

Журнал эволюционной биохимии и физиологии, 2022, T. 58, № 2, стр. 141-154
ВЛИЯНИЕ ИНТРАНАЗАЛЬНО ВВОДИМЫХ ИНСУЛИНА И ГАНГЛИОЗИДОВ НА МЕТАБОЛИЧЕСКИЕ ПОКАЗАТЕЛИ И АКТИВНОСТЬ ИНСУЛИНОВОЙ СИСТЕМЫ В ПЕЧЕНИ КРЫС С САХАРНЫМ ДИАБЕТОМ 2-ГО ТИПА
И. О. Захарова 1, *, Л. В. Баюнова 1, К. В. Деркач 1, И. О. Илясов 1, А. О. Шпаков 1, Н. Ф. Аврова 1
1 Федеральное государственное бюджетное учреждение науки Институт эволюционной физиологии и биохимии
им. И.М. Сеченова Российской академии наук
Санкт-Петербург, Россия
* E-mail: zakhar@iephb.ru
Поступила в редакцию 23.12.2021
После доработки 12.01.2022
Принята к публикации 13.01.2022
- EDN: WYHBAE
- DOI: 10.31857/S0044452922020085
Аннотация
Регуляция метаболизма глюкозы инсулином осуществляется как за счет прямого взаимодействия гормона с его сигнальной системой и транспортерами глюкозы в клетках периферических тканей, так и опосредованно через центральную нервную систему (ЦНС). При сахарном диабете 2-го типа (СД2) наблюдается недостаток инсулина в мозге вследствие нарушенного рецептор-опосредуемого транспорта гормона через гематоэнцефалический барьер, что приводит к дисфункциям пищевого поведения, термогенеза, метаболизма углеводов и жиров. Использование интраназально вводимого инсулина (ИВИ) позволяет увеличить его содержание в мозге. ИВИ в комплексе с различными сенсибилизаторами обеспечивает восстановление толерантности к глюкозе за более короткий срок при меньших дозах инсулина. Комбинированная терапия в виде ИВИ (0.5 МЕ/крысу/сутки) крысам с СД2 и суммарных ганглиозидов мозга теленка (6 мг/кг/сутки) использовалась впервые. Через 4 нед у животных с СД2 после совместного интраназального введения инсулина и ганглиозидов восстанавливалась толерантность к глюкозе, в то время как по отдельности препараты были менее эффективны. С помощью Вестерн-блоттинга было показано, что увеличение чувствительности к инсулину в печени может быть обусловлено значительным снижением экспрессии негативного регулятора инсулинового сигналинга протеинфосфотирозинфосфатазы РТР1В и увеличением степени фосфорилирования ключевых эффекторных протеинкиназ Akt по Ser473, GSK3β по Ser9 и р38-МАРК по Thr180/Tyr182. При этом ингибирующее фосфорилирование GSK3β по Ser9 может осуществляться как вследствие активации Akt-киназы, так и р38-МАРК, и вклад последней, как мы полагаем, более значимый. Поскольку инсулин и ганглиозиды при интраназальном введении непосредственно воздействовали на структуры мозга, компетентные в отношении регуляции периферической инсулиновой чувствительности и углеводного обмена в печени, сделан вывод о превалировании в этом случае центрального механизма их действия.
Гиподинамия и нарушение питания, обусловленное увеличением в рационе доли рафинированных и легкоусвояемых углеводов и насыщенных жиров, стали в последние годы одними из причин значительного прироста числа пациентов с абдоминальным ожирением, метаболическим синдромом и сахарным диабетом 2-го типа (СД2). Характерной чертой этих метаболических заболеваний является инсулиновая резистентность (ИР), представляющая собой снижение чувствительности тканей к инсулину, нарушенная толерантность к глюкозе, дислипидемия, усиление процессов воспаления [1, 2]. Инсулиновая сигнальная система, активность которой в наибольшей степени ослабляется при метаболическом синдроме и СД2, играет исключительно важную роль в функционировании различных органов и тканей – печени, скелетных мышц, почек, жировой ткани, а также мозга [3]. Важно отметить, что при длительно текущих, тяжелых формах СД2 нарушения в инсулиновом сигналинге выявляются не только на периферии, но и в центральной нервной системе (ЦНС), хотя причинно-следственные связи между центральной и периферической ИР до конца не изучены [4, 5]. В условиях ИР наблюдается снижение уровня инсулина в ЦНС, несмотря на повышение содержания инсулина в крови, что обусловлено ослаблением рецептор-опосредуемого транспорта инсулина через гематоэнцефалический барьер [6–8]. Предполагается, что ИР в мозге может развиваться на начальных стадиях метаболического синдрома и СД2 и предшествует развитию ИР на периферии [9]. Поскольку дисбаланс инсулиновой сигнальной системы мозга приводит к изменениям метаболизма и функций периферических органов, что выражается в нарушении пищевого поведения, термогенеза, метаболизма углеводов, жиров и аминокислот с разветвленной цепью, то нормализация ее активности при СД2 может предотвратить эти нарушения. Одним из подходов для компенсации дефицита инсулина в ЦНС является его интраназальное введение, которое обеспечивает доставку гормона непосредственно в мозг, в том числе к гипоталамическим нейронам, имеющим наибольшую поверхностную плотность инсулиновых рецепторов [10, 11]. В настоящее время для предотвращения центральной и периферической ИР апробируются различные подходы, в том числе включающие различные комбинации препаратов, повышающих чувствительность клеток-мишеней к инсулину, что позволяет снизить эффективные дозы инсулина и длительность курса лечения. Так, например, эффективность интраназально вводимого инсулина (ИВИ) при диабетической патологии повышается при его совместном использовании с С-пептидом проинсулина [12], что обусловлено образованием гетероолигомерного комплекса инсулина с С-пептидом, повышающим биодоступность инсулина и улучшающим его взаимодействие с инсулиновым рецептором [13]. Чувствительность инсулиновой системы к своему агонисту повышается также в присутствии ингибиторов протеинфосфотирозинфосфатаз РТР1В и ТСРТР, негативных регуляторов инсулинового сигналинга, причем эти ингибиторы усиливают не только инсулиновый, но и лептиновый сигналинг, что вносит значимый вклад в снижение ИР при СД2 [14, 15].
Перспективным может оказаться и совместное применение интраназально вводимого инсулина с различными нейропротекторами и противовоспалительными препаратами. Многочисленные экспериментальные данные показывают, что при СД2 и метаболическом синдроме в нейронах усиливаются окислительный стресс и стресс эндоплазматического ретикулума, запускаются апоптотические и провоспалительные процессы, и главными факторами здесь являются устойчивая гипергликемия и обусловленное ею повышение уровня гликированных белков [16–18]. Совместное использование инсулина с противовоспалительными препаратами широкого спектра действия могло бы повысить эффективность инсулиновой терапии для коррекции центральной и периферической ИР. В качестве одного из перспективных кандидатов на роль таких препаратов нами рассматриваются ганглиозиды, которые, как было показано ранее, являются не только нейропротекторами, но и гепатопротекторами [19–22]. Ганглиозиды, относящиеся к классу сложных гликосфинголипидов, участвуют в процессах межклеточного взаимодействия, синаптогенеза и нейротрансмиссии [23]. В соответствии с последними исследованиями, экзогенные ганглиозиды способны подавлять воспалительные процессы, модулируя активность микроглиальных клеток [24]. Учитывая тот факт, что они хорошо растворимы в водных растворах и не вызывают раздражения слизистой оболочки носа, интраназальное введение может использоваться для эффективной доставки ганглиозидов в мозг. Необходимо отметить, что ранее такой способ их введения не использовался. Однако успехи в доставке различных водорастворимых препаратов путем их интраназального введения, включая инсулин [25], более подходящие в условиях клиники по сравнению с интрацеребровентрикулярным введением, указывают на обоснованность такого подхода в случае применения ганглиозидов для воздействия на нейроны головного мозга.
Цель наших исследований состояла в изучении влияния интраназально вводимых инсулина и ганглиозидов как раздельно, так и совместно на гомеостаз глюкозы, уровень инсулина и инсулиновые сигнальные пути в печени крыс линии Wistar при СД2, который индуцировали высокожировой диетой (ВЖД) и низкой дозой стрептозотоцина (СТЗ).
МЕТОДЫ ИССЛЕДОВАНИЯ
Для экспериментов использовали самцов крыс линии Wistar, которых содержали в стандартных условиях вивария. Все процедуры проводили в соответствии с правилами, разработанными Комитетом по биоэтике ИЭФБ РАН (15.02.2018 г.), и требованиями, изложенными в документах “European Communities Council Directive 1986” (86/609/EEC) и “Guide for the Care and Use of Laboratory Animals”.
Подготовка препаратов для интраназальных инъекций. Экстракцию ганглиозидов из мозга теленка проводили по методу Фолча, как описано нами ранее [26]. Экстрагированные ганглиозиды дополнительно очищали на колонках Sephadex G-25 “Fine” (“Pharmacia”, Щвеция) [27]. Содержание ганглиозидов оценивали по реакции сиаловых кислот с резорциновым реагентом. Для интраназального введения ганглиозиды разводили в физиологическом растворе до конечной концентрации 1 мг/10 мкл, инсулин (#I5500, “Sigma”, США) растворяли в 0.1 М натрий-цитратном буфере (pH 4.5) до концентрации 0.5 МЕ/20 мкл. Совместное интраназальное введение инсулина и ганглиозидов проводили с интервалом 10 мин.
Экспериментальная модель диабета. Для моделирования СД2 использовали самцов крыс Wistar, возраст которых на начало эксперимента составил 2 мес. СД2 индуцировали ВЖД, подробно описанной в работе [28], и однократной инъекцией низкой дозы СТЗ (20 мг/кг), который вводили в/б через 11 нед после начала ВЖД. В среднем СД2 развивается у 60–80% животных, вследствие чего на начальном этапе количество животных всегда берется с полуторным запасом. В нашем случае изначально было взято 54 крысы для индуцирования диабета, из которых для дальнейших экспериментов отобрано 36 животных. Результаты глюкозотолерантного теста, проведенного через 4 нед после введения СТЗ, показали, что у крыс развилась среднетяжелая форма СД2 с выраженными нарушениями толерантности к глюкозе. Животные двух контрольных групп получали стандартный корм и однократную инъекцию 0.1 М натрий-цитратного буфера (pH 4.5) вместо СТЗ. Из пула крыс с подтвержденным развившимся СД2 были сформированы 4 диабетические группы по 9 крыс в каждой. В течение последующих четырех недель проводили ежедневные интраназальные введения следующих препаратов: Группа 1 (Контроль, К) – физиологический раствор, Группа 2 (Контроль + + Ганглиозиды + Инсулин, КГИ) – ганглиозиды (6 мг/кг/сутки) и инсулин (0.5 МЕ/крысу/сутки), Группа 3 (Диабет, Д) – физиологический раствор, Группа 4 (Диабет + Инсулин, ДИ) – инсулин (0.5 МЕ/крысу/сутки), Группа 5 (Диабет + Ганглиозиды, ДГ) – ганглиозиды (6 мг/кг), Группа 6 (Диабет + Ганглиозиды + Инсулин, ДГИ) – ганглиозиды (6 мг/кг/сутки) и инсулин (0.5 МЕ/крысу/сутки). По истечении 4-недельного лечения крыс наркотизировали с помощью хлоралгидрата (400 мг/кг, “Sigma”, США), проводили декапитацию, оценивали массу абдоминального и эпидидимального жира. Образцы печени замораживали на сухом льду сразу же после извлечения и хранили в холодильной камере при –80°С для последующей оценки содержания исследуемых белков и их фосфорилированных форм методом Вестерн-блоттинга и изучения экспрессии целевых генов методом ПЦР в реальном времени.
Глюкозотолерантный тест. За два дня до окончания лечения интраназально вводимыми инсулином и ганглиозидами чувствительность к глюкозе оценивали с помощью орального глюкозотолерантного теста (оГТТ), для чего крысам перорально через зонд вводили глюкозу (2 г/кг), уровень которой в крови измеряли до и через 15, 30, 60 и 120 мин после нагрузки сахаром с помощью глюкометра и тест-полосок “One Touch Ultra” (США). оГТТ является не только более щадящим по сравнению с ГТТ с в/б инъекцией глюкозы, но и позволяет связать повышение секреции инсулина в ответ на глюкозную нагрузку с изменением инкретинового статуса, который меняется при всасывании глюкозы в желудочно-кишечном тракте. Для верификации развития СД2 у животных на стадии индукции диабета использовали глюкозотолерантный тест, в ходе которого глюкозу в той же дозе вводили в/б и по такому же протоколу измеряли уровень глюкозы в крови.
Иммуноферментный анализ. Уровень инсулина в плазме крови крыс оценивали до и через 30, 60 и 120 мин после глюкозной нагрузки, используя набор “Rat Insulin ELISA kit” (“Mercodia”, Швеция) в соответствии с инструкцией производителя. Объем забираемой из хвостовой вены крови для определения инсулина в каждой точке составил 100 мкл, для определения уровня гормона с помощью ИФА на каждую пробу использовали по 10 мкл полученной плазмы.
Вестерн-блоттинг. Ткань печени гомогенизировали в соотношении 1: 20 в лизирующем буфере, содержащем 20 мM Tris-HCl (pH 7.5), 150 мM NaCl, 2 мM EDTA, 2 мM EGTA, 0.5% Triton X-100, 0.5% дезоксихолат натрия, 15 мM NaF, 10 мM глицерофосфат натрия, 10 мM пирофосфат натрия, 1 мM Na3VO4, 1 мM фенилметилсульфонил фторид (PMSF), 0.02% NaN3 и коктейль ингибиторов протеаз (“Roche”, США). Клеточные фрагменты и неразрушенные клетки осаждали центрифугированием 500 g × 10 мин (4°C). Концентрацию белка в пробах измеряли по модифицированному методу Лоури. Для вертикального электрофореза в камерах “Mini-Protean” (“Bio-Rad”, США) на 9–12% полиакриамидные гели загружали пробы, содержащие 25–30 мкг белка. В качестве стандарта молекулярного веса использовали окрашенные маркеры “Spectra Multicolour Broad Range Protein Ladder (10–260 кДа)” (“Thermo Fisher Scientific”, США). Перенос белков на нитроцеллюлозную мембрану (0.45 μM, “Amersham”, Великобритания) методом мокрого переноса осуществляли в минитрансблоттере (“Bio-Rad”, США) в буфере, содержащем 25 мМ Tris-HCl, 192 мМ глицин, 20% этанола (v/v), при постоянном напряжении 100 В. Для блокирования неспецифического связывания мембраны инкубировали 30 мин при комнатной температуре в буфере, содержащем 20 мМ Трис-HC1 (pH 7.6), 150 мМ NaCl, 5% обезжиренного молока (“Europek”, Россия), 0.1% Tween-20. Инкубацию мембран с растворами первичных антител проводили при +4°С на шейкере MR-1 (“Biosan”, Латвия) в течение ночи. Первичные антитела разводили в большинстве случаев в 20 мМ Трис-HCl (pH 7.6), 150 мМ NaCl, 5% БСА-V фракция (“Amresco”, США), 0.1% Tween-20 в соотношении (1: 1000). Антитела к pAkt(Ser473) (#4058), Akt (#9272), pGSK3β(Ser9) (#9322), GSK3β (#9315), p-p38-MAPK(Thr180/Tyr182) (#4511) и p38-MAPK (#8690) были произведены компанией “Cell Signaling Technology” (США), антитела к SOCS3 (#ab16030) – “Abcam” (Великобритания), PTP1B (#610139) – “BD Transduction Laboratories” (США), GAPDH (#NB600-502) – “Novus Biologicals” (США). После 3-кратной промывки 20 мМ Триc-HCl (pH 7.6), 150 мМ NaCl, 0.1% Tween-20 мембраны покрывали растворами вторичных антител, приготовленными на 5% обезжиренном молоке в том же буфере и инкубировали в течение 1 ч при комнатной температуре. В качестве вторичных антител использовали анти-кроличьи (#7074) или анти-мышиные (#7076) IgG (“Cell Signaling Technology”, США), конъюгированные с пероксидазой хрена (HRP), либо биотинилированные IgG(H+L) (#14708, #14709) и стрептавидин-HRP (#405210, “BioLegend”). Сигнал HRP усиливали коммерческим ECL (Novex, США) или аналогом ECL, приготовленным в лабораторных условиях (люминол – пара-кумаровая кислота – Н2O2). Хемилюминесцентное свечение фиксировали на голубой фотопленке (“Phenix, Research Products”, США). Для нормализации данных мембраны после стриппинга окрашивали антителами к GAPDH. Проявленные фотопленки визуализировали на сканере “Canon” (CanoScar 800F). Денситометрическая обработка данных проводилась с помощью программы Bio7.
ПЦР в реальном времени. Тотальную РНК экстрагировали реактивом RNA Extract (“Евроген”, Россия). Кодирующую ДНК синтезировали с помощью набора “MMLV RT Kit” того же производителя. Амплификацию проводили в смеси, содержащей 10 нг обратно транскрибированного продукта, по 0.4 мкМ прямого и обратного праймеров, используя среду qPCRmix-HS SYBR+LowROX (“Евроген”, Россия) на приборе “7500 Real-Time PCR System” (“Thermo Fisher Scientific Inc.”, США). Экспрессию генов, кодирующих инсулиновый рецептор (InsR) и референсные гены Aсtb и 18S-rRNA, определяли с помощью следующих праймеров: InsR – CTGGAGAACTGCTCGGTCATT (For) и GGCCATAGACACGGAAAAGAAG (Rev), Aсtb – GCGAGTACAACCTTCTTGCAG (For) и CTGACCCATACCCACCATCAC (Rev), 18S-rRNA – CTGGAGAACTGCTCGGTCATT (For) и GGCCATAGACACGGAAAAGAAG (Rev). Результаты ПЦР анализировали с помощью программного обеспечения 7500 Software v 2.0.6. и ExpressionSuite Software v1.0.3. Для нормализации данных вычисляли показатель ∆Ct, равный разности среднего геометрического Ct референсных генов (18S и актина B) и значения Ct для исследуемого транскрипта. Для оценки уровня экспрессии изучаемых генов проводили сравнение с контрольными образцами путем вычисления значения ∆∆Ct, равного разности ∆Ct контроля из ∆Ct образца.
Статистическая обработка. Анализ полученных данных проводили в программе “Prism”. Данные представляли как среднее значение ± SEM. Нормальность распределения проверяли с помощью критерия Шапиро–Уилка. Для сравнения двух выборок с нормальным распределением использовали t-критерий Стьюдента. Статистически значимыми считали отличия при уровне значимости p < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Влияние раздельного и совместного интраназального введения инсулина и ганглиозидов на метаболические показатели и экспрессию гена инсулинового рецептора в печени диабетических крыс
Введение низкой дозы СТЗ (20 мг/кг) крысам с ожирением (возраст 4.5 месяца) приводило к развитию среднетяжелой формы СД2 с выраженными нарушениями толерантности к глюкозе. На момент окончания эксперимента у СД2 крыс, достигших возраста 7 месяцев, наблюдали не только повышение уровня глюкозы натощак с 4.66 ± 0.13 мМ в контрольной группе (К) до 5.59 ± 0.13 мМ в диабетической группе (Д) с высоким уровнем достоверности (p < 0.001), но и увеличение содержания инсулина в крови с 0.56 ± 0.04 нг/мл (группа К) до 0.76 ± 0.04 нг/мл (группа Д) (p <0.01) (табл. 1). Через 2 ч после нагрузки глюкозой (2 г/кг, перорально) у крыс диабетической группы концентрация глюкозы в крови превышала 9 мМ. Животные с СД2 имели повышенную массу тела, что обусловлено накоплением жировых запасов, причем в равной степени увеличивалось содержание как абдоминального, так и эпидидимального жиров (табл. 1).
Таблица 1.
Влияние интраназального введения инсулина (0.5 МЕ/крысу/сутки) и ганглиозидов (6 мг/кг/сутки) на массу тела, количество жировой ткани и метаболические показатели у самцов крыс с СД2, вызванным высокожировой диетой и низкой дозой стрептозотоцина
| К, n = 9 | КГИ, n = 9 | Д, n = 9 | ДИ, n = 9 | ДГ, n = 9 | ДГИ, n = 9 | |
|---|---|---|---|---|---|---|
| Масса тела, г | 385.3 ± 14.8 | 382.2 ± 12.2 | 427.4 ±9.6* | 416.8 ± 15.9 | 400.5 ± 14.3 | 411.7 ± 8.3 |
| Масса жира, г | 11.01 ± 0.63 | 9.28 ± 1.32 | 19.31 ± 1.14*** | 16.31 ± 1.00** | 14.29 ± 1.88# | 16.69 ± 1.42** |
| Глюкоза натощак, мМ | 4.66 ± 0.13 | 5.43 ± 0.11** | 5.59 ±0.13*** | 4.92 ± 0.17# | 5.42 ± 0.15** | 5.12 ± 0.16*# |
| Глюкоза (оГТТ, 120 мин), мМa | 6.40 ± 0.28 | 5.48 ± 0.28* | 9.28 ± 0.36*** | 7.56 ± 0.40*# | 7.22 ± 0.32## | 7.02 ± 0.17### |
| Инсулин натощак, нг/мл | 0.56 ± 0.04 | 0.54 ± 0.05 | 0.76 ±0.04** | 0.57 ± 0.04# | 0.67 ± 0.11 | 0.53 ± 0.05# |
| Инсулин (оГТТ, 120 мин), нг/мл a | 1.01 ± 0.09 | 0.82 ± 0.09 | 2.38 ± 0.20*** | 1.41 ±0.21## | 1.71 ± 0.18** # | 1.48 ± 0.21# |
Примечание. a – уровни глюкозы и инсулина через 120 мин после глюкозной нагрузки при проведении оГТТ. Данные представлены как среднее±SEM. Различия значимы по сравнению с группой К при: * – p < 0.05;** – p < 0.01;*** – p < 0.001. Различия значимы по сравнению с группой Д при: # – p < 0.05; ## – p < 0.01; ### – p < 0.001.
Для лечения использовали монотерапию в виде интраназального введения инсулина (0.5 МЕ/крысу, ИВИ) или суммарных ганглиозидов мозга теленка (6 мг/кг, ИВГ), а также совместное введение ИВИ и ИВГ в указанных дозах в течение 4 нед. При интраназальном способе введения инсулин и ганглиозиды транспортируются преимущественно в мозг и в значимых количествах не поступают в периферические органы и ткани. Это позволяет изучать центральные эффекты этих соединений на метаболические процессы на периферии. У крыс в интактной контрольной группе и ИВИ – контрольной группе не выявлено различий уровня глюкозы натощак (4.84 ± 0.16 мМ и 5.09 ± ± 0.15 мМ, соответственно, p > 0.05), что свидетельствует об отсутствии влияния ИВИ на глюкозный гомеостаз в отличие от системного введения инсулина. Не было различий и в уровне глюкозы между интактным контролем и группой здоровых крыс, получавшей ИВГ (5.12 ± 0.1 мМ, p > 0.05 в сравнении с контролем). Достаточно неожиданно было то, что при введении контрольным крысам ИВИ и ИВГ (группа КГИ) уровень глюкозы натощак значимо возрастал в сравнении с контролем (табл. 1). При этом совместное введение ИВИ и ИВГ повышало у контрольных крыс скорость утилизации глюкозы, на что указывают рассчитанные значения AUC0–120 (интегрированная площадь под кривой “концентрация глюкозы (мМ)–время (мин)” для глюкозных кривых в интервале времени от 0 до 120 мин в оральном глюкозотолерантном тесте (рис. 1a), результатом чего было значительное снижение в сравнении с контролем уровня глюкозы через 120 мин после нагрузки глюкозой (табл. 1). Лечение диабетических крыс ИВГ и комбинацией ИВИ и ИВГ восстанавливало чувствительность к инсулину, о чем свидетельствуют сниженные в сравнении с группой Д значения AUC0–120 (интегрированная площадь под кривой “концентрация инсулина (нг/мл) – время (мин)” во временном промежутке от 0 до 120 мин после глюкозной нагрузки (рис. 1b). При этом в случае монотерапии ИВИ отмечали тенденцию к снижению, но различия с группой Д не были значимыми (p > 0.05).
Рис. 1.
Оценка влияния ИВИ (0.5 МЕ/крысу/сутки) и ИВГ (6 мг/кг/сутки) при раздельном и совместном введении на чувствительность к глюкозе и инсулину у крыс с СД2, вызванным высокожировой диетой и низкой дозой стрептозотоцина, при проведении орального глюкозотолерантного теста. Обозначения групп на графике: группа 1 (C) – контроль (К), группа 2 (CGI) – контроль + ганглиозиды + инсулин (КГИ), группа 3 (D) – диабет (Д), группа 4 (DI) – диабет + инсулин (ДИ), группа 5 (DG) – диабет + ганглиозиды (ДГ), группа 6 (DGI) – диабет + ганглиозиды + инсулин (ДГИ).
(a) – AUC0–120, интегрированная площадь под кривой “концентрация глюкозы (мМ) – время (мин)” для глюкозных кривых в течение 120 мин после нагрузки глюкозой. Данные представлены как среднее ± SEM (n = 9). (b) – AUC0–120, интегрированная площадь под кривой “концентрация инсулина (нг/мл) – время (мин)” для инсулиновых концентрационных кривых в течение 120 мин после нагрузки глюкозой. Данные представлены как среднее ± SEM (n = 5). Различия значимы по сравнению с контрольными крысами (К) при: а – p < 0.05; b – p < 0.01; c – p < 0.001. Различия значимы по сравнению с необработанными диабетическими крысами (Д) при: d – p < 0.05; e – p < 0.01; f – p < 0.001.

Поскольку чувствительность к инсулину может зависеть от количества инсулиновых рецепторов в тканях-мишенях, оценивали экспрессию гена инсулинового рецептора (InsR) в печени крыс. Показано, что экспрессия этого гена при СД2 не отличалась от таковой в контроле, но в группе диабетических крыс, обработанных ИВГ, она достоверно повышалась на 56% (p < 0.05 в сравнении с группами К и Д) (рис. 2). При этом экспрессия гена InsR в группах диабетических крыс, обработанных ИВИ или комбинацией ИВИ и ИВГ, по сравнению с группами К и Д не изменялась (рис. 2). Это указывает на то, что обнаруженное нами отчетливо выраженное повышение чувствительности к инсулину в группе ДГИ не может быть обусловлено изменением экспрессии гена инсулинового рецептора в печени, как одной из основных мишеней инсулина.
Рис. 2.
Оценка влияния ИВИ (0.5 МЕ/крысу/сутки) и ИВГ (6 мг/кг/сутки) при раздельном и совместном введении на экспрессию гена инсулинового рецептора (InsR) в печени крыс с СД2, вызванным высокожировой диетой и низкой дозой стрептозотоцина. Обозначения групп на графике: группа 1 (C) – контроль (К), группа 2 (CGI) – контроль + ганглиозиды + инсулин (КГИ), группа 3 (D) – диабет (Д), группа 4 (DI) – диабет + инсулин (ДИ), группа 5 (DG) – диабет + ганглиозиды (ДГ), группа 6 (DGI) – диабет + ганглиозиды + инсулин (ДГИ). Уровень экспрессии гена InsR нормировали по уровню экспрессии генов Actb и 18S rRNA. Данные представлены как среднее ± SEM (n = 9). Различия статистически значимы: а – p < 0.05 по сравнению с группой 1 (С) контрольной (К), d – p < 0.05 по сравнению с группой 3 (D) диабетической (Д).

Влияние раздельного и совместного интраназального введения инсулина и ганглиозидов на сигнальные пути, активируемые инсулином, в печени диабетических крыс
Поскольку ключевую роль в негативной регуляции инсулинового сигналинга в печени и других тканях играют протеинфосфотирозинфосфатаза РТР1В и супрессор цитокинового сигналинга SOCS3, то с помощью Вестерн-блоттинга и специфических антител к этим белкам определяли их содержание в ткани печени диабетических крыс с лечением ИВИ и ИВГ и без такового в сравнении с контрольными животными. Показано, что содержание белка SOCS3 в печени не менялось во всех изученных экспериментальных группах, в то время как уровень белка РТР1В снижался как у контрольных, так и у диабетических крыс, обработанных совместно ИВИ и ИВГ (рис. 3).
Рис. 3.
Влияние ИВИ (0.5 МЕ/крысу/сутки) и ИВГ (6 мг/кг/сутки) при раздельном и совместном введении на содержание белка РТР1В и SOCS3 в печени крыс с СД2, вызванным высокожировой диетой и низкой дозой стрептозотоцина. Обозначения групп на рисунке: группа 1 (C) – контроль (К), группа 2 (CGI) – контроль + ганглиозиды + инсулин (КГИ), группа 3 (D) – диабет (Д), группа 4 (DI) – диабет+инсулин (ДИ), группа 5 (DG) – диабет + ганглиозиды (ДГ), группа 6 (DGI) – диабет + ганглиозиды + инсулин (ДГИ). (a) – изменение экспрессии протеинфосфотирозинфосфатазы РТР1В, (b) – изменение экспрессии супрессора цитокинового сигналинга SOCS3. Данные представлены как среднее ± SEM (n = 9). Различия значимы по сравнению с контрольными крысами (К) при: а – p < 0.05; b – p < 0.01; c – p < 0.001. Различия значимы по сравнению с необработанными диабетическими крысами (Д) при: d – p < 0.05; e – p < 0.01; f – p < 0.001.
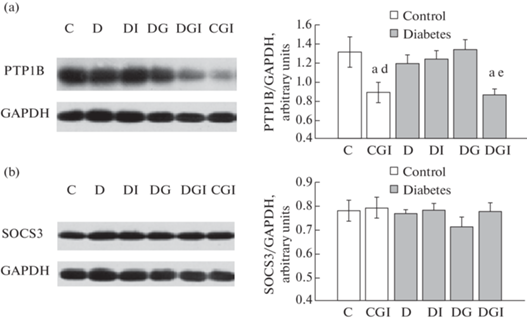
При этом в группе КГИ количество белка РТР1В было снижено на 34% в сравнении с интактным контролем, а в группе ДГИ отмечали снижение количества белка РТР1В как по отношению к контролю, так и к диабетической группе (Д), причем различий между группами КГИ и ДГИ по этому показателю выявлено не было (рис. 3a). Наблюдаемое снижение содержания РТР1В в печени может способствовать увеличению чувствительности гепатоцитов к инсулину. У контрольных крыс такая сенситизация, вероятно, обеспечивает повышение утилизации глюкозы, наблюдаемое нами в оральном глюкозотолерантном тесте (табл. 1), а в случае диабетических крыс опосредовать восстанавливающее действие комбинации ИВИ и ИВГ на нарушенную при СД2 толерантность к глюкозе (табл. 1, рис. 1a).
Основной мишенью инсулина является фермент серин/треониновая протеинкиназа Akt, которая в печени вовлечена в контроль выживаемости клеток, а также регулирует скорость утилизации глюкозы (инсулин-зависимый транспорт глюкозы в клетку), синтез гликогена и метаболизм липидов. В активированной инсулином или другими гормонами форме Akt-киназа фосфорилирована по остаткам Ser473 (основной сайт фосфорилирования) и(или) Thr308. Несмотря на развитие инсулиновой резистентности, уровень экспрессии фосфорилированной по остатку Ser473 Akt-киназы в печени диабетических крыс значимо не отличался от контрольной группы (рис. 4a). Лечение диабетических крыс ИВИ, ИВГ и совместно ИВИ и ИВГ приводило к достоверному повышению количества Ser473-фосфорилированной формы Akt-киназы в сравнении с группой К, а в группах ДГ и ДГИ также и в сравнении с группой Д (рис. 4a). Стимулирующий эффект на Ser473-фосфорилирование Akt-киназы в печени контрольных крыс оказывало также комбинированное применение ИВИ и ИВГ (p < 0.01 в сравнении с группой К) (рис. 4a).
Рис. 4.
Влияние ИВИ (0.5 МЕ/крысу/сутки) и ИВГ (6 мг/кг/сутки) при раздельном и совместном введении на уровень фосфорилирования протеинкиназ Akt, GSK3β и p38MAPK в печени крыс с СД2, вызванным высокожировой диетой и низкой дозой стрептозотоцина. Обозначения групп на рисунке: группа 1 (C) – контроль (К), группа 2 (CGI) – контроль + ганглиозиды + инсулин (КГИ), группа 3 (D) – диабет (Д), группа 4 (DI) – диабет+инсулин (ДИ), группа 5 (DG) – диабет + ганглиозиды (ДГ), группа 6 (DGI) – диабет + ганглиозиды + инсулин (ДГИ). (a) – фосфорилирование Akt-киназы по Ser473, (b) – фосфорилирование киназы GSK3β по Ser9, (c) –фосфорилирование p38-MAPK по Thr180/Tyr182. Данные представлены как среднее ± SEM (n = 9). Различия значимы по сравнению с контрольными крысами (К) при: а – p < 0.05; b – p < 0.01; c – p < 0.001. Различия значимы по сравнению с необработанными диабетическими крысами (Д) при: d – p < 0.05; e – p < 0.01; f – p < 0.001.

При активации Akt-киназы запускается каскад внутриклеточных реакций, ответственных за синтез гликогена, в основе чего лежит ингибирующее Akt-опосредуемое фосфорилирование фермента киназы гликогенсинтазы-3β (GSK3β) по остатку Ser9. С помощью Вестерн-блоттинга показано, что количество фосфорилированной формы GSK3β(Ser9) не в полной мере коррелирует с количеством Ser473-фосфорилированной формы Akt-киназы (рис. 4a, 4b). Лечение крыс одним ИВГ и совместно ИВИ и ИВГ приводило к повышению количества GSK3β(Ser9) в группах ДГ, ДГИ и КГИ, в то время как лечение одним ИВИ не влияло на фосфорилирование этого фермента, которое не отличалось от такового в контроле и в группе Д (рис. 4b). Помимо Akt-киназы фосфорилирование GSK3β в печени может осуществляться другими протеинкиназами, к числу которых принадлежит киназа р38-МАРК, относящаяся к семейству митогенактивируемых протеинкиназ, и активируемая при фосфорилировании по остаткам Thr180 и Tyr182. В отличие от Akt-киназы накопление фосфорилированной формы р38MAPK (Thr180/Tyr182) в группах КГИ, ДГ и ДГИ было выражено в еще большей степени (в среднем в три раза в сравнении с контролем и группой Д) (рис. 4c).
Необходимо отметить, что, как и в случае Akt-киназы, фосфорилирование р38-MAPK в печени диабетических крыс, обработанных одним ИВИ, не отличалась от такового в контрольной и диабетической группах (рис. 4c). Таким образом, выявленное нами ингибирующее фосфорилирование GSK3β по остатку Ser9 может осуществляться как вследствие активации Akt-киназы, так и р38-МАРК, причем вклад последней, как мы полагаем, более значимый.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Гипоталамус контролирует не только пищевое поведение и энергетический баланс, но и является важнейшим звеном, отвечающим за центральную регуляцию метаболизма глюкозы и чувствительности тканей к инсулину на периферии [29]. Тот факт, что при интрацеребровентрикулярном введении инсулина подавляется глюконеогенез в печени и снижается уровень глюкозы в крови, служит подтверждением важности инсулинового сигналинга в мозге для поддержания гомеостаза глюкозы [30]. Благодаря воздействию инсулина на компетентные к этому гормону гипоталамические сигнальные системы снижается тонус симпатической передачи к жировой ткани, что приводит к подавлению липолиза и стимулирует синтез жирных кислот и триглицеридов адипоцитами [31].
В условиях СД2 и метаболического синдрома, в результате нарушения транспорта инсулина через гематоэнцефалических барьер, в мозге наблюдается дефицит инсулина [32, 33]. Наряду с этим в условиях липотоксичности, усиления окислительного стресса и воспаления, характерных для СД2, ослабляется активность инсулиновой системы мозга. Одним из перспективных подходов для компенсации инсулинового дефицита в ЦНС и нормализации инсулинового сигналинга является интраназальный способ введения инсулина, позволяющий, минуя гематоэнцефалический барьер, доставить гормон в гипоталамус и другие отделы мозга. Показано, что однократная обработка с помощью ИВИ уже через 10–20 минут увеличивает концентрацию инсулина в мозге и спинномозговой жидкости без влияния на содержание инсулина в крови [10]. При длительной обработке с помощью ИВИ накопление инсулина в мозге позволяет добиться частичной нормализации метаболических показателей и восстановления функциональной активности сигнальных систем на периферии и в ЦНС при различных формах диабета [11, 12]. Однако на фоне продолжительного интраназального введения инсулина могут выявляться побочные эффекты, среди которых значимое повышение уровня тиреотропного гормона в крови и нарушение функционирования тиреоидной системы [11]. В целях минимизации негативных последствий ИВИ продолжается поиск препаратов, способных повысить эффективность ИВИ, снизить его дозы и оптимизировать продолжительность ИВИ терапии. Одними из кандидатов на роль таких соединений является α-токоферол, основной компонент витамина Е [34]. Определенные надежды связывают и с применением ганглиозидов мозга.
Ганглиозиды, обладающие нейропротекторными свойствами, впервые использовались нами in vivo для коррекции когнитивных нарушений у крыс с неонатальной моделью СД2, причем по эффективности они не уступали ИВИ [35]. Препарат суммарных ганглиозидов, экстрагированных из мозга быка, состоит из моносиалоганглиозида GM1 (19.5%), дисиалоганглиозидов GD1a (38%) и GD1b (11%) и трисиалоганглиозида GT1b (25%) [36]. В наших экспериментах in vitro все основные ганглиозиды мозга защищали культивируемые нейроны от эксайтотоксичности [19]. Yamamoto и Mohanan также показали, что нейропротекторным эффектом обладает как наиболее стабильный GM1, так и другие основные ганглиозиды мозга [37]. В отличие от инсулина, который даже при интраназальном способе доставки в мозг подвергается частичному протеолизу, полная деградация ганглиозидов осуществляется лишь в лизосомах. Можно предположить, что от ганглиозидов GD1a, GD1b и GT1b при их интраназальном введении под действием нейраминидазы, локализованной в плазматической мембране, отщепляется сиаловая кислота. В этом случае будет образовываться ганглиозид GM1, эффекты которого изучены в наибольшей степени, поскольку GM1-содержащая фракция ганглиозидов используется для большинства экспериментов in vitro и in vivo. Таким образом, ганглиозиды, прежде всего GM1, должны сохранять структурную целостность и биологическую активность при их интраназальном введении.
Разнообразные по характеру эффекты ганглиозидов связывают с их способностью встраиваться в липидные рафты плазматической мембраны, что приводит к изменению ее жидкостных свойств и состава микродоменов, в том числе вызывая изменения и в локализации сигнальных белков [38]. Модуляция функциональной активности сопряженных с G-белками рецепторов, а также рецепторов, наделенных тирозинкиназной активностью, имеет место в результате непосредственного взаимодействия ганглиозидов как с этими рецепторами, так и с находящимися с ними в комплексе регуляторными и адаптерными белками [21, 23, 39, 40]. В последние годы появились доказательства участия ганглиозидов в регуляции сигнальных путей инсулина [41, 42]. Инсулин вызывает гиперполяризацию гипоталамических нейронов, экспрессирующих агути-подобный пептид, через АТФ-зависимые калиевые (${\text{K}}_{{{\text{АТФ}}}}^{ + }$) каналы, что в дальнейшем при участии блуждающего нерва приводит к подавлению глюконеогенеза в печени [29]. Экзогенный ганглиозид GM1 также способен активировать ${\text{K}}_{{{\text{АТФ}}}}^{ + }$-каналы через посредство генерации мощного вазодилататора NO, что было показано на мезентериальной артерии [43]. Именно с индукцией вазодилатации связывают нейропротекторное действие GM1 при ишемии головного мозга. Влияние экзогенных ганглиозидов на ${\text{K}}_{{{\text{АТФ}}}}^{ + }$-каналы гипоталамических нейронов пока не исследовано. Однако эндогенные аналоги ганглиозидов, как было показано в проопиомеланокортин-экспрессирующих нейронах с нокаутом по одному из ферментов биосинтеза гликосфинголипидов, способны регулировать связывание фосфатидилинозитол-3-фосфата c SUR-1 субъединицей ${\text{K}}_{{{\text{АТФ}}}}^{ + }$-канала [44].
Нами показано, что 4-недельное лечение крыс с СД2 с помощью ИВИ и ИВГ восстанавливало толерантность к глюкозе, причем совместное введение препаратов было более эффективным (рис. 1). Одной из молекулярных причин этого, с нашей точки зрения, является повышение чувствительности паренхимы печени к инсулину. Так, у диабетических крыс, леченых совместно ИВИ и ИВГ, снижалось содержание белка, негативного регулятора инсулинового сигналинга – фосфатазы РТР1В, который, как известно, дефосфорилирует активированный гормоном инсулиновый рецептор и его субстраты IRS1 и IRS2, блокируя передачу инсулинового сигнала к внутриклеточным мишеням [45, 46]. При этом, однако, нами не было выявлено снижения экспрессии белка SOCS3, который также вовлечен в негативный контроль чувствительности клеток к инсулину, а также к адипокину лептину, чьи сигнальные пути тесно взаимосвязаны с таковыми инсулина [47]. Необходимо отметить, что семейство супрессоров цитокинового сигналинга, помимо SOCS3, включает ряд других представителей, в том числе SOCS7 [48]. На периферии SOCS7 вносит значимый вклад в регуляцию метаболизма глюкозы, и при делеции кодирующего его гена усиливается поглощение глюкозы тканями и нарушается толерантность к глюкозе [49]. Возможно, что при ИВГ и ИВИ меняется экспрессия белка SOCS7, что требует дальнейшего изучения.
Сигнальный путь, включающий инсулиновый рецептор, белок IRS1/2, фосфатидилинозитол-3-киназу и Akt-киназу, играет центральную роль в реализации эффектов инсулина в клетках-мишенях, включая гепатоциты [50]. В условиях СД2 и метаболического синдрома воспаление печеночной паренхимы, наблюдаемое в виде жировой трансформации или стеатогепатоза, приводит к развитию инсулиновой резистентности, но причиной этого может быть не изменение экспрессии и функциональной активности инсулиновых рецепторов, а модуляция активности нижележащих эффекторных звеньев инсулинового сигналинга, и в первую очередь сигнального пути, включающего фосфатидилинозитол-3-киназу и Akt-киназу [50, 51]. Показано, например, что выключение в печени мышей гена, кодирующего каталитическую субъединицу фосфатидилинозитол-3-киназу – p110α, снижает чувствительность мутантных животных к инсулину, нарушает толерантность к глюкозе, усиливает глюконеогенез, повышает уровень лептина в крови и препятствует реализации терапевтического эффекта метформина на липидный и углеводный обмен [52]. Нами в печени диабетических крыс не было выявлено снижения степени фосфорилирования Akt-киназы. Однако с учетом повышенного уровня инсулина в крови и сохранения экспрессии инсулинового рецептора в печени можно сделать вывод об ослаблении ответа этого фермента на стимуляцию инсулином, что является еще одним свидетельством в пользу инсулиновой резистентности в гепатоцитах. При этом лечение диабетических животных с помощью ИВГ и комбинации ИВИ и ИВГ повышало содержание в печени Ser473-фосфорилированной формы Akt-киназы и соотношение pAkt(Ser473)/Akt. Интересно, что этот эффект дублируется и у контрольных крыс, обработанных ИВИ и ИВГ. Более значимое повышение соотношения pAkt(Ser473)/Akt коррелирует с более выраженным снижением массы тела и жира, обусловленным интенсификацией липолиза в адипоцитах и(или) ингибированием липогенеза в печени. Помимо гепатоцитов, Akt-киназа также локализована в эндотелии прилегающих сосудов, контролируя кровяное давление посредством активации эндотелиальной изоформы NO-синтазы, что существенно для активации процесса регенерации печеночной ткани [53, 54]. Вследствие этого вызываемое ИВГ и ИВИ повышение активности Akt-киназы может оказывать вазидилататорный эффект на сосуды, питающие гепатоциты, и тем самым улучшать обменные процессы в них.
Одной из мишеней Akt-киназы является киназа GSK3β, которая вовлечена в метаболизм глюкозы и другие биохимические процессы. Вызываемое Akt-киназой фосфорилирование GSK3β в гепатоцитах приводит к ее инактивации, подавляя глюконеогенез и стимулируя депонирование глюкозы в виде гликогена, что в конечном итоге улучшает глюкозный гомеостаз и предотвращает гипергликемию. В свою очередь повышение активности GSK3β играет негативную роль в развитии метаболических нарушений при СД2 [55]. Фосфорилирование GSK3β может осуществляться и киназой p38-MAPK, которая играет двоякую роль в пролиферации гепатоцитов, регулируя в них окислительно-восстановительные процессы и контролируя продукцию активных форм кислорода [56]. Нами показано, что повышение фосфорилирования Akt-киназы в печени крыс групп ДГ и ДГИ, а также группы КГИ ассоциировано со значимым повышением фосфорилирования киназы GSK3β по остатку Ser9, что свидетельствует о функциональной связи между активностью этих ферментов. Необходимо однако отметить, что ингибирование GSK3β может быть результатом ее фосфорилирования другой киназой – p38MAPK, способной подавлять апоптоз и повышать выживаемость клеток, что удалось продемонстрировать для тимоцитов и нейронов [57]. В печени киназа p38-MAPK необходима для полноценной регенерации этого органа после частичной гепатэктомии и, наряду с этим, вовлечена в негативную регуляцию гепатоканцерогенеза [58, 59]. Тот факт, что профиль уровня фосфорилирования GSK3β по остатку Ser9 совпадает с изменением фосфорилирования киназы p38-MAPK, позволяет предположить, что она, как и Akt-киназа, также ответственна за инактивацию GSK3β в условиях лечения диабетических крыс с помощью ИВГ или совместно ИВГ и ИВИ. Более того, количественный анализ степени фосфорилирования позволяет предположить, что вклад киназы p38-MAPK в ингибирующее фосфорилирование GSK3β может быть больше, чем таковой Akt-киназы.
Тот факт, что ганглиозиды вводились нами интраназально и непосредственно воздействовали на структуры мозга, компетентные в отношении регуляции периферической инсулиновой чувствительности и углеводного обмена в печени, свидетельствует о превалировании в этом случае центрального механизма их действия. Так, показано, что ганглиозиды плохо проникают через гематоэнцефалический барьер как в прямом, так и в обратном направлении. В опытах с внутривенной инъекцией фторированного производного [18F]-GM1 обезьянам было установлено, что через 4 ч в мозг поступало не более 0.4% исходного количества ганглиозидов [60]. Эффективность совместного введения ганглиозидов с ИВИ может указывать на взаимоусиление их сигнальных путей в гипоталамусе и других отделах мозга. Молекулярные механизмы этого процесса являются предметом наших дальнейших исследований.
Таким образом, нами впервые показано, что интраназальное введение ганглиозидов и их комбинации с инсулином приводят к нормализации метаболических показателей у крыс с СД2, улучшая у них чувствительность к глюкозе и инсулину. Одними из молекулярных причин этого являются обнаруженные нами в печени диабетических крыс, леченых совместно ИВИ и ИВГ, снижение экспрессии фосфатазы PTP1B, негативного регулятора инсулинового сигналинга, повышение активности Akt-киназы и киназы p38-MAPK, а также ингибирование активности их субстрата – киназы GSK3β. Повышение активности основных компонентов инсулинового сигналинга в печени важно как для нормализации энергетического баланса при СД2, так и для подавления в печени воспалительных процессов и интенсификации ее регенерации.
Список литературы
Brøns C, Jensen CB, Storgaard H, Hiscock NJ, White A, Appel JS, Jacobsen S, Nilsson E, Larsen CM, Astrup A, Quistorff B, Vaag A (2009) Impact of short-term high-fat feeding on glucose and insulin metabolism in young healthy men. J Physiol 587: 2387–2397. https://doi.org/10.1113/jphysiol.2009.169078
Small L, Brandon AE, Turner N, Cooney GJ (2018) Modeling insulin resistance in rodents by alterations in diet: what have high-fat and high-calorie diets revealed? Am J Physiol Endocrinol Metab 314: E251-E265. https://doi.org/10.1152/ajpendo.00337.2017
Duarte AI, Moreira PI, Oliveira CR (2012) Insulin in central nervous system: more than just a peripheral hormone. J Aging Res 2012: 384017. https://doi.org/10.1155/2012/384017
Heni M, Kullmann S, Preissl H, Fritsche A, Häring HU (2015) Impaired insulin action in the human brain: causes and metabolic consequences. Nat Rev Endocrinol 11: 701–711. https://doi.org/10.1038/nrendo.2015.173
Scherer T, Sakamoto K, Buettner C (2021) Brain insulin signalling in metabolic homeostasis and disease. Nat Rev Endocrinol 17: 468–483. https://doi.org/10.1038/s41574-021-00498-x
Adam CL, Findlay PA, Aitken RP, Milne JS, Wallace JM (2012) In vivo changes in central and peripheral insulin sensitivity in a large animal model of obesity. Endocrinology 153: 3147–3157.https://doi.org/10.1210/en.2012-1134
Chua LM, Lim ML, Chong PR, Hu ZP, Cheung NS, Wong BS (2012) Impaired neuronal insulin signaling precedes Aβ42 accumulation in female AβPPsw/PS1ΔE9 mice. J. Alzheimers Dis 29: 783–791.https://doi.org/10.3233/JAD-2012-111880
Ruegsegger GN, Manjunatha S, Summer P, Gopala S, Zabeilski P, Dasari S, Vanderboom PM, Lanza IR, Klaus KA, Nair KS (2019) Insulin deficiency and intranasal insulin alter brain mitochondrial function: a potential factor for dementia in diabetes. FASEB J 33: 4458–4472. https://doi.org/10.1096/fj.201802043R
Shpakov AO, Derkach KV, Berstein LM (2015) Brain signaling systems in the Type 2 diabetes and metabolic syndrome: promising target to treat and prevent these diseases. Future Sci OA 1: FSO25. https://doi.org/10.4155/fso.15.23
Born J, Lange T, Kern W, McGregor GP, Bickel U, Fehm HL (2002) Sniffing neuropeptides: a transnasal approach to the human brain. Nat Neurosci 5: 514–516. https://doi.org/10.1038/nn849
Derkach KV, Bogush IV, Berstein LM, Shpakov AO (2015) The influence of intranasal insulin on hypothalamic-pituitary axis in normal and diabetic rats. Horm Metab Res 47: 916–924. https://doi.org/10.1055/s-0035-1547236
Derkach KV, Bondareva VM, Perminova AA, Shpakov AO (2019) C-peptide and insulin during combined intranasal administration improve the metabolic parameters and activity of the adenylate cyclase system in the hypothalamus, myocardium, and epididymal fat of rats with type 2 diabetes. Cell and Tissue Biol 13: 228–236. https://doi.org/10.1134/S1990519X1903003
Landreh M, Johansson J, Jörnvall H (2013) C-peptide: a molecule balancing insulin states in secretion and diabetes-associated depository conditions. Horm Metab Res 45: 769–773.https://doi.org/10.1055/s-0033-1347208
Galic S, Hauser C, Kahn BB, Haj FG, Neel BG, Tonks NK, Tiganis T (200) Coordinated regulation of insulin signaling by the protein tyrosine phosphatases PTP1B and TCPTP. Mol Cell Biol 25: 819–829. https://doi.org/10.1128/MCB.25.2.819-829.2005
Zhang ZY, Dodd GT, Tiganis T (2015) Protein Tyrosine Phosphatases in Hypothalamic Insulin and Leptin Signaling. Trends Pharmacol Sci 36: 661–674. https://doi.org/10.1016/j.tips.2015.07.003
Xu W, Caracciolo B, Wang HX, Winblad B, Backman L, Qiu C, Fratiglioni L (2010) Accelerated progression from mild cognitive impairment to dementia in people with diabetes. Diabetes 59: 2928–2935.https://doi.org/10.2337/db10-0539
Asslih S, Damri O, Agam G (2021) Neuroinflammation as a Common Denominator of Complex Diseases (Cancer, Diabetes Type 2, and Neuropsychiatric Disorders). Int J Mol Sci 22: 6138. https://doi.org/10.3390/ijms22116138
André C, Guzman-Quevedo O, Rey C, Rémus-Borel J, Clark S, Castellanos-Jankiewicz A, Ladeveze E, Leste-Lasserre T, Nadjar A, Abrous DN, Laye S, Cota D (2017) Inhibiting Microglia Expansion Prevents Diet-Induced Hypothalamic and Peripheral Inflammation. Diabetes 66: 908–919.https://doi.org/10.2337/db16-0586
Avrova NF, Victorov IV, Tyurin VA, Zakharova IO, Sokolova TV, Andreeva NA, Stelmaschuk EV, Tyurina YY, Gonchar VS (1998) Inhibition of glutamate-induced intensification of free radical reactions by gangliosides: possible role in their protective effect in rat cerebellar granule cells and brain synaptosomes. Neurochem Res 23: 945–952. https://doi.org/10.1023/a1021076220411
Gorria M, Huc L, Sergent O, Rebillard A, Gaboriau F, Dimanche-Boitrel MT, Lagadic-Gossmann D (2006) Protective effect of monosialoganglioside GM1 against chemically induced apoptosis through targeting of mitochondrial function and iron transport. Biochem Pharmacol 72: 1343–1353. https://doi.org/10.1016/j.bcp.2006.07.014
Zakharova IO, Sokolova TV, Vlasova YA, Furaev VV, Rychkova MP, Avrova NF (2014) GM1 ganglioside activates ERK1/2 and Akt downstream of Trk tyrosine kinase and protects PC12 cells against hydrogen peroxide toxicity. Neurochem Res 39: 2262–2275. https://doi.org/10.1007/s11064-014-1428-6
Nikolaeva S, Bayunova L, Sokolova T, Vlasova Y, Bachteeva V, Avrova N, Parnova R (2015) GM1 and GD1a gangliosides modulate toxic and inflammatory effects of E. coli lipopolysaccharide by preventing TLR4 translocation into lipid rafts. Biochim Biophys Acta 1851: 239–247. https://doi.org/10.1016/j.bbalip.2014.12.004
Sipione S, Monyror J, Galleguillos D, Steinberg N, Kadam V (2020) Gangliosides in the Brain: Physiology, Pathophysiology and Therapeutic Applications. Front Neurosci 14: 572965. https://doi.org/10.3389/fnins.2020.572965
Galleguillos D, Wang Q, Steinberg N, Shrivastava G, Dhami K, Rubinstein K, Giuliani F, Churchward M, Power C, Todd K, Sipione S (2020) Anti-inflammatory role of GM1 and modulatory effects of gangliosides on microglia functions. bioRxiv. https://doi.org/10.1101/2020.03.04.975862
Dholakia J, Prabhakar B, Shende P (2021) Strategies for the delivery of antidiabetic drugs via intranasal route. Int J Pharm 608: 121068. https://doi.org/10.1016/j.ijpharm.2021.121068
Zakharova IO, Avrova NF (2001) The effect of cold stress on ganglioside fatty acid composition and ganglioside-bound sialic acid content of rat brain subcellular fractions. J Therm Biol 26: 215–222. https://doi.org/10.1016/s0306-4565(00)00045-0
Vanier MT, Holm M, Ohman R, Svennerholm L (1971) Developmental profiles of gangliosides in human and rat brain. J Neurochem 18: 581–592. https://doi.org/10.1111/j.1471-4159.1971.tb11988.x
Derkach KV, Bondareva VM, Chistyakova OV, Berstein LM, Shpakov AO (2015) The Effect of Long-Term Intranasal Serotonin Treatment on Metabolic Parameters and Hormonal Signaling in Rats with High-Fat Diet/Low-Dose Streptozotocin-Induced Type 2 Diabetes. Int J Endocrinol 2015: 245459. https://doi.org/10.1155/2015/245459
Ruud J, Steculorum SM, Brüning J (2017) Neuronal control of peripheral insulin sensitivity and glucose metabolism. Nat Commun 8: 15259. https://doi.org/10.1038/ncomms15259
Obici S, Zhang BB, Karkanias G, Rossetti L (2002) Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med 8: 1376–1382. https://doi.org/10.1038/nm1202-798
Scherer T, O’Hare J, Diggs-Andrews K, Schweiger M, Cheng B, Lindtner C, Zielinski E, Vempati P, Su K, Dighe S, Milsom T, Puchowicz M, Scheja L, Zechner R, Fisher SJ, Previs SF, Buettner C (2011) Brain insulin controls adipose tissue lipolysis and lipogenesis. Cell Metab 13: 183–194. https://doi.org/10.1016/j.cmet.2011.01.008
Romanova IV, Derkach KV, Mikhrina AL, Sukhov IB, Mikhailova EV, Shpakov AO (2018) The Leptin, Dopamine and Serotonin Receptors in Hypothalamic POMC-Neurons of Normal and Obese Rodents. Neurochem Res 43: 821–837. https://doi.org/10.1007/s11064-018-2485-z
Derkach K, Zakharova I, Zorina I, Bakhtyukov A, Romanova I, Bayunova L, Shpakov A (2019) The evidence of metabolic-improving effect of metformin in Ay/a mice with genetically-induced melanocortin obesity and the contribution of hypothalamic mechanisms to this effect. PLoS One 14: e0213779. https://doi.org/10.1371/journal.pone.0213779
Zakharova IO, Bayunova LV, Zorina II, Sokolova TV, Shpakov AO, Avrova NF (2021) Insulin and α-Tocopherol Enhance the Protective Effect of Each Other on Brain Cortical Neurons under Oxidative Stress Conditions and in Rat Two-Vessel Forebrain Ischemia/Reperfusion Injury. Int J Mol Sci 220: 11768. https://doi.org/10.3390/ijms222111768
Sukhov IB, Lebedeva MF, Zakharova IO, Derkach KV, Bayunova LV, Zorina II, Avrova NF, Shpakov AO (2019) Intranasal administration of insulin and gangliosides improves spatial memory in rats with neonatal type 2 Diabetes Mellitus. Bull Exp Biol Medicine 168: 317–320. https://doi.org/10.1007/s10517-020-04699-8
Tettamanti G, Bonali F, Marchesini S, Zambotti V (1973) A new procedure for the extraction, purification and fractionation of brain gangliosides. Biochim Biophys Acta 296: 160–170. https://doi.org/10.1016/0005-2760(73)90055-6
Yamamoto HA, Mohanan PV (2003) Ganglioside GT1b and melatonin inhibit brain mitochondrial DNA damage and seizures induced by kainic acid in mice. Brain Res 964: 100–106. https://doi.org/10.1016/s0006-8993(02)04083-0
Nishio M, Fukumoto S, Furukawa K, Ichimura A, Miyazaki H, Kusunoki S, Urano T, Furukawa K (2004) Overexpressed GM1 suppresses nerve growth factor (NGF) signals by modulating the intracellular localization of NGF receptors and membrane fluidity in PC12 cells. J Biol Chem 279: 33368–33378. https://doi.org/10.1074/jbc.M403816200
Farooqui T, Franklin T, Pearl DK, Yates AJ (1997) Ganglioside GM1 enhances induction by nerve growth factor of a putative dimer of TrkA. J Neurochem 68: 2348–2355. https://doi.org/10.1046/j.1471-4159.1997.68062348.x
Prasanna X, Jafurulla M, Sengupta D, Chattopadhyay A (2016) The ganglioside GM1 interacts with the serotonin 1A receptor via the sphingolipid binding domain. Biochim Biophys Acta 1858: 2818–2826. https://doi.org/10.1016/j.bbamem.2016.08.009
Ji S, Tokizane K, Ohkawa Y, Ohmi Y, Banno R, Okajima T, Kiyama H, Furukawa K, Furukawa K (2016) Increased a-series gangliosides positively regulate leptin/Ob receptor-mediated signals in hypothalamus of GD3 synthase-deficient mice. Biochem Biophys Res Commun 479: 453–460. https://doi.org/10.1016/j.bbrc.2016.09.077
Inamori KI, Inokuchi JI (2020) Roles of Gangliosides in Hypothalamic Control of Energy Balance: New Insights. Int J Mol Sci 21: 5349. https://doi.org/10.3390/ijms21155349
Furian AF, Rattmann YD, Oliveira MS, Royes, LF, Marques MC, Santos AR, Mello CF (2009) Nitric oxide and potassium channels mediate GM1 ganglioside-induced vasorelaxation. Naunyn Schmiedebergs Arch Pharmacol 380: 487–495. https://doi.org/10.1007/s00210-009-0469-x
Dieterle V, Herzer S, Gröne H-J, Jennemann R, Nordström V (2020) Ganglioside deficiency in hypothalamic POMC neurons promotes body weight gain. Int J Obes 44: 510–524. https://doi.org/10.1038/s41366-019-0388-y
Eleftheriou P, Geronikaki A, Petrou A (2019) PTP1b Inhibition, A Promising Approach for the Treatment of Diabetes Type II. Curr Top Med Chem 19: 246–263. https://doi.org/10.2174/1568026619666190201152153
Villamar-Cruz O, Loza-Mejía MA, Arias-Romero LE, Camacho-Arroyo I (2021) Recent advances in PTP1B signaling in metabolism and cancer. Biosci Rep 41: BSR20211994. https://doi.org/10.1042/BSR20211994
Cao L, Wang Z, Wan W (2018) Suppressor of Cytokine Signaling 3: Emerging Role Linking Central Insulin Resistance and Alzheimer’s Disease. Front Neurosci 12: 417. https://doi.org/10.3389/fnins.2018.00417
Linossi EM, Calleja DJ, Nicholson SE (2018) Understanding SOCS protein specificity. Growth Factors 36: 104–117. https://doi.org/10.1080/08977194.2018.1518324
Banks AS, Li J, McKeag L, Hribal ML, Kashiwada M, Accili D, Rothman PB (2005) Deletion of SOCS7 leads to enhanced insulin action and enlarged islets of Langerhans. J Clin Invest 115: 2462–2471. https://doi.org/10.1172/JCI23853
Saltiel AR (2021) Insulin signaling in health and disease. J Clin Invest 131: e142241. https://doi.org/10.1172/JCI142241
Petersen MC, Shulman G (2018) Mechanisms of Insulin Action and Insulin Resistance. Physiol Rev 98: 2133-2223.https://doi.org/10.1152/physrev.00063.2017
Sopasakis V, Liu P, Suzuki R, Kondo T, Winnay J, Tran TT, Asano T, Smyth G, Sajan M, Farese RV, Kahn CR, Zhao JJ (2010) Specific roles of the p110alpha isoform of phosphatidylinsositol 3-kinase in hepatic insulin signaling and metabolic regulation. Cell Metab 11: 220–230. https://doi.org/10.1016/j.cmet.2010.02.002
Morales-Ruiz M, Cejudo-Martín P, Fernández-Varo G, Tugues S, Ros J, Angeli P, Rivera F, Arroyo V, Rodés J, Sessa WC, Jiménez W (2003) Transduction of the liver with activated Akt normalizes portal pressure in cirrhotic rats. Gastroenterology 125: 522–531. https://doi.org/10.1016/s0016-5085(03)00909-0
Morales-Ruiz M, Santel A, Ribera J, Jiménez W (2017) The Role of Akt in Chronic Liver Disease and Liver Regeneration. Semin Liver Dis 37: 11–16. https://doi.org/10.1055/s-0036-1597819
Bala A, Roy S, Das D, Marturi V, Mondal C, Patra S, Haldar PK, Samajdar G (2021) Role of Glycogen synthase kinase-3 in the etiology of Type 2 Diabetes Mellitus: A review. Curr Diabetes Rev.https://doi.org/10.2174/1573399817666210730094225
Tormos AM, Taléns-Visconti R, Nebreda A R, Sastre J (2013) p38 MAPK: a dual role in hepatocyte proliferation through reactive oxygen species. Free Radic Res 47: 905–916.https://doi.org/10.3109/10715762.2013.821200
Thornton TM, Pedraza-Alva G, Deng B, Wood CD, Aronshtam A, Clements JL, Sabio G, Davis RJ, Matthews DE, Doble B, Rincon M (2008) Phosphorylation by p38 MAPK as an alternative pathway for GSK3beta inactivation. Science 320: 667–670.https://doi.org/10.1126/science.1156037
Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, Karin M (2008) Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell 14: 156–165.https://doi.org/10.1016/j.ccr.2008.06.016
Rius-Pérez S, Tormos AM, Pérez S, Finamor I, Rada P, Valverde ÁM, Nebreda AR, Sastre J, Taléns-Visconti R (2019) p38α deficiency restrains liver regeneration after partial hepatectomy triggering oxidative stress and liver injury. Sci Rep 9: 3775. https://doi.org/10.1038/s41598-019-39428-3
Revunov E, Johnström P, Arakawa R, Malmquist J, Jucaite A, Defay T, Takano A, Schou M (2020) First Radiolabeling of a Ganglioside with a Positron Emitting Radionuclide: In Vivo PET Demonstrates Low Exposure of Radiofluorinated GM1 in Non-human Primate Brain. ACS Chem Neurosci. 11: 1245–1249. https://doi.org/10.1021/acschemneuro.0c00161
Дополнительные материалы отсутствуют.
Инструменты
Журнал эволюционной биохимии и физиологии