

Журнал физической химии, 2019, T. 93, № 1, стр. 137-141
Необычное поведение флуоресцеина в условиях электрохимического окисления в водном фосфатном буферном раствореО. К. Лебедева, В. С. Снытко, И. И. Кузнецова, Д. Ю. Культин, А. Н. Захаров, Л. М. Кустов
О. К. Лебедева a, *, В. С. Снытко a, И. И. Кузнецова a, Д. Ю. Культин a, А. Н. Захаров a, Л. М. Кустов a, b
a Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
b Институт органической химии им. Н.Д. Зелинского РАН
119991 Москва, Россия
* E-mail: lebedeva@general.chem.msu.ru
Поступила в редакцию 25.05.2018
Аннотация
Проведено электрохимическое и каталитическое окисление пероксидом водорода флуоресцеина (ФЛ) в фосфатном буферном растворе. Показано, что в исследованном интервале потенциалов адсорбции ФЛ не происходит, т.е. исключается гетерогенный характер окисления, однако генерирование окислителя обусловлено электрохимическим процессом, связанным с образованием активных промежуточных частиц in situ. Установлено, что существует граница концентрации пероксида водорода, ниже которой окисления ФЛ не происходит. Выявлена зависимость скорости реакции гомогенного окисления ФЛ пероксидом водорода от состояния поверхности платинового катализатора.
Ксантеновые красители, к которым относится флуоресцеин (ФЛ), широко используются в текстильной промышленности, для декоративных целей, в биологических исследованиях. ФЛ находит аналитическое применение в определении пероксидов, в том числе и органических [1]. Для удаления флуоресцеина из воды эффективным является окисление [2]. ФЛ в водных растворах существует в различных формах в зависимости от рН: катионной (в кислой среде), нейтральной (хиноидная форма, лактон, цвиттер‑ион) и в щелочной среде как анион или дианион [3, 4]. ФЛ – электроактивное вещество и может быть окислен или восстановлен на различных электродах [5, 6]. Современные процессы окисления, использующие передовые электрохимические подходы, разработаны для предотвращения загрязнения окружающей среды, в особенности водных потоков [7, 8]. Частицы-окислители могут быть получены из таких анионов, как сульфат, фосфат, карбонат, борат. Важно отметить, что образование стабильных окислителей не всегда является прямым электрохимическим процессом, но чаще всего включает как электрохимическую, так и химическую стадию. Так, по мнению авторов [8], в фосфатных электролитах электрохимическая стадия приводит к образованию пероксофосфат-радикала. Дальнейшая рекомбинация позволяет получить более устойчивый пероксодифосфат-ион. Взаимодействие пероксофосфат-радикала с радикалом ОН• может приводить к образованию монопероксофосфорной кислоты Н3РО5. Образование различных окислителей возможно также из воды. В работе [9] сделана попытка термодинамической оценки образования радикалов OH• и их реакционной способности в окислении органических веществ. Согласно [10], в кислой среде на платине образование гидроксил-радикалов происходит при потенциале 2.8 В, образование пероксида водорода – при потенциале 1.78 В, пероксигидроксил-радикала – при потенциале 1.7 В. В нейтральной и щелочной среде потенциалы этих процессов ниже, и для образования радикала OH• потенциал составляет 1.89 В [11]. Широко также используют химическое окисление ФЛ пероксидом водорода в присутствии различных гомогенных и гетерогенных катализаторов [2, 12, 13].
Представляется актуальным использовать фосфатный буфер как электролитическую среду, позволяющую получить различные окислители in situ. ФЛ – удобное модельное вещество, позволяющее контролировать процессы, происходящие при анодных воздействиях. Исследование окисления ФЛ в электрохимических условиях в фосфатном буферном растворе (рН 7.2) также интересно, поскольку это значение рН соответствует различным биологическим жидкостям (например, крови). При этом значении рН ФЛ находится в нейтральной форме. Нейтральная форма ФЛ характеризуется малым значением квантового выхода [4], что дает возможность исследовать изменение его концентрации методом спектроскопии в УФ- и видимой области. В последние десятилетия ФЛ находит применение в известной тестовой реакции на содержание кислородных радикалов (“Oxygen Radical Absorption Capacity” – ORAC), которая широко применяется для определения антиоксидантов в пищевой промышленности [14]. Однако метод не лишен недостатков и нуждается в усовершенствовании, например, в уменьшении длительности определения и порога чувствительности, что может быть решено при исследовании поведения ФЛ в выбранных условиях.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве катализатора и электрода-катализатора использовали платинированную платину с истинной поверхностью S = 40 см2. Истинную поверхность определяли электрохимическим методом по водородной области циклической вольтамперометрии (ЦВА) в 0.5 М серной кислоте по известной методике [15]. Электрохимические исследования проводили в трехэлектродной ячейке с разделенным катодно-анодным пространством. Рабочим электродом служила платинированная платина, вспомогательным – платиновая пластина, электродом сравнения – насыщенный хлорсеребряный электрод (хсэ). Фоновым раствором служил фосфатный буфер, приготовленный из 0.5 М Na2HPO4, 0.5 M NaH2PO4 (“х.ч.”) и 0.5 М H2SO4. Электрохимические исследования проводили с помощью потенциостата Autolab PGSTAT302N. Скорость развертки составляла 50 мВ/с. Интервал исследуемых потенциалов от –0.6 до 2.4 В (хсэ). Перед исследованиями раствор продували аргоном. Электрод-катализатор перед каждым исследованием подвергали многократной катодно-анодной чистке при силе тока I = ±100 мА. Раствор ФЛ готовили непосредственно перед каждым исследованием. Концентрацию ФЛ варьировали в пределах 0.001–0.01 мМ. Концентрацию ФЛ определяли по градуировочному графику при длине волны максимума λ = 470 нм. Для всех спектрофотометрических измерений использовали прибор Shimadzu UV-3600Plus. Спектр ФЛ в фосфатном буфере соответствует литературным данным [4]. Растворы для каталитического окисления ФЛ готовили из пероксида водорода с концентрацией 33%. Концентрацию определяли титрованием стандартизированным по щавелевой кислоте раствором перманганата калия в кислой среде.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Методом ЦВА определяли адсорбцию ФЛ на платине в фосфатном буферном растворе (рис. 1). Анодный и катодный ход ЦВА в буфере отличается от ЦВА в серной кислоте. На анодной стационарной кривой (второй и последующие циклы), снятой в интервале потенциалов от –0.6 до 2.4 В (хсэ), четко видны три пика хемосорбированного водорода. В области потенциалов, соответствующих выделению газообразного кислорода (Е = = 1.0–1.1 В) в серной кислоте, в фосфатном буфере обнаружено медленное возрастание тока с выходом на стационарное значение. Такую форму ЦВА наблюдали в работе [16] для растворов с буферной емкостью 100 ммоль. На основании необратимости катодного и анодного хода ЦВА в исследуемой области потенциалов можно заключить, что преимущественно адсорбируется дигидрофосфат-анион с последующей диссоциацией на протон и гидрофосфат-анион [17].
Рис. 1.
ЦВА платинового электрода в растворе фосфатного буфера (сплошная линия) и в растворе фосфатного буфера в присутствии флуоресцеина С = = 1.8 × 10–5 М (пунктирная линия). Скорость развертки потенциала 50 мВ/с. Электрод сравнения – хлорсеребряный.
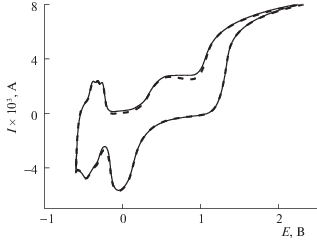
Исследование адсорбции ФЛ в условиях разомкнутой цепи проводили при потенциале Е = = 0.92 В (хсэ). При введении ФЛ в раствор фосфатного буфера при потенциале 0.92 В адсорбционных сдвигов потенциала не наблюдается даже при значительных временах адсорбции (до 1 ч). ЦВА с ФЛ совпадают с фоновой кривой (рис. 1). Оба этих факта свидетельствуют об отсутствии адсорбции ФЛ на поверхности платины в выбранных условиях.
Проведено электрохимическое окисление ФЛ в фосфатном буфере при постоянном токе в интервале от 8 до 100 мА и при постоянных значениях потенциала в интервале от 1.5 до 2.4 В. Относительное уменьшение (C/Co) во времени концентрации ФЛ от силы анодного тока представлено на рис. 2. При увеличении потенциала ток возрастает от 10 до 80 мА. Увеличение значения потенциалов окисления до 2.4 В не приводит к возрастанию конверсии ФЛ по сравнению с экспериментом при 2.2 В. Это, возможно, связано с тем, что полученный ток окисления для указанных потенциалах практически одинаков (75–78 мА). В то же время увеличение тока (зависимость С/Со от тока) сопровождается возрастанием конверсии (рис. 3). Поэтому для дальнейших исследований были выбраны значения Е = 2.2 В и I = 100 мA.
Рис. 2.
Зависимости изменения относительной концентрации флуоресцеина (С/Со) от времени при различных анодных токах: 1 – 8, 2 – 16, 3 – 40, 4 – 100 мА.
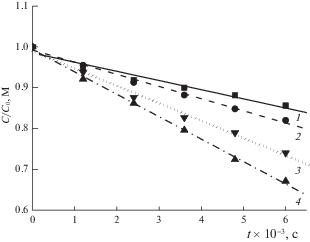
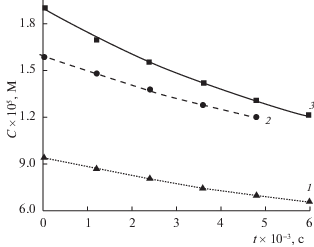
Рис. 3.
Зависимости изменения концентрации флуоресцеина от времени при анодном окислении при потенциале Е = 2.2 В при разных начальных концентрациях флуоресцеина (С × 106, М): 1 – 9.5, 2 – 16, 3 – 19.
Согласно данным рис. 1 и 2, окисление ФЛ происходит не на поверхности электрода, а в объеме электролита. Роль потенциала или тока сводится к генерированию окислителей в процессе электролиза воды или ионов гидрофосфата. Из воды могут быть получены пероксид водорода или радикал OH•, который может рекомбинировать с образованием пероксида водорода [18]. Другие окислители могут быть получены из гидрофосфат-аниона или дигидрофосфат-аниона по реакциям [8]:
Электрокаталитическое окисление тетрабромфлуоресцеина и дихлорфлуоресцеина [19] на платине в щелочной среде в области потенциалов до кислородного перенапряжения подчиняется кинетическому уравнению первого порядка. Эффективная константа скорости для процесса электрокаталитического окисления дихлорфлуоресцеина составила (1.9 ± 0.2) × 10–8 с–1, а для тетрабромфлуоресцеина (1.5 ± 0.2) × 10–8 с–1. В реакции образуются минеральные продукты [19].
В обзоре [20] рассмотрена роль генерируемых в электрохимическом процессе радикалов OH• и пероксида водорода в гетерогенном окислении органических веществ до минеральных продуктов. Высокий потенциал (E = 2.8 В) делает радикал OH• неселективным окислителем.
Влияние концентрации ФЛ на начальную скорость гомогенного окисления в условиях электрохимической генерации частиц-окислителей в фосфатном буфере при постоянном потенциале Е = 2.2 В показано на рис. 3. Формальный порядок реакции по концентрации ФЛ (n = 1.2) установлен по величинам начальных скоростей при постоянной исходной концентрации пероксида водорода. Таким образом, кинетическое уравнение, описывающее гомогенное окисление ФЛ электрохимически генерируемыми частицами-окислителями имеет следующий вид:
где 9 × 10–4 – эффективная константа скорости.Полученные результаты свидетельствуют о сложном многостадийном процессе окисления ФЛ на платине в фосфатном буфере.
Согласно данным 1Н и 13С ЯМР, при окислении ФЛ при I = 100 мA в течение 12 ч органических продуктов в растворе не образуется. Степень конверсии ФЛ в этих условиях, определенная по результатам измерения УФ-спектров, составляет >90%.
Среди окислителей, генерируемых в анодном процессе, может образовываться пероксид водорода. Пероксид водорода часто используют для окисления различных органических веществ [21]. Механизм разложения пероксида водорода сложный и включает как гетерогенные, так и гомогенные стадии. Гомогенная стадия окисления органического вещества зависит от природы органического вещества и катализатора [21].
С целью установления особенностей поведения ФЛ в условиях гомогенного окисления пероксидом водорода без наложения тока и в присутствии платины, было изучено окисление ФЛ при неизменной его концентрации в фосфатном буфере, но при различных начальных концентрациях Н2О2 (рис. 4). В таких же условиях было изучено поведение ФЛ в зависимости от величины площади поверхности платинового катализатора при неизменной начальной концентрации ФЛ и пероксида водорода (рис. 5). Как видно из приведенных данных, в обоих случаях наблюдается линейный ход зависимости концентрации ФЛ от времени, что однозначно свидетельствует о нулевом порядке реакции по ФЛ.
Рис. 4.
Зависимости изменения концентрации флуоресцеина (Со = 19 × 10–6 М) от времени при окислении пероксидом водорода в присутствии платинового катализатора (s = 40 см2) без наложения тока при начальных концентрациях пероксида водорода: 1 – 0.04, 2 – 0.083, 3 – 0.11, 4 – 0.138 М.
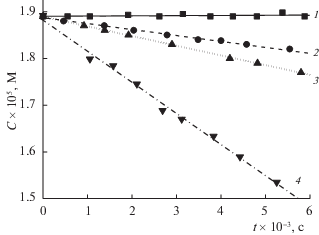
Рис. 5.
Зависимости изменения концентрации флуоресцеина (Со = 19 × 10–6 М) от времени при окислении пероксидом водорода (Со = 0.14 М) без наложения тока при разных значениях величины поверхности платинового катализатора: 1 – 0.002, 2 – 0.73, 3 – 40 см2.
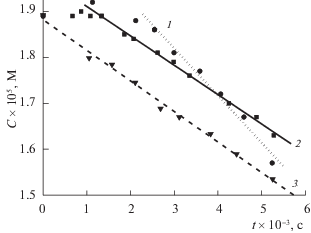
Увеличение концентрации пероксида водорода приводит к увеличению степени конверсии ФЛ. Однако, как видно из рис. 4, окисление ФЛ начинается только по достижении некоторого порогового значения концентрации пероксида водорода. С другой стороны, зависимость концентрации ФЛ от времени при постоянной концентрации пероксида водорода зависит от площади поверхности платинового электрода. С учетом того, что ФЛ не адсорбируется на электроде, следует вывод о каталитическом поведении металла. Очевидно, платиновый электрод, не находящийся под током, косвенно участвует в изучаемом процессе как гетерогенный катализатор, генерируя на поверхности активные частицы-окислители и влияя, таким образом, на кинетику окисления ФЛ.
Другая интересная особенность окисления ФЛ в рассмотренных условиях обнаруживается при изменении площади поверхности электрода-катализатора. На рис. 5 видно, что, начиная с некоторой величины площади поверхности платины, появляется своеобразный индукционный эффект. Поскольку индукционный период (τ) возрастает по мере уменьшения площади поверхности (s) катализатора на котором не наблюдается адсорбции ФЛ:
можно сделать вывод, что процесс его окисления действительно происходит не на поверхности металла, а в объеме. Тем не менее, это не исключает влияния платины на кинетику окисления ФЛ, поскольку именно на поверхности металла происходит генерирование частиц окислителя.
Таким образом, на основании данных по окислению ФЛ в водном фосфатном буферном растворе при постоянном токе и при постоянном потенциале обнаружено необычное кинетическое поведение субстрата. По результатам работы можно сделать следующие выводы
1. В исследованном интервале потенциалов адсорбция ФЛ не происходит, что исключает гетерогенный характер окисления. Это означает, что окисление ФЛ происходит гомогенно, однако генерирование окислителя обусловлено электрохимическим процессом, связанным с образованием активных промежуточных частиц in situ.
2. Существует граница концентрации пероксида водорода, ниже которой окисления ФЛ не происходит, что может указывать на электрохимическое происхождение окислителя.
3. Указанные выше особенности подтверждает зависимость скорости реакции окисления ФЛ пероксидом водорода от площади поверхности платинового катализатора.
Установлено, что окислители, принимающие участие в гомогенном окислении ФЛ, генерируются гетерогенно на поверхности платинового катализатора.
Исследование было выполнено при поддержке Российского научного фонда (проект № 14-50-00126).
Список литературы
Яблоцкий К.В., Шеховцова Т.Н. // Журн. аналит. химии. 2010. Т. 65. С. 676 (обзор).
Pirillo S., Einschlag F.S.G., Ferreira M.L. et al. // J. Mol. Catalysis B: Enzymatic. 2010. V. 66. P. 63.
Queiroz N.L., Nascimento J.A.M., Nascimento M.L. et al. // Electroanalysis, 2017. V. 29. P. 489.
Sjoback R., Nygren J., Kubista M. // Spectrochim. Acta, Part A. 1995. V. 51. P. L7–L21.
Wang B., Ma Y., Wang S., Zhang L. et al. // J. Mater. Chem. C. 2014. P. 4423.
Compton R., Page D., Sealy G. // J. Electroanalyt. Chem. 1984. V. 163. P. 65.
Sirés I., Brillas E., Oturan M.A., Rodrigo M.A. et al. // Environ Sci. Pollut. Res. Int. 2014. V. 21. P. 8336. (A review).
Cañizares P., Sáez C., Sánchez-Carretero A., Rodrigo M. // J. Appl. Electrochem . 2009. V. 39. P. 2143.
Jaimes R., Vazquez-Arenas J., González I., Galván M. // Electrochim. Acta. 2017. V. 229. P. 345.
Loures C., Alcântara M., Filho H., et al. // Int. Rev. Chem. Eng. 2013. V. 5. P. 102.
Sawyer D.T., Roberts J.L. // Acc. Chem. Res. 1988. V. 21. P. 469.
Li Q., Wu G., Cullen D. A., More K. L. et al. // ACS Catal. 2014. V. 4. P. 3193.
Кузнецова Е.А. Способ окисления органических соединений в присутствии пероксида водорода. Патент RU 2 301 790(13). 2007.
Bisby R., Brooke R., Navaratnam S. // Food Chem. 2008. V. 108. P. 1002.
Практикум по электрохимии / Под ред. Б.Б. Дамаскина. М.: Высшая школа, 1991. С. 198.
Daubinger P., Kieninger J., Unmüssig T., Urbanab G.A. // Phys. Chem. Chem. Phys. 2014. V. 16. P. 8392.
Gisbert R., Garcнa G., Koper M. // Electrochim. Acta. 2010. V. 55. P. 7961.
Латимер В.М. Окислительные состояния элементов и их потенциалы в водных растворах. М.: Изд-во иностр. лит., 1954. С. 47.
Bogdanovsky G.A., Vidovich G.L., Kultin D.Yu. et al. // Applied Catalysis A: General. 2002. V. 232. P. 137.
Martínez-Huitle C., Andrade L. // Quim. Nova. 2011. V. 34. P. 850.
Miller C., Valentine R. // Wat. Res. 1999. V. 33. P. 2805.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии