обладающие нейротропным, в том числе противосудорожным или нейропротекторным действием. Так, K-клетки кишечника секретируют глюкозозависимый инсулинотропный полипептид (GIP), стимулирующий выделение инсулина в ответ на рецепцию жиров и углеводов. Рецепторы этого пептида обнаружены в головном мозге [83] и в том числе в гиппокампе, где их активация усиливает пролиферацию прогениторных клеток субгранулярного слоя зубчатой извилины [84]. Повышение экспрессии рецепторов GIP отмечается на разных стадиях литий-пилокарпиновой модели височной эпилепсии [85], а их агонисты показали нейропротекторный эффект в данной модели [86]. Вместе с тем отмечено, что уровень этого пептида в плазме крови растет после внутрижелудочного введения мышам SCFA [87].
I-клетки тонкого кишечника выделяют в кровь холецистокинин (CCK), пептидный гормон, стимулирующий выделение желчи и панкреатического сока, а также вызывающий чувство сытости. При этом нужно отметить, что как рецепторы к холецистокинину, так и сами холецистокинин-положительные интернейроны обнаружены в мозге, в том числе, в гиппокампе [88]. Этот факт существенно затрудняет оценку вклада этого гормона в реализацию кишечно-мозговых взаимодействий. Тем не менее уменьшение плотности CCK-иммунореактивных волокон наблюдается в зубчатой извилине в модели височной эпилепсии [89]. Отмечено, что у GF-мышей концентрация CCK в кишечнике снижена [90].
Пептид нейротензин, секретируемый N-клетками тонкого кишечника, также имеет центральный пул. Аналоги нейротензина обладают антиконвульсантным действием [91]. L-клетки толстого кишечника выделяют еще два пептидных гормона – глюкогоноподобный пептид-1 (GLP-1) и пептид YY (PYY). В отдельных исследованиях установлено, что данные пептиды обладают выраженным влиянием на мозг и могут играть существенную роль в патогенезе эпилепсии. В частности, пептид YY является естественным лигандом рецепторов нейропептида Y (Y1R и Y2R). Активация этих рецепторов оказывает противосудорожный и нейропротекторный эффект в экспериментальных моделях эпилепсии [92], а нарушение экспрессии Y1R и Y2R в мозге отмечается у пациентов с фокальной кортикальной дисплазией – распространенной причиной фармакорезистентной эпилепсии [93]. В свою очередь, активация центральных рецепторов GLP-1 усиливает долговременную потенциацию в гиппокампе, стимулирует нейрогенез и оказывает нейропротекторное действие [94]. Отмечено, что у GLP-1R-нокаутных мышей развиваются более тяжелые судороги и нейрональные повреждения после введения конвульсанта каината [95]. Уровень экспрессии GLP-1 коррелирует с изменениями состава кишечной микробиоты. Применение пробиотика Lactobacillus reuteri усиливает секрецию GLP-1 у здоровых людей после введения глюкозы [96]. Использование галактоолигосахаридов увеличивает секрецию GLP-1 и PYY [97].
Степень проникновения обсуждаемых пептидов через гематоэнцефалический барьер остается малоизученной. Тем не менее показано, что при наличии центральных рецепторов GIP мРНК самого пептида в мозге не обнаружено [83]. Вероятно, проникновение через гематоэнцефалический барьер происходит в циркумвентрикулярных областях. Это предположение доказано, в частности, для GLP-1 [98] и для нейротензина [99]. Кроме того, для многих нейротропных кишечных пептидов показано опосредованная реализация их влияния на структуры мозга через активацию собственных рецепторов на афферентных волокнах вагуса [100, 101].
Увеличение синтеза и высвобождение GLP-1 и PYY может происходить под действием, SCFA, выделяемых микробиотой [103–104].
Регуляция активности гипоталамо-гипофизарно-надпочечниковой системы. Выраженность припадков у животных в различных моделях эпилепсии также существенно зависит от уровня гормонов стресса [105, 106]. При этом стресс-реактивность связана с составом микробиоты (обзор – [107]). GF-мыши показывают повышенную стресс-реактивность (увеличение уровня кортикостерона и АКТГ) в условиях иммобилизации, заражение их Bifidobacterium infantis нивелирует эти нарушения [30]. У чувствительных к стрессу GF-крыс линии F344 в условиях умеренного стресса (при помещении в новое пространство) также отмечается более выраженное увеличение уровня кортикостерона в крови, изменения экспрессии генов кортикотропин-рилизинг-фактора в гипоталамусе и глюкокортикоидных рецепторов в гиппокампе [108].
Пре- и пробиотики могут влиять на уровень стероидных гормонов и их динамику при стрессе. У людей пребиотики галактоолигосахариды снижали уровень кортизола в слюне, измеренный сразу после пробуждения [109]. Схожие результаты были получены у испытуемых, принимавших комбинированный препарат пробиотиков из Lactobacillus helveticus R0052 и Bifidobacterium longum R0175, при измерении кортизола в моче [110]. Еще одно исследование было проведено с использованием бактерии Bifidobacterium longum 1714, которая рассматривается как психобиотик, т.е. пробиотик, способный влиять на поведение и когнитивные функции. В ситуации стресса (испытуемые опускали руки в ледяную воду) люди, принимавшие данный препарат в течение четырех недель, показали меньшее увеличение кортизола в слюне по сравнению с нелечеными испытуемыми [111]. В эксперименте, проведенном на мышах, введение Bifidobacterium breve CCFM1025 в течение 5 нед уменьшало изменения, вызванные непредсказуемым стрессом: поведенческие нарушения, увеличение уровня кортикостерона и TNFα в крови, уровня IL-1β в гиппокампе, изменения продукции кортикотропин-рилизинг-фактора в гипоталамусе и экспрессии генов глюкокортикоидных рецепторов в гиппокампе [112]. Было показано также, что стресс-индуцированное увеличение уровня кортикостерона может быть ослабленно введением SCFA [113]. При этом нужно отметить, что связь между гипоталамо-гипофизарно-надпочечниковой системой и кишечной микробиотой носит двунаправленный характер, поскольку стресс в свою очередь влияет на состояние и видовой состав микробиоты, что было выявлено у взрослых животных [114, 115] и у потомства стрессированных самок [116].
4) Прямое влияние микробных метаболитов на нейроны мозга
Еще одним путем взаимодействия кишечной микробиоты с мозгом является прямое влияние микробных метаболитов на нейроны мозга. К таким соединениям можно отнести, в частности, короткоцепочечные жирные кислоты (SCFA–ацетат, пропионат, бутират и др. [117]), которые производятся микробиотой в толстом кишечнике при ферментации пищевых волокон, причем микроорганизмы различаются по способности продуцировать SCFA, а состав микробиоты зависит от диеты [118, 119]. SCFA могут непосредственно действовать на нейроны, что было показано на культуре клеток, выделенных из узловатого ганглия вагуса [120]. Также возможно прямое, не опосредованное вегетативной нервной системой, действие SCFA на клетки ЦНС, поскольку, попав в кровь, они могут в ограниченном количестве проникать в мозг, влияя на активность центральных нейронов [121]. SCFA являются лигандами рецепторов, активируемых пролифераторами пероксисом (PPARs) [122], о которых речь пойдет ниже.
III. РЕЦЕПТОРЫ, АКТИВИРУЕМЫЕ ПРОЛИФЕРАТОРОМ ПЕРОКСИСОМ, И ИХ ЛИГАНДЫ КАК ВОЗМОЖНЫЕ РЕГУЛЯТОРЫ ФОРМИРОВАНИЯ ЭПИЛЕПТИЧЕСКИХ ПРОЦЕССОВ В МОЗГЕ
Как уже было сказано выше, важную роль в кишечно-мозговых взаимодействиях и механизмах нейропротекторного действия модификации кишечной микробиоты могут играть SCFA, которые являются естественными лигандами рецепторов, активируемых пролифератором пероксисом (PPARs). PPARs – это ядерные транскрипционные факторы, регулирующие экспрессию целого ряда генов, участвующих в обмене углеводов и липидов, в развитии воспалительных реакций и других процессах, включая клеточную дифференцировку и апоптоз [123–125].
Семейство PPARs включает три типа рецепторов: PPARα, PPAR β/δ и PPARγ [126], которые отличаются друг от друга по распределению в тканях, некоторой специфичности лигандов и физиологических функций [127]. Экспрессия PPARs выявлена в различных клетках организма, включая кишечник и мозг. В клетках нервной системы она обнаружена у разных биологических видов: крыс [128], мышей и человека [129]. PPARs обнаружены в нейронах [130–133], олигодендроцитах [133, 134], микроглии [135] и астроцитах [136, 137]. При этом отмечается региональная специфичность экспрессии PPARα, PPARγ и PPAR β/δ в мозге [128]. В частности, экспрессия PPARγ повышена в таких областях, как кора, обонятельный бугорок, ядра таламуса и ретикулярная формация [128].
PPARs представляют большой интерес как один из основных интерфейсов взаимодействия кишечной микробиоты и центральных регуляторных систем организма (рис. 1).
Рис. 1.
Схема основных путей оси кишечник–мозг в условиях эпилептогенеза и роль PPARs в них. I – нервно-проводниковый путь; II – иммунный путь; III – нейроэндокринный путь; IV – прямой метаболитный путь; SCFA – короткоцепочечные жирные кислоты; CCK – холецистокинин; GLP-1 – глюкогоноподобный пептид-1; GIP – глюкозозависимый инсулинотропный полипептид; PYY – пептид YY; IL-1 – интерлейкин 1; IL-6 – интерлейкин 6; IL-17 – интерлейкин 17; TNFα – фактор некроза опухоли α; PPARs – рецепторы, активируемые пероксисомным пролифератором.
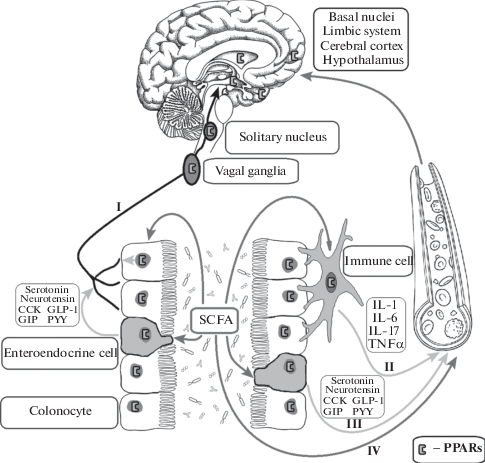
Прежде всего, важно отметить высокую степень экспрессии рецепторов как в кишечнике, так и в головном мозге [127]. Кроме того, как было сказано выше [123], PPARs могут быть активированы SCFA микробиоты кишечника, причем как в кишечнике, так и в головном мозге. PPARs также могут играть существенную роль в организации нервно-проводникового пути кишечно-мозговых взаимодействий на всех уровнях: окончаний афферентных волокон в кишечнике, периферических ганглиев вагуса, ядра солитарного тракта и его проекций в различных областях мозга. Так, активация в кишечнике PPARα эндогенным агонистом олеоилэтаноламином приводит к изменениям в уровне экспрессии рецепторов дофамина в стриатуме только в условиях интактного вагуса [138], что указывает на вовлеченность PPARα в регуляцию активности афферентных волокон блуждающего нерва. Доказано, что экспрессия PPARγ в нижнем ганглии вагуса связана с характером питания [139]. Показано также, что стимуляция блуждающего нерва после моделирования ишемического инсульта у крыс увеличивает экспрессию PPARγ в перифокальных областях коры [140]. Эти данные указывают на то, что микробиота за счет активация PPARs может модулировать активность афферентов блуждающего нерва, что, учитывая доказанную эффективность VNS-терапии, может сказываться на эпилептогенезе. Вместе с тем PPARs могут регулировать синтез кишечных пептидов, предположительно вовлеченных в патогенез эпилепсии, поскольку показано, что увеличение синтеза и высвобождение GLP-1 и PYY может происходить под действием SCFA, выделяемых микробиотой [103–104]. Выявлено также, что выделение GLP-1 в кишечнике стимулируется агонистами PPARβ/δ [141]. Наконец, наличие экспрессии PPARγ в гипоталамусе, гипофизе и надпочечниках и ее увеличение в модели иммунного стресса (ЛПС-индуцированной лихорадки) [142] указывают на возможную вовлеченность PPARs в стресс-модулирующие эффекты микробиоты кишечника, в том числе, при эпилепсии.
Большой интерес представляет тот факт, что активация PPARs приводит к подавлению воспалительных процессов [143] – важного патофизиологического механизма эпилепсии [144]. Свидетельство того, что PPARs играют роль в воспалении, было продемонстрировано на мышах, нокаутных по гену PPARα. Макрофаги этих мышей демонстрируют повышенный ответ на действие провоспалительных стимулов [145]. Лиганд-индуцированная активация PPARα и PPARγ ингибирует продукцию многих медиаторов воспаления (провоспалительных цитокинов IL-1, IL-6, TNF-α и других) в иммунных и иных клетках организма [146], включая микроглию [147] и астроциты [148]. Эти эффекты могут быть опосредованы через ингибирование экспрессии провоспалительных генов, в частности, NF-κB и AP-1 [149]. С другой стороны, активация PPARα приводит к усилению экспрессии гена противовоспалительного цитокина – естественного антагониста рецепторов IL-1 (IL-1ra) [150], способного ослаблять развитие судорог и сопутствующих нарушений поведения при эпилепсии [71].
Еще одним возможным механизмом участия PPARs в эпилептогенезе может быть их вероятное влияние на уровень глутамата в синаптической щели: было показано, что PPARγ усиливает продукцию астроглиального транспортера глутамата GLT-1 (EAAT2) [151], необходимого для удаления глутамата из синаптической щели.
Нейропротекторное действие агонистов PPARγ было показано в модели острых судорог, индуцированных пентилентетразолом [147] и в хронической литий-пилокарпиновой модели височной эпилепсии [153–155]. В частности, было выявлено, что использование агониста PPARγ росиглитазона в литий-пилокарпиновой модели эпилепсии предотвращает когнитивный дефицит, активацию астроцитов и развитие спонтанных рецидивирующих судорог [153, 154], снижает воспалительную реакцию микроглии [155, 156], уменьшает оксидативный стресс [155], ингибирует воспалительные реакции в гиппокампе [155] и подавляет NMDA-опосредованные эпилептиформные разряды [157]. Также показано, что фенофибрат и безафибрат, агонисты PPARα, проявляют противосудорожные свойства в пентилентеразоловой и литий-пилокарпиновой моделях [158, 159].
Эти данные позволяют рассматривать агонисты PPAR как перспективные препараты для лечения фармакорезистентной эпилепсии.
ЗАКЛЮЧЕНИЕ
Несмотря на активно проводимые исследования, значительная часть больных эпилепсией страдает фармакорезистентыми формами заболевания. Опубликованные данные указывают на возможную вовлеченность кишечно-мозговых взаимодействий в модуляцию эпилептогенеза. Появляются данные о нейропротекторных свойствах конкретных про- и пребиотиков в регуляции эпилептических процессов. Систематизируются данные о путях и механизмах, опосредующих влияние кишечной микробиоты на мозг. В рамках данной проблемы одним из наиболее перспективных направлений является исследование свойств лигандов рецепторов, активируемых пролифератором пероксисом. Благодаря своим противовоспалительным и нейропротекторным свойствам PPARs могут быть потенциальными терапевтическими мишенями для лечения эпилепсии [160], а их экспрессия в органах и тканях указывает на возможную вовлеченность в реализацию кишечно-мозговых взаимодействий, в том числе, в условиях эпилептогенеза.
При этом ряд актуальных вопросов в этом направлении остается нерешенным. В частности, основные экспериментальные исследования в моделях эпилепсии выполнены с применением агонистов PPARα и PPARγ, но действие агонистов PPARβ/δ на процессы эпилептогенеза остается малоизученным. В литературе практически отсутствуют данные о динамике экспрессии генов PPARα, PPARγ и PPARβ/δ в клетках нервной системы в ходе эпилептогенеза. Аналогично, накоплено недостаточно данных об экспрессии PPARs в толстом кишечнике и периферической нервной системе при применении их агонистов, модификации кишечной микробиоты, а также в условиях эпилептогенеза.
Понимание данных вопросов необходимо для разработки эффективных схем терапии эпилепсии и судорожных состояний, в связи с чем они должны быть разрешены в ходе дальнейших исследований.
Список литературы
Sander J.W. (2003) The epidemiology of epilepsy revisited. CurrOpinNeurol. 16: 165–170. https://doi.org/10.1097/01.wco.0000063766.15877.8e
Mayer E.A. Knight R., Mazmanian S.K., Cryan J.F., Tillisch K. (2014) Gut Microbes and the Brain: Paradigm Shift in Neuroscience. J Neurosci 34: 15490–15496. https://doi.org/10.1523/JNEUROSCI.3299-14.2014
Jena A., Montoya C.A., Mullaney J.A., Dilger R.N., Young W., McNabb W.C., Roy N.C. (2020) Gut-Brain Axis in the Early Postnatal Years of Life: A Developmental Perspective. Front Integr Neurosci 14:44. https://doi.org/10.3389/fnint.2020.00044
Moysidou C.M., Owens R.M. (2021) Advances in modelling the human microbiome–gut–brain axis in vitro. Biochem Soc Transact 49 (1):187–201. https://doi.org/10.1042/BST20200338
Shaikh M.F., Lee C.Y., Chen W.N., Shaikh F.A. (2020) The Gut-Brain-Axis on the Manifestation of Depressive Symptoms in Epilepsy: An Evidence-Driven Hypothesis. Front Pharmacol 11: 465. https://doi.org/10.3389/fphar.2020.00465
Iannone L.F., Preda A., Blottière H.M., Clarke G., Albani D., Belcastro V., Carotenuto M., Cattaneo A., Citraro R., Ferraris C., Ronchi F., Luongo G., Santocchi E., Guiducci L., Baldelli P., Iannetti P., Pedersen S., Petretto A., Provasi S., Selmer K., Spalice A., Tagliabue A., Verrotti A., Segata N., Zimmermann J., Minetti C., Mainardi P., Giordano C., Sisodiya S., Zara F., Russo E., Striano P. (2019) Microbiota-gut brain axis involvement in neuropsychiatric disorders. Expert Rev Neurother 19: 1037–1050. https://doi.org/10.1080/14737175.2019.1638763
De Caro C., Iannone L.F., Citraro R., Striano P., De Sarro G., Constanti A., Cryan J.F., Russo E. (2019) Can we ‘seize’ the gut microbiota to treat epilepsy? Neurosci Biobehav Rev 107: 750–764. https://doi.org/10.1016/j.neubiorev.2019.10.002
Jiang C., Li G., Huang P., Liu Z., Zhao B. (2017) The Gut Microbiota and Alzheimer’s Disease. J Alzheimer’s Dis 58: 750–764. https://doi.org/10.3233/JAD-161141
Cattaneo A., Cattane N., Galluzzi S., Provasi S., Lopizzo N., Festari C., Ferrari C., Guerra U.P., Paghera B., Muscio C., Bianchetti A., Volta G.D., Turla M., Cotelli M.S., Gennuso M., Prelle A., Zanetti O., Lussignoli G., Mirabile D., Bellandi D., Gentile S., Belotti G., Villani D., Harach T., Bolmont T., Padovani A., Boccardi M., Frisoni G.B. (2017) Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol Aging 49: 60–68. https://doi.org/10.1016/j.neurobiolaging.2016.08.019
Parashar A., Udayabanu M. (2017) Gut microbiota: Implications in Parkinson’s disease. Parkinsonism Relat. Disord. 38. https://doi.org/10.1016/j.parkreldis.2017.02.002.
Forsyth C.B., Shannon K.M., Kordower J.H., Voigt R.M., Shaikh M., Jaglin J.A., Estes J.D., Dodiya H.B., Keshavarzian A. (2011) Increased Intestinal Permeability Correlates with Sigmoid Mucosa alpha-Synuclein Staining and Endotoxin Exposure Markers in Early Parkinson’s Disease. PLoS One 6: e28032. https://doi.org/10.1371/journal.pone.0028032
Hilton D., Stephens M., Kirk L., Edwards P., Potter R., Zajicek J., Broughton E., Hagan H., Carroll C. (2014) Accumulation of α-synuclein in the bowel of patients in the pre-clinical phase of Parkinson’s disease. Acta Neuropathol 127: 235–41. https://doi.org/10.1007/s00401-013-1214-6
Mangiola F. (2016) Gut microbiota in autism and mood disorders. World J Gastroenterol 22. https://doi.org/10.3748/wjg.v22.i1.361
Liang S., Wu X., Hu X., Wang T., Jin F. (2018) Recognizing Depression from the Microbiota–Gut–Brain Axis. Int J Mol Sci 19: 1592. https://doi.org/10.3390/ijms19061592
Chu F., Shi M., Lang Y., Shen D., Jin T., Zhu J., Cui L. (2018) Gut Microbiota in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis: Current Applications and Future Perspectives. Mediators Inflamm 2018:8168717. https://doi.org/10.1155/2018/8168717
Dahlin M., Prast-Nielsen S. (2019) The gut microbiome and epilepsy. EBioMedicine 44: 741–746. https://doi.org/10.1016/j.ebiom.2019.05.024
Chen C.-H., Lin C.-L., Kao C.-H. (2015) Irritable Bowel Syndrome Increases the Risk of Epilepsy. Medicine (Baltimore) 94: e1497. https://doi.org/10.1097/MD.0000000000001497
Pittayanon R., Lau J.T., Yuan Y., Leontiadis G.I., Tse F., Surette M., Moayyedi P. (2019) Gut Microbiota in Patients With Irritable Bowel Syndrome–A Systematic Review. Gastroenterology 157: 97–108. https://doi.org/10.1053/j.gastro.2019.03.049
Peng A., Qiu X., Lai W., Li W., Zhang L., Zhu X., He S., Duan J., Chen L. (2018) Altered composition of the gut microbiome in patients with drug-resistant epilepsy. Epilepsy Res 147: 102–107. https://doi.org/10.1016/j.eplepsyres.2018.09.013
Xie G., Zhou Q., Qiu C.-Z., Dai W.-K., Wang H.-P., Li Y.-H., Liao J.-X., Lu X.-G., Lin S.-F., Ye J.-H., Ma Z.-Y., Wang W.-J. (2017) Ketogenic diet poses a significant effect on imbalanced gut microbiota in infants with refractory epilepsy. World J Gastroenterol 23: 6164–6171. https://doi.org/10.3748/wjg.v23.i33.6164
Gómez-Eguílaz M., Ramón-Trapero J.L., Pérez-Martínez L., Blanco J.R. (2018) The beneficial effect of probiotics as a supplementary treatment in drug-resistant epilepsy: a pilot study. Benef Microbes 9: 875–881. https://doi.org/10.3920/BM2018.0018
Paoli A., Mancin L., Bianco A., Thomas E., Mota J.F., Piccini F. (2019) Ketogenic Diet and Microbiota: Friends or Enemies? Genes (Basel) 10: 534. https://doi.org/10.3390/genes10070534
Linefeed M., Eng A., Darban H., Bjerkner A., Zetterström C.K., Allander T., Andersson B., Borenstein E., Dahlin M., Prast-Nielsen S. (2019) The ketogenic diet influences taxonomic and functional composition of the gut microbiota in children with severe epilepsy.Biofilms Microbiomes 5: 5. https://doi.org/10.1038/s41522-018-0073-2
Shen W., Gaskins H.R., McIntosh M.K. (2014) Influence of dietary fat on intestinal microbes, inflammation, barrier function and metabolic outcomes. J Nutr Biochem 25: 270–280. https://doi.org/10.1016/j.jnutbio.2013.09.009
Olson C.A., Vuong H.E., Yano J.M., Liang Q.Y., Nusbaum D.J., Hsiao E.Y. (2018) The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell 173: 1728–1741.e13. https://doi.org/10.1016/j.cell.2018.04.027
Eor J.Y., Tan P.L., Son Y.J., Kwak M.J., Kim S.H. (2021) Gut microbiota modulation by both Lactobacillus fermentum MSK 408 and ketogenic diet in a murine model of pentylenetetrazole-induced acute seizure. Epilepsy Res 169: 106506. https://doi.org/10.1016/j.eplepsyres.2020.106506
Nishino R., Mikami K., Takahashi H., Tomonaga S., Furuse M., Hiramoto T., Aiba Y., Koga Y., Sudo N. (2013) Commensal microbiota modulate murine behaviors in a strictly contamination-free environment confirmed by culture-based methods. Neurogastroenterol Motil 25: 521–528. https://doi.org/10.1111/nmo.12110
Heijtz R.D., Wang S., Anuar F., Qian Y., Bjorkholm B., Samuelsson A., Hibberd M.L., Forssberg H., Pettersson S. (2011) Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci USA 108: 3047–3052. https://doi.org/10.1073/pnas.1010529108
Sudo N., Chida Y., Aiba Y., Sonoda J., Oyama N., Yu X.-N., Kubo C., Koga Y. (2004) Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J Physiol 558: 263–275. https://doi.org/10.1113/jphysiol.2004.063388
Konno R., Oowada T., Ozaki A., Iida T., Niwa A., Yasumura Y., Mizutani T. (1993) Origin of D-alanine present in urine of mutant mice lacking D-amino-acid oxidase activity. Am J Physiol Liver Physiol 265: G699–G703. https://doi.org/10.1152/ajpgi.1993.265.4.G699
Kapur J. (2018) Role of <scp>NMDA</scp> receptors in the pathophysiology and treatment of status e pilepticus. Epilepsia Open 3: 165–168. https://doi.org/10.1002/epi4.12270
Chapman A.G. (1998) Chapter 24 Glutamate receptors in epilepsy. https://doi.org/10.1016/s0079-6123(08)60449-5
Cryan J.F., O’Riordan K.J., Cowan C.S.M., Sandhu K.V., Bastiaanssen T.F.S., Boehme M., Codagnone M.G., Cussotto S., Fulling C., Golubeva A.V., Guzzetta K.E., Jaggar M., Long-Smith C.M., Lyte J.M., Martin J.A., Molinero-Perez A., Moloney G., Morelli E., Morillas E., O’Connor R., Cruz-Pereira J.S., Peterson V.L., Rea K., Ritz N.L., Sherwin E., Spichak S., Teichman E.M., van de Wouw M., Ventura-Silva A.P., Wallace-Fitzsimons S.E., Hyland N., Clarke G., Dinan T.G. (2019) The Microbiota-Gut-Brain Axis. Physiol Rev 99: 1877–2013. https://doi.org/10.1152/physrev.00018.2018
Hoban A.E., Stilling R.M., Moloney G., Shanahan F., Dinan T.G., Clarke G., Cryan J.F. (2018) The microbiome regulates amygdala-dependent fear recall. Mol Psychiatry 23:1134–1144. https://doi.org/10.1038/mp.2017.100
Luczynski P., Whelan S.O., O’Sullivan C., Clarke G., Shanahan F., Dinan T.G., Cryan J.F. (2016) Adult microbiota-deficient mice have distinct dendritic morphological changes: differential effects in the amygdala and hippocampus. Eur J Neurosci 44: 2654–2666. https://doi.org/10.1111/ejn.13291
Collins J., Borojevic R., Verdu E.F., Huizinga J.D., Ratcliffe E.M. (2014) Intestinal microbiota influence the early postnatal development of the enteric nervous system. Neurogastroenterol Motil 26: 98–107. https://doi.org/10.1111/nmo.12236
Chinna Meyyappan A., Forth E., Wallace C.J.K., Milev R. (2020) Effect of fecal microbiota transplant on symptoms of psychiatric disorders: a systematic review. BMC Psychiatry 20: 299. https://doi.org/10.1186/s12888-020-02654-5
He Z., Cui B.-T., Zhang T., Li P., Long C.-Y., Ji G.-Z., Zhang F.-M. (2017) Fecal microbiota transplantation cured epilepsy in a case with Crohn’s disease: The first report. World J Gastroenterol 23: 3565–3568. https://doi.org/10.3748/wjg.v23.i19.3565
Bercik P., Denou E., Collins J., Jackson W., Lu J., Jury J., Deng Y., Blennerhassett P., Macri J., McCoy K.D., Verdu E.F., Collins S.M. (2011) The Intestinal Microbiota Affect Central Levels of Brain-Derived Neurotropic Factor and Behavior in Mice. Gastroenterology 141: 599–609. https://doi.org/10.1053/j.gastro.2011.04.052
Iughetti L., Lucaccioni L., Fugetto F., Predieri B., Berardi A., Ferrari F. (2018) Brain-derived neurotrophic factor and epilepsy: a systematic review. Neuropeptides 72: 23–29. https://doi.org/10.1016/j.npep.2018.09.005
Desbonnet L., Garrett L., Clarke G., Kiely B., Cryan J.F., Dinan T.G. (2010) Effects of the probiotic Bifidobacterium infantis in the maternal separation model of depression. Neuroscience 170: 1179–1188. https://doi.org/10.1016/j.neuroscience.2010.08.005
Bagdy G., Kecskemeti V., Riba P., Jakus R. (2007) Serotonin and epilepsy. J Neurochem 100: 857–873. https://doi.org/10.1111/j.1471-4159.2006.04277.x
During D. (1993) Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 341: 1607–1610. https://doi.org/10.1016/0140-6736(93)90754-5
Janik R., Thomason L.A.M., Stanisz A.M., Forsythe P., Bienenstock J., Stanisz G.J. (2016) Magnetic resonance spectroscopy reveals oral Lactobacillus promotion of increases in brain GABA, N-acetyl aspartate and glutamate. Neuroimage 125: 988–995. https://doi.org/10.1016/j.neuroimage.2015.11.018
Doelken M.T., Stefan H., Pauli E., Stadlbauer A., Struffert T., Engelhorn T., Richter G., Ganslandt O., Doerfler A., Hammen T (2008) 1H-MRS profile in MRI positive- versus MRI negative patients with temporal lobe epilepsy. Seizure 17: 490–497. https://doi.org/10.1016/j.seizure.2008.01.008
Savignac H.M., Corona G., Mills H., Chen L., Spencer J.P.E., Tzortzis G., Burnet P.W.J. (2013) Prebiotic feeding elevates central brain derived neurotrophic factor, N-methyl-d-aspartate receptor subunits and d-serine. Neurochem Int 63: 756–764. https://doi.org/10.1016/j.neuint.2013.10.006
Williams S., Chen L., Savignac H.M., Tzortzis G., Anthony D.C., Burnet P.W. (2016) Neonatal prebiotic (BGOS) supplementation increases the levels of synaptophysin, GluN2A-subunits and BDNF proteins in the adult rat hippocampus. Synapse 70: 121–124.https://doi.org/10.1002/syn.21880
Bravo J.A., Forsythe P., Chew M.V., Escaravage E., Savignac H.M., Dinan T.G., Bienenstock J., Cryan J.F. (2011) Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc Natl Acad Sci 108: 16050–5. https://doi.org/10.1073/pnas.1102999108
Sperk G., Furtinger S., Schwarzer C., Pirker S. (2004) GABA and Its Receptors in Epilepsy Adv Exp Med Biol 548: 92–103. https://doi.org/10.1007/978-1-4757-6376-8_7
Jaglin M., Rhimi M., Philippe C., Pons N., Bruneau A., Goustard B., Daugé V., Maguin E., Naudon L., Rabot S. (2018) Indole, a Signaling Molecule Produced by the Gut Microbiota, Negatively Impacts Emotional Behaviors in Rats. Front Neurosci 12: 216. https://doi.org/10.3389/fnins.2018.00216
Vazquez E., Barranco A., Ramirez M., Gruart A., Delgado-Garcia J.M., Jimenez M.L., Buck R., Rueda R. (2016) 51. Dietary 2’-Fucosyllactose Enhances Operant Conditioning and Long-Term Potentiation via Gut-Brain Communication through the Vagus Nerve in Rodents. PLoS One 11: e0166070. https://doi.org/10.1371/journal.pone.0166070
Perez-Burgos A., Wang B., Mao Y.-K., Mistry B., Neu-feld K.-A.M., Bienenstock J., Kunze W. (2013) Psychoactive bacteria Lactobacillus rhamnosus (JB-1) elicits rapid frequency facilitation in vagal afferents. Am J Physiol Liver Physiol 304: G211–G220. https://doi.org/10.1152/ajpgi.00128.2012
Sgritta M., Dooling S.W., Buffington S.A., Momin E.N., Francis M.B., Britton R.A., Costa-Mattioli M. (2019) Mechanisms Underlying Microbial-Mediated Changes in Social Behavior in Mouse Models of Autism Spectrum Disorder. Neuron 101: 246–259.e6. https://doi.org/10.1016/j.neuron.2018.11.018
Lulic D., Ahmadian A., Baaj A.A., Benbadis S.R., Vale F.L. (2009) Vagus nerve stimulation. Neurosurg Focus 27: E5. https://doi.org/10.3171/2009.6.FOCUS09126
Bonaz B., Picq C., Sinniger V., Mayol J.F., Clarençon D. (2013) Vagus nerve stimulation: from epilepsy to the cholinergic anti-inflammatory pathway. Neurogastroenterol Motil 25: 208–221. https://doi.org/10.1111/nmo.12076
Beekwilder J.P., Beems T. (2010) Overview of the Clinical Applications of Vagus Nerve Stimulation. J Clin Neurophysiol 27: 130–138. https://doi.org/10.1097/WNP.0b013e3181d64d8a
Hornby P.J. (2001) II. Excitatory amino acid receptors in the brain-gut axis. Am J Physiol Liver Physiol 280: G1055–G1060. https://doi.org/10.1152/ajpgi.2001.280.6.G1055
De Vadder F., Grasset E., Mannerås Holm L., Karsenty G., Macpherson A.J., Olofsson L.E., Bäckhed F. (2018) Gut microbiota regulates maturation of the adult enteric nervous system via enteric serotonin networks. Proc Natl Acad Sci USA 115: 6458–6463. https://doi.org/10.1073/pnas.1720017115
Breit S., Kupferberg A., Rogler G., Hasler G. (2018) Vagus Nerve as Modulator of the Brain–Gut Axis in Psychiatric and Inflammatory Disorders. Front Psychiatry 9: 44. https://doi.org/10.3389/fpsyt.2018.00044
Sarkar A., Lehto S.M., Harty S., Dinan T.G., Cryan J.F., Burnet P.W.J. (2016) Psychobiotics and the Manipulation of Bacteria–Gut–Brain Signals. Trends Neurosci 39: 763–781. https://doi.org/10.1016/j.tins.2016.09.002
Wang S.-Z., Yu Y.-J., Adeli K. (2020) Role of Gut Microbiota in Neuroendocrine Regulation of Carbohydrate and Lipid Metabolism via the Microbiota-Gut-Brain-Liver Axis. Microorganisms 8: 527. https://doi.org/10.3390/microorganisms8040527
Mazmanian S.K., Liu C.H., Tzianabos A.O., Kasper D.L. (2005) An Immunomodulatory Molecule of Symbiotic Bacteria Directs Maturation of the Host Immune System. Cell 122: 107–118. https://doi.org/10.1016/j.cell.2005.05.007
Ivanov I.I., Frutos R. de L., Manel N., Yoshinaga K., Rifkin D.B., Sartor R.B., Finlay B.B., Littman D.R. (2008) Sp6fic Microbiota Direct the Differentiation of IL-17-Producing T-Helper Cells in the Mucosa of the Small Intestine. Cell Host Microbe 4: 337–349. https://doi.org/10.1016/j.chom.2008.09.009
Gaboriau-Routhiau V., Rakotobe S., Lécuyer E., Mulder I., Lan A., Bridonneau C., Rochet V., Pisi A., De Paepe M., Brandi G., Eberl G., Snel J., Kelly D., Cerf-Bensussan N. (2009) The Key Role of Segmented Filamentous Bacteria in the Coordinated Maturation of Gut Helper T Cell Responses. Immunity 31: 677–689. https://doi.org/10.1016/j.immuni.2009.08.020
Furusawa Y., Obata Y., Fukuda S., Endo T.A., Nakato G., Takahashi D., Nakanishi Y., Uetake C., Kato K., Kato T., Takahashi M., Fukuda N.N., Murakami S., Miyauchi E., Hino S., Atarashi K., Onawa S., Fujimura Y., Lockett T., Clarke J.M., Topping D.L., Tomita M., Hori S., Ohara O., Morita T., Koseki H., Kikuchi J., Honda K., Hase K., Ohno H. (2013) Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504: 446–450. https://doi.org/10.1038/nature12721
Round J.L., Lee S.M., Li J., Tran G., Jabri B., Chatila T.A., Mazmanian S.K. (2011) The Toll-Like Receptor 2 Pathway Establishes Colonization by a Commensal of the Human Microbiota. Science 332: 974–977. https://doi.org/10.1126/science.1206095
Lavelle E.C., Murphy C., O’Neill L.A.J., Creagh E.M. (2010) The role of TLRs, NLRs, and RLRs in mucosal innate immunity and homeostasis. Mucosal Immunol 3: 17–28.https://doi.org/10.1038/mi.2009.124
Qin F., Wu H., Li X., Han J. (2020) Correlation between changes in gut flora and serum inflammatory factors in children with noninfectious diarrhea. J Int Med Res 48: 300060519896154. https://doi.org/10.1177/0300060519896154
Li G., Bauer S., Nowak M., Norwood B., Tackenberg B., Rosenow F., Knake S., Oertel W.H., Hamer H.M. (2011) Cytokines and epilepsy. Seizure 20: 249–256. https://doi.org/10.1016/j.seizure.2010.12.005
Vezzani A., Viviani B. (2015) Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 96: 70–82. https://doi.org/10.1016/j.neuropharm.2014.10.027
Dyomina A.V., Zubareva O.E., Smolensky I.V., Vasilev D.S., Zakharova M.V., Kovalenko A.A., Schwarz A.P., Ischenko A.M., Zaitsev A.V. (2020) Anakinra Reduces Epileptogenesis, Provides Neuroprotection, and Attenuates Behavioral Impairments in Rats in the Lithium–Pilocarpine Model of Epilepsy. Pharmaceuticals 13: 340. https://doi.org/10.3390/ph13110340
Mazarati A.M., Pineda E., Shin D., Tio D., Taylor A.N., Sankar R. (2010) Comorbidity between epilepsy and depression: Role of hippocampal interleukin-1β. Neurobiol Dis 37: 461–467. https://doi.org/10.1016/j.nbd.2009.11.001
Yano J.M., Yu K., Donaldson G.P., Shastri G.G., Ann P., Ma L., Nagler C.R., Ismagilov R.F., Mazmanian S.K., Hsiao E.Y. (2015) Indigenous Bacteria from the Gut Microbiota Regulate Host Serotonin Biosynthesis. Cell 161: 264–276. https://doi.org/10.1016/j.cell.2015.02.047
Spohn S.N., Mawe G.M. (2017) Non-conventional features of peripheral serotonin signalling – the gut and beyond. Nat Rev Gastroenterol Hepatol 14: 412–420. https://doi.org/10.1038/nrgastro.2017.51
Gershon M.D. (2013) 5-Hydroxytryptamine (serotonin) in the gastrointestinal tract. Curr Opin Endocrinol Diabetes Obes 20: 14–21. https://doi.org/10.1097/MED.0b013e32835bc703
León-Ponte M., Ahern G.P., O’Connell P.J. (2007) Serotonin provides an accessory signal to enhance T-cell activation by signaling through the 5-HT7 receptor. Blood 109: 3139–3146. https://doi.org/10.1182/blood-2006-10-052787
Jones L.A., Sun E.W., Martin A.M., Keating D.J. (2020) The ever-changing roles of serotonin. Int J Biochem Cell Biol 125: 105776. https://doi.org/10.1016/j.biocel.2020.105776
Reigstad C.S., Salmonson C.E., III J.F.R., Szurszewski J.H., Linden D.R., Sonnenburg J.L., Farrugia G., Kashyap P.C. (2015) Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J 29: 1395–1403. https://doi.org/10.1096/fj.14-259598
Wikoff W.R., Anfora A.T., Liu J., Schultz P.G., Lesley S.A., Peters E.C., Siuzdak G. (2009) Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci USA 106: 3698–3703. https://doi.org/10.1073/pnas.0812874106
Murugesan A., Rani M.R.S., Hampson J., Zonjy B., Lacuey N., Faingold C.L., Friedman D., Devinsky O., Sainju R.K., Schuele S., Diehl B., Nei M., Harper R.M., Bateman L.M., Richerson G., Lhatoo S.D. (2018) Serum serotonin levels in patients with epileptic seizures. Epilepsia 59: e91–e97. https://doi.org/10.1111/epi.14198
Young L.W., Darios E.S., Watts S.W. (2015) An immunohistochemical analysis of SERT in the blood–brain barrier of the male rat brain. Histochem Cell Biol 144: 321–329. https://doi.org/10.1007/s00418-015-1343-1
Van Vliet E.A., Aronica E., Gorter J.A. (2015) Blood–brain barrier dysfunction, seizures and epilepsy. Semin Cell Dev Biol 38: 26–34. https://doi.org/10.1016/j.semcdb.2014.10.003
Usdin T.B., Mezey E., Button D.C., Brownstein M.J., Bonner T.I. (1993) Gastric inhibitory polypeptide receptor, a member of the secretin-vasoactive intestinal peptide receptor family, is widely distributed in peripheral organs and the brain. Endocrinology 133: 2861–2870. https://doi.org/10.1210/endo.133.6.8243312
Nyberg J. (2005) Glucose-Dependent Insulinotropic Polypeptide Is Expressed in Adult Hippocampus and Induces Progenitor Cell Proliferation. J Neurosci 25: 1816–1825. https://doi.org/10.1523/JNEUROSCI.4920-04.2005
Figueiredo C.P., Pamplona F.A., Mazzuco T.L., Aguiar A.S., Walz R., Prediger R.D.S. (2010) Role of the glucose-dependent insulinotropic polypeptide and its receptor in the central nervous system: therapeutic potential in neurological diseases. Behav Pharmacol 21: 394–408. https://doi.org/10.1097/FBP.0b013e32833c8544
Tian M.-J., Wang R.-F., Hölscher C., Mi R.-L., Yuan Z.-Y., Li D.-F., Xue G.-F. (2019) The novel GLP-1/GIP dual receptor agonist DA3-CH is neuroprotective in the pilocarpine-induced epileptogenesis rat model. Epilepsy Res 154: 97–106. https://doi.org/10.1016/j.eplepsyres.2019.05.008
Lin H.V., Frassetto A., Kowalik Jr. E.J., Nawrocki A.R., Lu M.M., Kosinski J.R., Hubert J.A., Szeto D., Yao X., Forrest G., Marsh D.J. (2012) Butyrate and Propionate Protect against Diet-Induced Obesity and Regulate Gut Hormones via Free Fatty Acid Receptor 3-Independent Mechanisms. PLoS One 7: e35240. https://doi.org/10.1371/journal.pone.0035240
Fasano C., Rocchetti J., Pietrajtis K., Zander J.-F., Manseau F., Sakae D.Y., Marcus-Sells M., Ramet L., Morel L.J., Carrel D., Dumas S., Bolte S., Bernard V., Vigneault E., Goutagny R., Ahnert-Hilger G., Giros B., Daumas S., Williams S., El Mestikawy S. (2017) Regulation of the Hippocampal Network by VGLUT3-Positive CCK-GABAergic Basket Cells. Front Cell Neurosci 11: 11: 140. https://doi.org/10.3389/fncel.2017.00140
Sun C., Sun J., Erisir A., Kapur J. (2014) Loss of cholecystokinin-containing terminals in temporal lobe epilepsy. Neurobiol Dis 62: 44–55. https://doi.org/10.1016/j.nbd.2013.08.018
Woods S.E., Leonard M.R., Hayden J.A., Brophy M.B., Bernert K.R., Lavoie B., Muthupalani S., Whary M.T., Mawe G.M., Nolan E.M., Carey M.C., Fox J.G. (2015) Impaired cholecystokinin-induced gallbladder emptying incriminated in spontaneous “black” pigment gallstone formation in germfree Swiss Webster mice. Am J Physiol Liver Physiol 308: G335–G349. https://doi.org/10.1152/ajpgi.00314.2014
Lee H.-K., Zhang L., Smith M.D., White H.S., Bulaj G. (2009) Glycosylated Neurotensin Analogues Exhibit Sub-picomolar Anticonvulsant Potency in a Pharmacoresistant Model of Epilepsy. ChemMedChem 4: 400–405. https://doi.org/10.1002/cmdc.200800421
Corvino V., Marchese E., Giannetti S., Lattanzi W., Bonvissuto D., Biamonte F., Mongiovì A.M., Michetti F., Geloso M.C. (2012) The neuroprotective and neurogenic effects of neuropeptide Y administration in an animal model of hippocampal neurodegeneration and temporal lobe epilepsy induced by trimethyltin. J Neurochem 122: 415–426. https://doi.org/10.1111/j.1471-4159.2012.07770.x
Li L., Deng J., Liu C., Luo H., Guan Y., Zhou J., Qi X., Li T., Xu Z.D., Luan G.-M. (2016) Altered expression of neuropeptide Y receptors caused by focal cortical dysplasia in human intractable epilepsy. Oncotarget 7: 15329–15338. https://doi.org/10.18632/oncotarget.7855
Salcedo I., Tweedie D., Li Y., Greig N.H. (2012) Neuroprotective and neurotrophic actions of glucagon-like peptide-1: an emerging opportunity to treat neurodegenerative and cerebrovascular disorders. Br J Pharmacol 166: 1586–1599. https://doi.org/10.1111/j.1476-5381.2012.01971.x
During M.J., Cao L., Zuzga D.S., Francis J.S., Fitzsimons H.L., Jiao X., Bland R.J., Klugmann M., Banks W.A., Drucker D.J., Haile C.N. (2003) Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat Med 9: 1173–1179. https://doi.org/10.1038/nm919
Simon M.-C., Strassburger K., Nowotny B., Kolb H., Nowotny P., Burkart V., Zivehe F., Hwang J.-H., Stehle P., Pacini G., Hartmann B., Holst J.J., MacKenzie C., Bindels L.B., Martinez I., Walter J., Henrich B., Schloot N.C., Roden M. (2015) Intake of Lactobacillus reuteri Improves Incretin and Insulin Secretion in Glucose-Tolerant Humans: A Proof of Concept. Diabetes Care 38: 1827–1834. https://doi.org/10.2337/dc14-2690
Overduin J., Schoterman M.H.C., Calame W., Schonewille A.J., Ten Bruggencate S.J.M. (2013) Dietary galacto-oligosaccharides and calcium: effects on energy intake, fat-pad weight and satiety-related, gastrointestinal hormones in rats. Br J Nutr 109: 1338–1348. https://doi.org/10.1017/S0007114512003066
Kastin A.J., Akerstrom V., Pan W. (2002) Interactions of Glucagon-Like Peptide-1 (GLP-1) with the Blood-Brain Barrier. J Mol Neurosci 18: 7–14. https://doi.org/10.1385/JMN:18: 1–2:07
Banks W.A., Wustrow D.J., Cody W.L., Duff Davis M., Kastin A.J. (1995) Permeability of the blood-brain barrier to the neurotensin8–13 analog NT1. Brain Res 695: 59–63. https://doi.org/10.1016/0006-8993(95)00836-F
Krieger J.-P., Langhans W., Lee S.J. (2015) Vagal mediation of GLP-1’s effects on food intake and glycemia. Physiol Behav 152: 372–380. https://doi.org/10.1016/j.physbeh.2015.06.001
Koda S., Date Y., Murakami N., Shimbara T., Hanada T., Toshinai K., Niijima A., Furuya M., Inomata N., Osuye K., Nakazato M. (2005) The Role of the Vagal Nerve in Peripheral PYY3–36-Induced Feeding Reduction in Rats. Endocrinology 146: 2369–2375. https://doi.org/10.1210/en.2004-1266
Samuel B.S., Shaito A., Motoike T., Rey F.E., Backhed F., Manchester J.K., Hammer R.E., Williams S.C., Crowley J., Yanagisawa M., Gordon J.I. (2008) Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci USA 105: 16767–16772. https://doi.org/10.1073/pnas.0808567105
Bindels L.B., Dewulf E.M., Delzenne N.M. (2013) GPR43/FFA2: physiopathological relevance and therapeutic prospects. Trends Pharmacol Sci 34: 226–232. https://doi.org/10.1016/j.tips.2013.02.002
Larraufie P., Martin-Gallausiaux C., Lapaque N., Dore J., Gribble F.M., Reimann F., Blottiere H.M. (2018) SCFAs strongly stimulate PYY production in human enteroendocrine cells. Sci Rep 8: 74. https://doi.org/10.1038/s41598-017-18259-0
Taher T.R., Salzberg M., Morris M.J., Rees S., O’Brien T.J. (2005) Chronic Low-Dose Corticosterone Supplementation Enhances Acquired Epileptogenesis in the Rat Amygdala Kindling Model of TLE. Neuropsychopharmacology 30: 1610–1616. https://doi.org/10.1038/sj.npp.1300709
Karst H., Bosma A., Hendriksen E., Kamphuis W., de Kloet E.R., Joëls M. (1997) Effect of Adrenalectomy in Kindled Rats. Neuroendocrinology 66: 348–359. https://doi.org/10.1159/000127258
De Weerth C. (2017) Do bacteria shape our development? Crosstalk between intestinal microbiota and HPA axis. Neurosci Biobehav Rev 83: 458–471. https://doi.org/10.1016/j.neubiorev.2017.09.016
Crumeyrolle-Arias M., Jaglin M., Bruneau A., Vancassel S., Cardona A., Daugé V., Naudon L., Rabot S. (2014) Absence of the gut microbiota enhances anxiety-like behavior and neuroendocrine response to acute stress in rats. Psychoneuroendocrinology 42: 207–217. https://doi.org/10.1016/j.psyneuen.2014.01.014
Schmidt K., Cowen P.J., Harmer C.J., Tzortzis G., Er-rington S., Burnet P.W.J. (2015) Prebiotic intake reduces the waking cortisol response and alters emotional bias in healthy volunteers. Psychopharmacology (Berl) 232: 1793–1801. https://doi.org/10.1007/s00213-014-3810-0
Messaoudi M., Lalonde R., Violle N., Javelot H., Desor D., Nejdi A., Bisson J.-F., Rougeot C., Pichelin M., Cazaubiel M., Cazaubiel J.-M. (2011) Assessment of psychotropic-like properties of a probiotic formulation (Lactobacillus helveticus R0052 and Bifidobacterium longum R0175) in rats and human subjects. Br J Nutr 105: 755–764. https://doi.org/10.1017/S0007114510004319
Allen A.P., Hutch W., Borre Y.E., Kennedy P.J., Temko A., Boylan G., Murphy E., Cryan J.F., Dinan T.G., Clarke G. (2016) Bifidobacterium longum 1714 as a translational psychobiotic: modulation of stress, electrophysiology and neurocognition in healthy volunteers. Transl Psychiatry 6: e939. https://doi.org/10.1038/tp.2016.191
Tian P., O’Riordan K.J., Lee Y., Wang G., Zhao J., Zhang H., Cryan J.F., Chen W. (2020) Towards a psychobiotic therapy for depression: Bifidobacterium breve CCFM1025 reverses chronic stress-induced depressive symptoms and gut microbial abnormalities in mice. Neurobiol Stress 12: 100216. https://doi.org/10.1016/j.ynstr.2020.100216
Van de Wouw M., Boehme M., Lyte J.M., Wiley N., Strain C., O’Sullivan O., Clarke G., Stanton C., Dinan T.G., Cryan J.F. (2018) Short-chain fatty acids: microbial metabolites that alleviate stress-induced brain-gut axis alterations. J. Physiol. 596: 4923–4944. https://doi.org/10.1113/JP276431
Bailey M.T., Dowd S.E., Galley J.D., Hufnagle A.R., Allen R.G., Lyte M. (2011) Exposure to a social stressor alters the structure of the intestinal microbiota: Implications for stressor-induced immunomodulation. Brain Behav Immun 25: 397–407. https://doi.org/10.1016/j.bbi.2010.10.023
Bailey M.T., Dowd S.E., Parry N.M.A., Galley J.D., Schauer D.B., Lyte M. (2010) Stressor Exposure Disrupts Commensal Microbial Populations in the Intestines and Leads to Increased Colonization by Citrobacter rodentium. Infect Immun 78: 1509–1519. https://doi.org/10.1128/IAI.00862-09
Golubeva A.V., Crampton S., Desbonnet L., Edge D., O’Sullivan O., Lomasney K.W., Zhdanov A.V., Crispie F., Moloney R.D., Borre Y.E., Cotter P.D., Hyland N.P., O’Halloran K.D., Dinan T.G., O’Keeffe G.W., Cryan J.F. (2015) Prenatal stress-induced alterations in major physiological systems correlate with gut microbiota composition in adulthood. Psychoneuroendocrinology 60: 58–74. https://doi.org/10.1016/j.psyneuen.2015.06.002
Russo R., Cristiano C., Avagliano C., De Caro C., La Rana G., Raso G.M., Canani R.B., Meli R., Calignano A. (2018) Gut-brain Axis: Role of Lipids in the Regulation of Inflammation, Pain and CNS Diseases. Curr Med Chem 25: 3930–3952. https://doi.org/10.2174/0929867324666170216113756
Holmes E., Li J.V., Marchesi J.R., Nicholson J.K. (2012) Gut Microbiota Composition and Activity in Relation to Host Metabolic Phenotype and Disease Risk. Cell Metab 16: 559–564. https://doi.org/10.1016/j.cmet.2012.10.007
Morrison D.J., Preston T. (2016) Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 7: 189–200. https://doi.org/10.1080/19490976.2015.1134082
Goswami C., Iwasaki Y., Yada T. (2018) Short-chain fatty acids suppress food intake by activating vagal afferent neurons. J Nutr Biochem 57: 130–135. https://doi.org/10.1016/j.jnutbio.2018.03.009
Frost G., Sleeth M.L., Sahuri-Arisoylu M., Lizarbe B., Cerdan S., Brody L., Anastasovska J., Ghourab S., Hankir M., Zhang S., Carling D., Swann J.R., Gibson G., Viardot A., Morrison D., Louise Thomas E., Bell J.D. (2014) The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat Commun 5: 3611. https://doi.org/10.1038/ncomms4611
Oh H.Y.P., Visvalingam V., Wahli W. (2019) The PPAR–microbiota–metabolic organ trilogy to fine-tune physiology. FASEB J 33: 9706–9730. https://doi.org/10.1096/fj.201802681RR
Zolezzi J.M., Santos M.J., Bastías-Candia S., Pinto C., Godoy J.A., Inestrosa N.C. (2017) PPARs in the central nervous system: roles in neurodegeneration and neuroinflammation. Biol Rev 92: 2046–2069. https://doi.org/10.1111/brv.12320
Hong F., Pan S., Guo Y., Xu P., Zhai Y. (2019) PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules 24: 2545. https://doi.org/10.3390/molecules24142545
Cullingford T.E. (2004) The ketogenic diet; fatty acids, fatty acid-activated receptors and neurological disorders. Prostaglandins, Leukot Essent Fat Acids 70: 253–264. https://doi.org/10.1016/j.plefa.2003.09.008
Berger J., Moller D.E. (2002) The Mechanisms of Action of PPARs. Annu Rev Med 53: 409–435. https://doi.org/10.1146/annurev.med.53.082901.104018
Grygiel-Górniak B. (2014) Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications – a review. Nutr J 13: 17. https://doi.org/10.1186/1475-2891-13-17
Moreno S., Farioli-Vecchioli S., Cerù M. (2004) Immunolocalization of peroxisome proliferator-activated receptors and retinoid x receptors in the adult rat CNS. Neuroscience 123: 131–145. https://doi.org/10.1016/j.neuroscience.2003.08.064
Warden A., Truitt J., Merriman M., Ponomareva O., Jameson K., Ferguson L.B., Mayfield R.D., Harris R.A. (2016) Localization of PPAR isotypes in the adult mouse and human brain. Sci Rep 6: 27618. https://doi.org/10.1038/srep27618
Xing G.Q., Zhang L.X., Zhang L., Heynen T., Yoshikawa T., Smith M., Weiss S., Deterawadleigh S. (1995) Rat PPARδ Contains a CGG Triplet Repeat and Is Prominently Expressed in the Thalamic Nuclei. Biochem Biophys Res Commun 217: 1015–1025. https://doi.org/10.1006/bbrc.1995.2871
Braissant O., Foufelle F., Scotto C., Dauça M., Wahli W. (1996) Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 137: 354–366. https://doi.org/10.1210/endo.137.1.8536636
Krémarik-Bouillaud P., Schohn H., Dauça M. (2000) Regional distribution of PPARβ in the cerebellum of the rat. J Chem Neuroanat 19: 225–232. https://doi.org/10.1016/S0891-0618(00)00065-X
Woods J.W., Tanen M., Figueroa D.J., Biswas C., Zycband E., Moller D.E., Austin C.P., Berger J.P. (2003) Localization of PPARδ in murine central nervous system: expression in oligodendrocytes and neurons. Brain Res 975: 10–21. https://doi.org/10.1016/S0006-8993(03)02515-0
Granneman J., Skoff R., Yang X. (1998) Member of the peroxisome proliferator-activated receptor family of transcription factors is differentially expressed by oligodendrocytes. J Neurosci Res 51: 563–573. https://doi.org/10.1002/(SICI)1097-4547(19980301)51:5<563::AID-JNR3>3.0.CO;2-D
Bernardo A., Ajmone-Cat M.A., Levi G., Minghetti L. (2003) 15-Deoxy-Δ12,14-prostaglandin J2 regulates the functional state and the survival of microglial cells through multiple molecular mechanisms. J Neurochem 87: 742–751. https://doi.org/10.1046/j.1471-4159.2003.02045.x
Cristiano L., Bernardo A., Cerù M.P. (2001) Peroxisome proliferator-activated receptors (PPARs) and peroxisomes in rat cortical and cerebellar astrocytes. J Neurocytol 30: 671–683. https://doi.org/10.1023/A:1016525716209
Cullingford T.E., Bhakoo K., Peuchen S., Dolphin C.T., Patel R., Clark J.B. (2002) Distribution of mRNAs Encoding the Peroxisome Proliferator-Activated Receptor α, β, and γ and the Retinoid X Receptor α, β, and γ in Rat Central Nervous System. J Neurochem 70: 1366–375. https://doi.org/10.1046/j.1471-4159.1998.70041366.x
Hankir M.K., Seyfried F., Hintschich C.A., Diep T.-A., Kleberg K., Kranz M., Deuther-Conrad W., Tellez L.A., Rullmann M., Patt M., Teichert J., Hesse S., Sabri O., Brust P., Hansen H.S., de Araujo I.E., Krügel U., Fenske W.K. (2017) Gastric Bypass Surgery Recruits a Gut PPAR-α-Striatal D1R Pathway to Reduce Fat Appetite in Obese Rats. Cell Metab 25: 335–344. https://doi.org/10.1016/j.cmet.2016.12.006
Liu C., Bookout A.L., Lee S., Sun K., Jia L., Lee C., Udit S., Deng Y., Scherer P.E., Mangelsdorf D.J., Gautron L., Elmquist J.K. (2014) PPARγ in Vagal Neurons Regulates High-Fat Diet Induced Thermogenesis. Cell Metab 19: 722–730. https://doi.org/10.1016/j.cmet.2014.01.021
Ying Jiang Y., Li L., Liu B., Zhang Y., Chen Q., Chang-qing Li.C. (2015) PPARγ Upregulation Induced by Vagus Nerve Stimulation Exerts Anti-Inflammatory Effect in Cerebral Ischemia/Reperfusion Rats. Med Sci Monit 21: 268–275. https://doi.org/10.12659/MSM.891407
Daoudi M., Hennuyer N., Borland M.G., Touche V., Duhem C., Gross B., Caiazzo R., Kerr–Conte J., Pattou F., Peters J.M., Staels B., Lestavel S. (2011) PPARβ/δ Activation Induces Enteroendocrine L Cell GLP-1 Production. Gastroenterology 140: 1564–1574. https://doi.org/10.1053/j.gastro.2011.01.045
Liu Y.L., Shi J.X., Lu J., Che Z.Q., Zhu H.L., Hou Y.Q., Yin Y.L., Zhao S.J., Ding B.Y., Liu H.M. (2010) Up-regulated expression of peroxisome proliferator-activated receptor γ in the hypothalamic–pituitary–adrenal axis of weaned pigs after Escherichia coli lipopolysaccharide challenge. Vet J 184: 230–235. https://doi.org/10.1016/j.tvjl.2009.02.010
Villapol S. (2018) Roles of Peroxisome Proliferator-Activated Receptor Gamma on Brain and Peripheral Inflammation. Cell Mol Neurobiol 38: 121–132. https://doi.org/10.1007/s10571-017-0554-5
Rana A., Musto A.E. (2018) The role of inflammation in the development of epilepsy. J Neuroinflammat15: 144. https://doi.org/10.1186/s12974-018-1192-7
Crisafulli C., Cuzzocrea S. (2009) he role of endogenous and exogenous ligands for the peroxisome proliferator-activated receptor alpha (PPAR-α) in the regulation of inflammation in macrophages. Shock 32: 62–73. https://doi.org/10.1097/SHK.0b013e31818bbad6
Ricote M., Glass C. (2007) PPARs and molecular mechanisms of transrepression. Biochim Biophys Acta – Mol Cell Biol Lipids 1771: 926–935. https://doi.org/10.1016/j.bbalip.2007.02.013
Xu J., Storer P.D., Chavis J.A., Racke M.K., Drew P.D. (2005) Agonists for the peroxisome proliferator-activated receptor-α and the retinoid X receptor inhibit inflammatory responses of microglia. J Neurosci Res 81: 403–411. https://doi.org/10.1002/jnr.20518
Xu J., Chavis J.A., Racke M.K., Drew P.D. (2006) Peroxisome proliferator-activated receptor-α and retinoid X receptor agonists inhibit inflammatory responses of astrocytes. J Neuroimmunol 176: 95–105. https://doi.org/10.1016/j.jneuroim.2006.04.019
Daynes R.A., Jones D.C. (2002) Emerging roles of PPARS in inflammation and immunity. Nat Rev Immunol 2: 748–759. https://doi.org/10.1038/nri912
Stienstra R., Mandard S., Tan N.S., Wahli W., Trautwein C., Richardson T.A., Lichtenauer-Kaligis E., Kersten S., Müller M. (2007) The Interleukin-1 receptor antagonist is a direct target gene of PPARα in liver. J Hepatol 46: 869–877. https://doi.org/10.1016/j.jhep.2006.11.019
Zhao C., Jiang M., Zhang L., Hu Y., Hu Z., Zhang M., Qi J., Su A., Lou N., Xian X., Zhang J., Li W., Zhang M. (2019) Peroxisome proliferator-activated receptor gamma participates in the acquisition of brain ischemic tolerance induced by ischemic preconditioning via glial glutamate transporter 1 in vivo and in vitro. J Neurochem 151: 608–625. https://doi.org/10.1111/jnc.14824
Adabi Mohazab R., Javadi-Paydar M., Delfan B., Dehpour A.R. (2012) Possible involvement of PPAR-gamma receptor and nitric oxide pathway in the anticonvulsant effect of acute pioglitazone on pentylenetetrazole-induced seizures in mice. Epilepsy Res 101: 28–35. https://doi.org/10.1016/j.eplepsyres.2012.02.015
Hong S., Xin Y., HaiQin W., GuiLian Z., Ru Z., ShuQin Z., HuQing W., Li Y., Ning B., YongNan L. (2013) The PPARγ agonist rosiglitazone prevents neuronal loss and attenuates development of spontaneous recurrent seizures through BDNF/TrkB signaling following pilocarpine-induced status epilepticus. Neurochem Int 63: 405–412. https://doi.org/10.1016/j.neuint.2013.07.010
Hong S., Xin Y., HaiQin W., GuiLian Z., Ru Z., ShuQin Z., HuQing W., Li Y., Yun D. (2012) The PPARγ agonist rosiglitazone prevents cognitive impairment by inhibiting astrocyte activation and oxidative stress following pilocarpine-induced status epilepticus. Neurol Sci 33: 559–566. https://doi.org/10.1007/s10072-011-0774-2
Sun H., Huang Y., Yu X., Li Y., Yang J., Li R., Deng Y., Zhao G. (2008) Peroxisome proliferator-activated receptor gamma agonist, rosiglitazone, suppresses CD40 expression and attenuates inflammatory responses after lithium pilocarpine-induced status epilepticus in rats. Int J Dev Neurosci 26: 505–515. https://doi.org/10.1016/j.ijdevneu.2008.01.009
Peng J., Wang K., Xiang W., Li Y., Hao Y., Guan Y. (2019) Rosiglitazone polarizes microglia and protects against pilocarpine-induced status epilepticus. CNS Neurosci Ther 25: 1363–1372. https://doi.org/10.1111/cns.13265
Wong S.-B., Cheng S.-J., Hung W.-C., Lee W.-T., Min M.-Y. (2015) Rosiglitazone Suppresses In Vitro Seizures in Hippocampal Slice by Inhibiting Presynaptic Glutamate Release in a Model of Temporal Lobe Epilepsy. PLoS One 10: e0144806. https://doi.org/10.1371/journal.pone.0144806
Saha L., Bhandari S., Bhatia A., Banerjee D., Chakrabarti A. (2014) Anti-kindling Effect of Bezafibrate, a Peroxisome Proliferator-activated Receptors Alpha Agonist, in Pentylenetetrazole Induced Kindling Seizure Model. J Epilepsy Res 4: 45–54. https://doi.org/10.14581/jer.14011
Porta N., Vallée L., Lecointe C., Bouchaert E., Staels B., Bordet R., Auvin S. (2009) Fenofibrate, a peroxisome proliferator-activated receptor-α agonist, exerts anticonvulsive properties. Epilepsia 50: 943–948. https://doi.org/10.1111/j.1528-1167.2008.01901.x
Łukawski K., Gryta P., Łuszczki J., Czuczwar S.J. (2016) Exploring the latest avenues for antiepileptic drug discovery and development. Expert Opin Drug Discov 11: 369–382. https://doi.org/10.1517/17460441.2016.1154840
Дополнительные материалы отсутствуют.
Инструменты
Журнал эволюционной биохимии и физиологии