

Журнал физической химии, 2019, T. 93, № 10, стр. 1584-1600
Палладий-кобальтовые катализаторы на углеродном носителе в гидродехлорировании хлорбензола
С. В. Клоков a, *, Е. С. Локтева a, Е. В. Голубина a, К. И. Маслаков a, О. Я. Исайкина a, М. В. Тренихин b, c
a Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
b Омский государственный технический университет
Омск, Россия
c Российская академия наук, Сибирское отделение, Омский научный центр
Омск, Россия
* E-mail: servadklokov@gmail.com
Поступила в редакцию 15.03.2019
После доработки 15.03.2019
Принята к публикации 09.04.2019
Аннотация
Пиролизом опилок, пропитанных раствором нитратов палладия и кобальта, синтезированы биметаллические и монометаллические композитные материалы, содержащие ультрамикропористый углерод, схожий по структуре с активированным углем, и наночастицы Pd0, CoO и, возможно, PdCoO2. По данным ПЭМ, распределение металлсодержащих частиц по размеру в биметаллических катализаторах несколько шире, чем в монометаллических, а средний размер частиц больше. Часть металлсодержащих частиц расположена на поверхности носителя, другие запрессованы в его матрицу или покрыты углеродной оболочкой. Биметаллические катализаторы проявили высокую стабильность и активность в реакции газофазного гидродехлорирования (ГДХ) хлорбензола с образованием преимущественно бензола, причем конверсия хлорбензола на всех кобальтсодержащих катализаторах была выше 85% во всем исследованном температурном диапазоне (150–300°С). По данным РФЭС, в ходе процесса ГДХ происходит хлорирование металлов и частичное разрушение углеродной оболочки металлических частиц под действием хлороводорода, сопровождающееся выходом частиц активных металлов на поверхность катализатора. Введение кобальта способствует снижению степени дезактивации палладийсодержащих центров катализаторов и обеспечивает дополнительные активные центры на основе СоО.
Хлорорганические соединения (ХОС) входят в число опаснейших органических загрязнителей из-за их негативного экологического и биологического воздействия. Однако ХОС до сих пор широко используются в промышленности; кроме того, значительное количество отходов накоплено до вступления в силу 17 мая 2004 г. Стокгольмской конвенции о стойких органических загрязнителях. Одним из наиболее эффективных и ресурсосберегающих способов утилизации ХОС является каталитическое гидродехлорирование (ГДХ).
Наиболее часто в ГДХ используют катализаторы на основе переходных металлов: Pd, Pt, Ni [1, 2], а в качестве носителей хорошо подходят устойчивые к действию НСl углеродные материалы [3].
Биметаллические катализаторы, включающие благородный и неблагородный металл, обычно более устойчивы к отравлению HCl [4, 5]. В работе [6] методом РФЭС показано, что железо в составе PdFe катализаторов подвергается хлорированию хлороводородом, выделяющимся при ГДХ ССl4, и таким образом защищает от окисления палладиевые активные центры. Кроме того, добавление второго благородного или переходного металла к катализатору может способствовать повышению активности [7, 8] или селективности по отношению к определенным продуктам реакции [9]. Так, в работах [7, 8] Ni и Fe применяли в качестве модификаторов катализатора Pd/Al2O3 в реакции ГДХ хлорбензола. Установлено, что небольшая добавка неблагородного металла (до 1 : 1 мольн.) повышает эффективность и стабильность биметаллических катализаторов по сравнению с Pd/Al2O3. Одной из причин может служить изменение дисперсности активной фазы в результате взаимодействия между двумя металлами, поскольку ГДХ относят к структурно-чувствительным реакциям. Так, средний размер частиц биметаллических катализаторов PdPt/C, приготовленных в работе [10], составляет 0.6–1.0 нм, что заметно меньше, чем в составе монометаллических Pt/C и Pd/C, содержащих частицы размером 1.7–1.8 нм. При этом эффективность биметаллических катализаторов в ГДХ дихлорметана оказалась выше по сравнению с монометаллическими аналогами.
Ранее мы показали, что катализаторы Pd/C [11, 12] и Co/C [13], полученные малостадийным способом, включающим пропитку древесных опилок солью металла с дальнейшим пиролизом опилок в инертной атмосфере, обладают хорошими каталитическими свойствами в гидродехлорировании хлорбензола в паровой фазе. Палладиевые катализаторы проявили также высокую эффективность в мультифазном ГДХ гексахлорбензола [11]. В настоящей работе похожим способом приготовлены биметаллические катализаторы PdCo/C, проведено исследование их физико-химических свойств и тестирование в парофазном ГДХ хлорбензола, а также сопоставление с монометаллическими Pd/C и Co/C системами.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление катализаторов
Для получения PdCo/C применяли метод, описанный в [12] для синтеза Pd/C. В качестве исходного материала для получения носителя использовали опилки березы повислой (Betula pendula, фракция 0.25–0.5 мм), источником палладия служил раствор Pd(NO3)2 (“Аурат”, Россия, [Pd] = 464.6 г/л), а источником кобальта – соль Co(NO3)2 · 6H2O (“Реахим”, Россия, “ч.д.а.”).
Для приготовления биметаллических образцов навеску предварительно высушенных опилок смачивали раствором солей кобальта и палладия в количестве, немного превышающем влагоемкость опилок, и выдерживали в течение 3 суток.
Затем проводили пиролиз влажных опилок, пропитанных солями, в инертной атмосфере (N2, 15 мл/мин) в проточном кварцевом реакторе при 450°C в течение 4 ч.
Методика приготовления и свойства образцов Pd/C и Co/C описаны соответственно в [12] и [13].
Целевое содержание палладия во всех катализаторах составляло 1 масс. %, содержание кобальта варьировали от 0.5 до 2 мас. %.
Физико-химические исследования катализаторов
Удельную поверхность образцов измеряли методом низкотемпературной адсорбции-десорбции азота на приборе Autosorb-1 (“Quantachrome”, США). Расчет удельной площади поверхности, объема и среднего диаметра пор образцов проводили методами BET, BJH и NLDFT.
Морфологию образцов изучали методом сканирующей электронной микроскопии (СЭМ) на приборе JEOL JSM-6390LA (JEOL Ltd., Япония), оснащенном приставкой для локального элементного анализа методом энергодисперсионной рентгеновской спектроскопии (ЭДС).
Снимки просвечивающей электронной микроскопии высокого разрешения (ПЭМ ВР) получали при ускоряющем напряжении 200 кВ на приборе JEM 2100 (JEOL Ltd., Япония), оснащенном приставкой для локального элементного анализа методом ЭДС.
Спектры КР получали на спектрометре Horiba Jobin Yvon LabRam HR 800 UV (Франция), дифракционная решетка 300 мм–1, совмещенном с микроскопом Olympus BX41 (объектив 50×). Использовали зеленое излучение (514.532 нм) ионного аргонового лазера мощностью 10 мВт, время экспозиции составляло 50 c, разрешение спектра 3.5 см–1.
Спектры РФЭС регистрировали на спектрометре Axis Ultra DLD (“Kratos Analytical Limited”, Великобритания) с использованием источника монохроматического излучения Al Kα (hν = = 1486.6 эВ, 150 Вт). Предварительную калибровку шкалы прибора производили по пикам основных уровней чистых золота и меди Au 4f7/2 (83.96 эВ) и Cu 2p3/2 (932.62 эВ). Энергия пропускания анализатора составляла 160 эВ для обзорных спектров и 40 эВ для спектров высокого разрешения. Образцы наносили на двустороннюю адгезионную ленту. Для компенсации заряда образцов использовали нейтрализатор. Спектры калибровали по энергии связи низкоэнергетической компоненты спектра C1s электронов, которую принимали равной 284.5 эВ. Фон неупругих потерь электронов вычитали по методу Ширли. Спектры высокого разрешения были разложены на компоненты с использованием программного обеспечения CasaXPS.
Содержание Pd и Co в катализаторах определяли методом атомно-абсорбционной спектроскопии (ААС) на приборе iCE 3000 Series AA Spectrometer (Thermo Scientific, США) с использованием атомизации в пламени. Данные обрабатывали с помощью программного обеспечения SOLAAR. Металлы переводили в растворимую форму методом озоления: углеродный носитель сжигали при прокаливании образца на воздухе (30 мин при 600°C, 2 ч при 1000°C), остаток растворяли в царской водке при кипячении с обратным холодильником в течение 2 ч и разбавляли дистиллированной водой до содержания металлов ~2–6 мг/л.
Температурно-программированное восстановление водородом (ТПВ) проводили на анализаторе хемосорбции УСГА-101 (ООО “Унисит”, Россия), пропуская через кварцевый реактор с образцом смесь 5% H2 + 95% Ar (30 мл/мин), при нагревании от 30 до 600°С со скоростью 10 K/мин. Изменение состава газовой смеси во время анализа фиксировали с помощью детектора по теплопроводности (температура 60°С). Сигнал детектора калибровали по восстановлению NiO.
Каталитическое ГДХ хлорбензола
Газофазное ГДХ хлорбензола проводили в проточном трубчатом кварцевом реакторе с неподвижным слоем катализатора. Катализатор (8 мг) помещали в реактор между слоями кварцевого фильтра (“Whatman QM-A”) и нагревали в токе водорода до нужной температуры. Хлорбензол (ХБ) подавали в реактор снизу из барботера, пропуская через него водород. Реакцию проводили в изотермическом режиме при фиксированных температурах в интервале от 150 до 300°С. После достижения постоянной конверсии при заданной температуре подачу смеси ХБ + H2 прекращали, в реактор подавали водород и поднимали температуру до следующего значения.
Реакционную смесь анализировали методом газожидкостной хроматографии на приборе Agilent 6890N (капиллярная колонка DBWAX, 30 м, детектор пламенно-ионизационный).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Содержание элементов по данным ААС, СЭМ-ЭДС и РФЭС
В работе сравнивали два монометаллических образца Pd/C и Со/С и два биметаллических композита PdCo(1.1)/C и PdCo(0.7)/C, где индексы 1.1 и 0.7 показывают массовое соотношение металлов по данным ААС.
Поскольку формирование катализатора происходит в ходе пиролиза древесных опилок, пропитанных раствором солей металлов (Pd и/или Co), причем носитель формируется из опилок, важно определить общее содержание и распределение металлов в объеме и на поверхности образцов. В табл. 1 сопоставлены данные о составе образцов, полученные разными методами. Видно, что общее содержание палладия, определенное методом ААС, увеличивается в ряду Pd/C < < PdCo(1.1)/C < PdCo(0.7)/C, а содержание кобальта в PdCo(0.7)/C в два раза выше, чем в PdCo(1.1)/C, и немного выше, чем в Co/C.
Таблица 1.
Содержание элементов (в мас. %) в Pd/C, PdCo/C и Co/C и по данным ААС, СЭМ-ЭДС и РФЭС
| Метод | Элемент | Содержание элементов, мас. % | |||
|---|---|---|---|---|---|
| Pd/C | PdCo(1.1)/C | PdCo(0.7)/C | Co/C | ||
| ААС | Pd | 0.56 | 0.85 | 1.07 | – |
| Co | – | 0.77 | 1.48 | 1.30 | |
| СЭМ-ЭДС | C | 83.24 | 82.43 | 77.21 | 84.58 |
| O | 14.02 | 15.60 | 14.54 | 13.45 | |
| Pd | 1.44 | 1.56 | 2.39 | – | |
| Co | – | 0.29 | 4.36 | 1.70 | |
| Ca | 0.60 | 0.12 | 0.85 | 0.13 | |
| Si | 0.01 | не обнаружено | 0.15 | не обнаружено | |
| Na | 0.52 | не обнаружено | 0.29 | 0.14 | |
| Mg | 0.17 | не обнаружено | 0.21 | не обнаружено | |
| РФЭС до катализа1 | C | 84.06 | 84.22 | 83.55 | 87.04 |
| O | 12.29 | 11.43 | 12.25 | 10.64 | |
| Pd | 3.15 | 3.38 | 2.39 | – | |
| Co | – | 0.69 | 1.56 | 1.97 | |
| Ca | 0.50 | 0.28 | 0.25 | 0.35 | |
| РФЭС после катализа2 | C | 85.45 | 86.90 | 79.64 | 84.94 |
| O | 10.62 | 8.81 | 10.92 | 9.23 | |
| Pd | 2.09 | 1.67 | 2.39 | – | |
| Co | – | 1.11 | 4.47 | 2.94 | |
| Ca | 0.50 | 0.44 | 0.81 | 1.18 | |
| Si | 0.59 | 0.29 | 0.76 | 0.44 | |
| Cl | 0.75 | 0.78 | 1.01 | 1.27 | |
Содержание Pd на поверхности Pd-содержащих образцов, определенное методами РФЭС и СЭМ, выше в 2–5.5 раза по сравнению с его содержанием в объеме, найденным методом ААС. Поверхностная концентрация Co, определенная методом СЭМ-ЭДС, превышает объемную более чем в 3 раза только для образца PdCo(0.7)/C. Обогащение кобальтом поверхности образца Co/C, найденное методами СЭМ-ЭДС и РФЭС, не так значительно. В составе PdCo(1.1)/C содержание Со на поверхности, найденное методом РФЭС, практически совпадает с объемным, найденным ААС, а его поверхностное содержание по методу СЭМ-ЭДС в два раза ниже объемного. Следует учитывать, что методы СЭМ-ЭДА и РФЭС – локальные, и некоторые колебания могут наблюдаться в зависимости от исследованной области, но в целом приведенные результаты свидетельствуют об обогащении поверхности всех образцов палладием. Распределение кобальта на поверхности и в объеме зависит от состава образца.
Методы РФЭС и СЭМ-ЭДС позволяют определить присутствие микропримесей, которые обычно имеются в составе исходных опилок и могут оставаться в углеродных материалах, полученных при пиролизе. По данным РФЭС, во всех образцах присутствует примесь Ca (<0.5 мас. %), а по данным СЭМ-ЭДС, также Na и Mg. Соли или оксиды этих металлов могут выступать в качестве промоторов каталитического действия в ГДХ, например, уменьшать степень дезактивации активного компонента путем связывания HCl. По данным СЭМ-ЭДС, в составе Pd/C и PdCo(0.7)/C содержится больше Са, Na и Mg по сравнению с другими образцами.
Структурные и текстурные свойства катализаторов
На рис. 1 представлены изотермы адсорбции-десорбции N2 на образцах Pd/C, PdCo/C и Co/C. Изотерма адсорбции N2 (рис. 1) образца Pd/C соответствует II типу по классификации ИЮПАК [14], характерному для непористых или макропористых тел. Схожий вид имеют на начальном участке изотермы адсорбции N2 на образцах PdCo(1.1)/C и Co/C, однако при p/p0 > 0.6 адсорбция на этих образцах достигает максимального значения и далее при повышении давления не меняется. Изотерма адсорбции азота на PdCo(0.7)/C более соответствует I типу, характерному для микропористых тел; она содержит отчетливую точку перегиба между значениями p/p0, составляющими 0.1 и 0.2. Отметим, что для всех образцов наблюдается необратимый характер изотерм, десорбционная ветвь сильно отличается от адсорбционной. Такой вид кривых адсорбции/десорбции с открытым гистерезисом наблюдается в материалах, характеризующихся наличием пор с очень узким горлышком, размер которого сравним с размером молекулы адсорбируемого газа (0.364 нм для азота) [13, 15]. Так, открытый гистерезис наблюдали при адсорбции на углеродных молекулярных ситах Takeda 3A [16]. В ходе адсорбции при температуре жидкого азота диффузия молекул N2 в таких узких горлышках происходит очень медленно, что затрудняет достижение истинного равновесия при измерении адсорбции–десорбции; количество адсорбированного газа в процессе измерения десорбционной ветви увеличивается за счет все еще проходящего заполнения пор. Ранее изотермы адсорбции–десорбции азота с открытым гистерезисом наблюдали для систем Pd/C, полученных пиролизом опилок по аналогичной методике [12]. Таким образом, моно- и биметаллические Co-содержащие образцы, полученные пиролизом пропитанных солями опилок, включают углеродные материалы, пористая система которых преимущественно образована ультрамикропорами с узким устьем.

Неравновесный характер изотерм адсорбции-десорбции делает весьма условным расчет удельной площади поверхности полученных материалов. Расчет по методу БЭТ (табл. 2) на основании адсорбционных ветвей изотерм дает величины 148 м2/г (для Pd/C) и более. Расчеты с помощью NLDFT (Non-local Density Functional Theory) и методом Дубинина–Радушкевича (DR), который лучше учитывает вклад микропор, дают в 1.5–2.5 раза большие значения удельной площади поверхности по сравнению с методом БЭТ. Значения удельной поверхности, определенные методами DR и NLDFT, примерно совпадают для образцов PdCo(1.1)/C и Co/C, для которых изотермы адсорбции азота имеют схожий вид, в то время как для образцов Pd/C и PdCo(0.7)/C, для которых изотермы адсорбции азота сильно различаются, SDR < SNLDFT.
Таблица 2.
Площадь удельной поверхности катализаторов Pd/C, PdCo/C и Co/C, рассчитанная по методам BET, DR и NLDFT (r – средний радиус пор)
| Образец | SBET, м2/г | SDR, м2/г | SNLDFT, м2/г | r, Å | Vmicro DR, см3/г | VNLDFT, см3/г |
|---|---|---|---|---|---|---|
| Pd/C | 148 ± 15 | 280 ± 28 | 411 ± 41 | 16.4 | 0.0995 | 0.1121 |
| PdCo(1.1)/C | 210 ± 21 | 362 ± 36 | 386 ± 39 | 13.7 | 0.1286 | 0.1427 |
| PdCo(0.7)/C | 164 ± 16 | 277 ± 28 | 385 ± 39 | 15.5 | 0.0985 | 0.1041 |
| Co/C | 261 ± 26 | 432 ± 43 | 436 ± 44 | 12.8 | 0.1537 | 0.1587 |
Сравнение результатов расчета методом DR, характеризующим объем микропор, и методом NLDFT, учитывающим все типы пор, позволяет вычислить вклад микропористости в структуре образцов. Соотношение Vmicro DR/VNLDFT возрастает в ряду Pd/C (0.89) < PdCo(1.1)/C (0.90) < < PdCo(0.7)/C (0.95) < Co/C (0.97). Такие значения соотношения Vmicro DR/VNLDFT характерны для микропористых материалов. Действительно, средние значения размеров пор составляют менее двух нанометров.
На основании полученных данных можно полагать, что в результате пиролиза опилок, пропитанных солями металлов, образуются углеродные материалы, включающие закрытые ультрамикропоры и/или микропоры с узкими устьями, т.е. угли низкой степени активации. Природа образующегося углеродного материала существенным образом зависит от природы металла, присутствующего при пиролизе.
По литературным данным, добавление металлов 8 группы IV периода Периодической таблицы облегчает протекание пиролиза биомассы. Так, в [17] добавление хлорида Fe, Co или Ni во время пиролиза смеси биомассы и полимерных отходов приводило к повышению выхода газообразных продуктов и теплоты сгорания продуктов при относительно низких температурах пиролиза, причем оптимальной добавкой служил CoCl2. Снижение энергии активации совместного пиролиза биомассы и полимерных отходов в присутствии соединений кобальта отмечено также в работе [18]. В результате добавления металлов Ni, Fe и Co [19, 20] заметно увеличивается доля H2 (с 1.5 до 15% в присутствии Ni/ZSM-5) в составе газообразных продуктов пиролиза.
Структурные особенности катализаторов изучали методом спектроскопии комбинационного рассеяния (КР).
Спектры КР всех полученных в работе композитов, приведенные на рис. 2, содержат как G-линию при 1580 см–1, относящуюся к растяжению связей C–C в гексагональных кольцах [21], так и D-линии, D1 при 1325 см–1 и D2 при 1620 см–1, обусловленные образованием “дырок” в π-валентной зоне, которые активируют двойное резонансное рассеяние единичных фононов, исходящих из центра зоны Бриллюэна в графитовой сетке. Присутствие обоих типов линий характерно для активированного угля (АУ), но не для графита, КР-спектры которых также показаны на рис. 2. В спектрах КР всех катализаторов наблюдается повышение интенсивности фона при сдвигах >1000 см–1, вызванное флуоресценцией [22] и типичное для углей или лигноцеллюлозных материалов, подвергнутых мягкому пиролизу [23]. Следовательно, по своей природе углерод в полученных образцах сходен с АУ и отличается от более упорядоченного графита.

Соотношение интенсивностей линий D и G (ID/IG) [24] позволяет оценить степень графитизации углеродного материала: при ее повышении снижается интенсивность линии D и соотношение ID/IG. Для всех образцов (рис. 2) ID/IG попадает в интервал значений, характерных для активированных углей, от 0.5 до 1.0 [25]. Наибольшее соотношение наблюдается для образца Pd/C, а наименьшее для Со/С. Таким образом, степень графитизации снижается в ряду Pd/C > PdCo(1.1)/C ≈ PdCo(0.7)/C > > Со/С. Эти данные коррелируют с различиями в форме изотерм адсорбции.
В КР-спектрах всех металлсодержащих образцов видны малоинтенсивные пики в области 500–850 см–1, которые не характерны для углеродных материалов. В спектре образца Co/C присутствует пик при 675–710 см–1, который можно отнести к оксиду кобальта [26]. В спектрах КР Pd-содержащих образцов есть небольшой пик при 790 см–1, который может относиться к гидроксиду кальция [27]. Для двух образцов Pd/C и PdCo(0.7)/C, в которых, по данным СЭМ-ЭДС, содержание кальция выше по сравнению с PdCo(1.1)/Cи Co/C, пик при 790 см–1 более интенсивный. Так как содержание Pd, Co и зольных примесей невелико (менее 1.5 мас. %), интенсивность этих пиков существенно ниже по сравнению с интенсивностью пиков углерода.
Текстуру и однородность состава полученных образцов исследовали также методом СЭМ-ЭДС. На рис. 3 представлены снимки СЭМ и карты распределения элементов на соответствующих участках.
Рис. 3.
Снимки СЭМ и карты распределения элементов в опилках (а, б), Pd/C (г–ж), PdCo(1.1)/C (з–м), PdCo(0.7)/C (н–c) и Co/C (т–х).

Сравнение снимков СЭМ показывает, что все полученные углеродные материалы сохраняют текстуру, характерную для опилок. Наряду с углеродом и металлами, все материалы содержат кислород, что может свидетельствовать о неполной карбонизации биомассы в ходе пиролиза или о присутствии на поверхности заметного количества кислородсодержащих функциональных групп.
Исследование природы носителя методом РФЭС
Природу углеродного носителя характеризовали также методом РФЭС.
На рис. 4а приведены участки спектров РФЭС ВР всех катализаторов, соответствующие C1s-электронам; видно, что они мало различаются. Основной пик с энергией связи 284.5 эВ, соответствует углероду в sp2-состоянии [28]. Имеется также небольшое плечо в области 286–289 эВ, которое обусловлено как асимметрией основного пика, так и небольшим вкладом от различных связей углерода с кислородом. Это подтверждается спектрами РФЭС O1s-электронов (рис. 4б); все они содержат только два характерных пика с энергиями связей 531.3 и 533.2 эВ, которые соответствуют двойным и одинарным связям атомов кислорода с атомами углерода, причем соотношение интенсивностей этих пиков (около 0.7) практически не зависит от типа образца. Такие связи могут отражать наличие на поверхности образцов кислорода или углекислого газа, адсорбированных из воздуха, или иных кислородсодержащих функциональных групп, но возможно также присутствие кислорода в углеродной структуре, обусловленное неполным пиролизом древесной биомассы.
Рис. 4.
Спектры РФЭС ВР катализаторов Pd/C, PdCo/C и Co/C а) С1s, б) O1s, в) Pd3d, г) Co2p-электронов до (сплошная линия) и после (пунктирная линия) каталитических испытаний в ГДХ ХБ; д) Cl2p-электронов после каталитических испытаний.
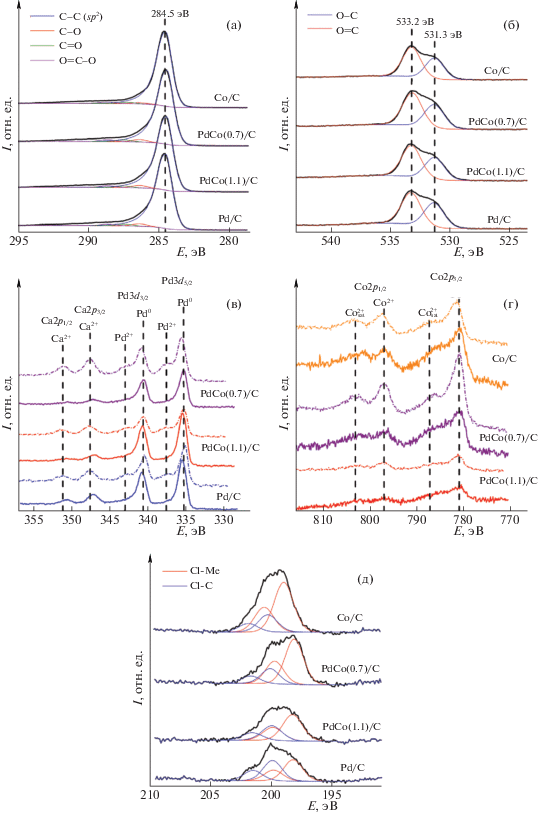
По результатам СЭМ-ЭДС и РФЭС видно, что концентрация кислорода на поверхности всех образцов не превышает 15%, как и в составе активированных углей, получаемых при температуре 400–500°C [29]. По данным СЭМ-ЭДС (табл. 1), содержание кислорода падает в ряду PdCo(1.1)/C ≈ ≈ PdCo(0.7)/C > Pd/C > Co/C, а соотношение C/O растет в этом же ряду. Однако содержание кислорода, определенное методом РФЭС, изменяется иным образом: оно снижается в ряду Pd/C ≈ ≈ PdCo(0.7)/C > PdCo(1.1)/C > Co/C. Различие обусловлено глубиной анализа, характерной для каждого из методов. Согласно обоим методам, наименьшее количество кислорода найдено в образце Co/C, что коррелирует с данными КР-спектроскопии и адсорбционными о том, что этот образец содержит углеродный материал с более высокой, а Pd/C – с меньшей степенью карбонизации по сравнению с PdCo(1.1)/C и PdCo(0.7)/C.
На основе разложения на компоненты РФЭС спектров C1s мы оценили содержание углерода в различных химических состояниях (табл. 3).
Таблица 3.
Доля атомов углерода в различных химических состояниях по данным РФЭС
| Образец | Доля атомов С в состояниях с определенной энергией связи | |||
|---|---|---|---|---|
| С–С (sp2) 284.5 эВ |
C–O 286.3 эВ |
C=O 287.4 эВ |
O=C–O 288.6 эВ |
|
| Pd/C | 95.0 | 2.2 | 1.5 | 1.3 |
| PdCo(1.1)/C | 94.4 | 2.4 | 1.5 | 1.7 |
| PdCo(0.7)/C | 94.4 | 2.3 | 1.6 | 1.7 |
| Co/C | 95.7 | 1.2 | 1.5 | 1.6 |
Видно, что во всех образцах примерно 95% атомов углерода связаны между собой, и лишь небольшая часть (не более 5%) – с атомами кислорода. Такая малая доля связей С–О, по-видимому, может быть удовлетворительно объяснена присутствием кислорода или CO2, адсорбированных из воздуха физически или химически с образованием функциональных групп на поверхности углеродного материала.
Таким образом, данные низкотемпературной адсорбции N2, КР-спектроскопии и РФЭС подтверждают получение в ходе пиролиза древесных опилок, пропитанных солями палладия и кобальта, углеродных материалов, содержащих преимущественно аморфный углерод и схожих по структуре с активированным углем. Однако степень карбонизации, тип пористой системы, особенно содержание ультрамикропор и степень их открытия несколько различаются в зависимости от природы и количества металлов, присутствовавших при пиролизе. По-видимому, соли кобальта способствуют активации углерода путем открытия некоторой части ультраузких устьев пор, что проявляется как в случае Co/C, так и для биметаллических PdCo/C систем.
Исследование электронного состояния металлов и их распределения по поверхности
Как видно из рис. 3, во всех образцах распределение Со и Pd по поверхности образцов равномерное, нигде не наблюдается участков, обогащенных или обедненных одним из металлов.
Особенностью образцов Pd/C, полученных пиролизом пропитанных солью палладия опилок в инертной атмосфере, является восстановленное состояние палладия [11, 12]. Электронное состояние нанесенных металлов в монометаллических и биметаллических композитах изучали методами РФЭС и ТПВ.
РФЭС спектры Pd3d-электронов всех образцов (рис. 4в) содержат дублеты узких асимметричных линий с энергией связи компоненты Pd3d5/2-электронов, равной 335.3 эВ. Наблюдаемые небольшие отклонения от этого положения могут быть вызваны неравномерной подзарядкой образцов в условиях анализа. Положение этого пика характерно для Pd0 [30]. Таким образом, на поверхности Pd/C и обоих биметаллических материалов содержится преимущественно Pd0. Однако детальный анализ спектров высокого разрешения показал, что на поверхности всех палладийсодержащих катализаторов можно выделить вклад Pd2+, величины которого показаны в табл. 4. При увеличении общего содержания палладия (табл. 1 и табл. 4) вклад окисленного палладия увеличивается, от 5% в Pd/C до 11% в составе PdCo(0.7)/C. Однако повышение содержания окисленного палладия не коррелирует с содержанием палладия на поверхности, определенным методом РФЭС, что свидетельствует о том, что присутствие Pd2+ обусловлено не окислением палладия на воздухе, а его неполным восстановлением из соли в ходе пиролиза.
Таблица 4.
Доля атомов палладия и кобальта в металлическом и окисленном состояниях на поверхности Pd/C, PdCo/C и Co/C до и после каталитических испытаний (%), содержание хлора в образцах, по данным РФЭС
| Образец | Pd0, % | Pd2+, % | Co0, % | Co2+, % | Co3+, % | Cl−Ме1, % | Cl−C, % | τ, мин | α × 10–2, мас. % | γ × 10–2, мас. % |
|---|---|---|---|---|---|---|---|---|---|---|
| Pd/C | 95 | 5 | – | – | – | – | – | – | ||
| PdCo(1.1)/C | 92 | 8 | 0 | 100 | 0 | – | – | – | ||
| PdCo(0.7)/C | 89 | 11 | 0 | 100 | 0 | – | – | – | ||
| Co/C | – | – | 0 | 100 | 0 | – | – | – | ||
| После каталитических испытаний | ||||||||||
| Pd/C | 81 | 19 | – | – | – | 51 | 49 | 1035 | 2.22 | 1.06 |
| PdCo(1.1)/C | 77 | 23 | 1 | 93 | 6 | 64 | 36 | 699 | 4.29 | 1.54 |
| PdCo(0.7)/C | 77 | 23 | 1 | 74 | 25 | 75 | 25 | 1372 | 3.31 | 0.48 |
| Co/C | – | – | 0 | 97 | 3 | 74 | 26 | 752 | 7.50 | 2.55 |
Спектры РФЭС ВР Co2p-электронов (рис. 4г) по форме идентичны для всех Со-содержащих композитов, а интенсивность пиков увеличивается при повышении концентрации Co. Энергия связи Co2p3/2-электронов (781.0 эВ) и наличие в спектрах интенсивных сателлитов, характерных для Со2+, указывают на присутствие кобальта в составе оксида CoO как в биметаллических образцах PdCo(0.7)/C и PdCo(1.1)/C, так и в монометаллическом Co/C [31]. Металлический кобальт и Co3+ в образцах не найдены. Можно предположить, что кобальт в катализаторах в условиях пиролиза не восстанавливается, либо после завершения пиролиза быстро подвергается окислению кислородом воздуха с образованием CoО. Металлический палладий более устойчив на воздухе и окислению не подвергается.
Чтобы определить, присутствует ли в объеме образцов окисленный палладий, а также найти условия восстановления кобальта из оксида, образцы изучали методом ТПВ.
На рис. 5 приведены профили ТПВ всех образцов. Профиль ТПВ Pd/C содержит один пик с максимумом при 515°C, который может быть обусловлен взаимодействием углерода с водородом с образованием метана при каталитическом действии Pd. Пиков, относящихся к восстановлению палладия из оксида, появление которых возможно в интервале от 60 до 200°С [32, 33], на ТПВ профилях палладийсодержащих образцов не наблюдается. Следовательно, как на поверхности, так и в объеме этого образца содержится преимущественно Pd0, а восстановление незначительного количества Pd2+, найденного на поверхности методом РФЭС, не заметно в профилях ТПВ или происходит в ходе подготовке образца к анализу. Восстановление окисленного палладия в нанесенных катализаторах может происходить при комнатной температуре во время продувания системы смесью водорода с инертным газом, однако в этом случае далее образуется гидрид палладия, который разлагается при температурах около 80°C, а характерных обратных пиков в профилях ТПВ исследуемых образцов не наблюдается. В литературе отмечается, что мелкодисперсные частицы палладия не склонны к образованию гидрида [34, 35], так что можно ожидать высокой дисперсности металлических частиц в полученных нами композитах.
Рис. 5.
Профили ТПВ катализаторов Pd/C, PdCo/C и Co/C (пунктирная линия – после хранения на воздухе в течение 1.5 лет).

На ТПВ профиле Co/C имеются два пика, малоинтенсивный с максимумом при 365°С, и широкий интенсивный пик с максимумом в районе 460°С. Второй пик преимущественно вызван образованием метана, а первый можно отнести к восстановлению CoO; по литературным данным, восстановление CoO до Co наблюдается при 300–450°С [36]. Стоит отметить, что при хранении на воздухе в течение полутора лет не происходит заметных изменений в ТПВ профиле образца Co/C: температура начала пика метанирования не изменяется (300°С), его максимум немного смещается вправо (470°С), а максимум пика, соответствующего восстановлению CoO, незначительно смещается в сторону низких температур (355°С).
На основании результатов ТПВ можно заключить, что в образцах отсутствует Со3+. Если бы Co3O4 имелся в объеме этих образцов, на профилях ТПВ следовало ожидать присутствия пика его восстановления в интервале 200–300°C [36], чего мы не наблюдали. Этот результат согласуется с данными РФЭС, свидетельствующими, что на поверхности всех кобальтсодержащих образцов Со3+ отсутствует.
На профиле образца PdCo(0.7)/C можно выделить асимметричный пик с максимумом при 440°С, соответствующий взаимодействию углеродного материала с водородом. Его асимметричность может быть вызвана наложением пика восстановления CoO, которое происходит в диапазоне температур 330–380°С, на пик, соответствующий образованию метана при взаимодействии углерода с Н2. Возможно также, что в этом образце присутствуют формы СоО, более прочно связанные с углеродным материалом, например, частицы CoO, покрытые углеродной оболочкой, восстановление которых происходит при повышенных температурах. Заметим, что в профиле этого образца пик взаимодействия углерода с водородом и его максимум сдвинуты в область низких температур по сравнению с профилем ТПВ Co/C, что, скорее всего, связано с каталитическим действием палладия.
Длительное (1.5 года) хранение образца PdCo(0.7)/C на воздухе приводит к следующим изменениям: 1) температура начала большого асимметричного пика, который был единственным в профиле ТПВ исходного образца, сдвигается к низким температурам (менее 240°С); 2) на восходящем склоне появляется небольшой пик с максимумом при 310°С, соответствующий восстановлению СоО. По-видимому, в ходе длительного хранения происходит модификация структуры композита, приводящая к появлению некоторой фракции оксида кобальта, способного к восстановлению в условиях ТПВ. Этот пик несколько сдвинут в низкотемпературную область по сравнению с наблюдаемым на профиле ТПВ Co/C. Снижение температуры восстановления неблагородного металла в присутствии палладия часто наблюдали в литературе [37], оно свидетельствует о плотном контакте между обоими металлами. Максимум асимметричного пика, обусловленного образованием метана, сдвинут в область более высоких температур по сравнению с наблюдаемыми в профилях ТПВ PdCo(0.7)/C и Co/C; положение этого максимума примерно совпадает с максимумами пиков метанирования в профилях ТПВ образцов PdCo(1.1)/C и Pd/C.
Профиль ТПВ образца PdCo(1.1)/C схож с профилем Pd/C и не содержит пиков восстановления оксида кобальта, по-видимому, в связи с его малым содержанием. После длительного хранения образца на воздухе пик, соответствующий взаимодействию углеродного материала с водородом, смещается в сторону низких температур (максимум сдвигается с 485 до 460°С). Происходящие изменения подтверждают наблюдение, сделанное при изучении образцов методом низкотемпературной адсорбции азота, о неравновесности полученных образцов. По-видимому, в ходе хранения происходит структурная перестройка композитов, приводящая к большей доступности некоторой доли СоО для восстановления в условиях ТПВ.
Обычно профили ТПВ предшественников нанесенных металлсодержащих катализаторов содержат несколько пиков восстановления металла, связанных с присутствием мелких и крупных частиц оксида металла, сильно или слабо связанных с поверхностью. Однако в наших Со-содержащих образцах, содержащих относительно большое количество Со, присутствует только один пик, который можно отнести к восстановлению СоО. Для обоих образцов поглощение водорода, оцененное по площади этого пика (50 и 74 мкмоль/г для Co/C и PdCo(0.7)/C соответственно), существенно меньше, чем необходимо для восстановления всего количества кобальта, присутствующего в образцах по данным ААС, в предположении, что весь металл находится в виде СоО (220 мкмоль/г и 251 мкмоль/г). При исследовании катализаторов на углеродных носителях методом ТПВ пик поглощения водорода, связанный с восстановлением СоО, может накладываться на пики, обусловленные поглощением водорода и выделением метана в ходе взаимодействия углерода с Н2 [38]. Возможно, что в образцах присутствует одна или несколько дополнительных форм СоО, например, более прочно связанных с носителем, пики восстановления которых сливаются с широким пиком, обусловленным выделением метана. Не исключено, что часть частиц CoO покрыта прочной углеродной оболочкой и начинает восстанавливаться по мере удаления этой оболочки за счет газификации в виде метана при действии H2.
Размеры частиц и степень взаимодействия металлов оценивали методом ПЭМ.
На рис. 6 представлены снимки ПЭМ катализаторов Pd/C, PdCo/C и Co/C, гистограммы, характеризующие распределение частиц по размеру, и средневзвешенный размер частиц.
Рис. 6.
Снимки ПЭМ и распределения частиц по размеру (d) в катализаторах Pd/C (а–в), PdCo(1.1)/С (г–е), PdCo(0.7)/С (ж–и) и Co/C (к–м).
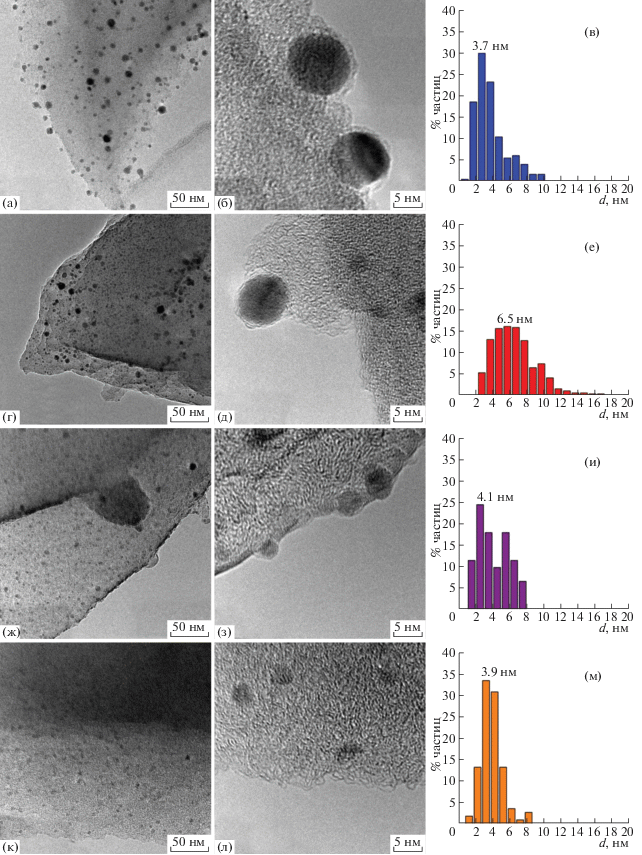
На основании различной контрастности частиц и локализации на углеродной матрице на приведенных снимках можно полагать, что в полученных композитах частицы металлов расположены как на поверхности, так и в объеме углеродного материала. Частицы, расположенные на поверхности, покрыты тонкой оболочкой из аморфного углерода толщиной 1 нм и менее. Для частиц Pd характерны межплоскостные расстояния 2.25–2.27 Å (эталонные дифрактограммы ICDD № 00-001-1310 и 00-046-1043), в то время как расчетные значения для некоторых палладийсодержащих частиц в составе образца Pd/C составили 2.26–2.37 Å. Повышенное значение межплоскостных расстояний (2.34–2.37 Å) может быть связано с образованием PdCx на поверхности частиц Pd [39] в ходе пиролиза древесных опилок в присутствии Pd. Средний размер частиц металла (3.7 нм) в монометаллических образцах минимальный (3.7 и 3.9 нм для Pd/C и Сo/C соответственно, а минимальная ширина распределения частиц по размерам характерна для образцов с высоким содержанием Со (Со/С и PdCo(0.7)/C соответственно).
В образце Co/C контрастность кобальтсодержащих частиц к углеродному материалу невысокая из-за окисленного состояния кобальта. Кристаллическая решетка частиц не наблюдается, так как, возможно, они находятся в аморфном состоянии. Похожий вид имеют частицы металлов в биметаллических образцах (PdCo(1.1)/C и PdCo(0.7)/C).
На снимках ПЭМ ВР биметаллических образцов присутствуют как кристаллические частицы, так и частицы без видимых признаков кристалличности. Однозначно определить, какой из металлов (или оба) содержит каждая частица, не представляется возможным. Поэтому распределения частиц по размеру для образцов PdCo(1.1)/C и PdCo(0.7)/C строили с учетом всех наблюдаемых методом ПЭМ металлических частиц. В образце PdCo(1.1)/C, содержащем относительно небольшое количество кобальта, средний размер частиц (6.5 нм) выше, чем в PdCo(0.7)/C (4.1 нм). В отдельных случаях в образце PdCo(0.7)/C (рис. 6з) наблюдались наночастицы с межплоскостным расстоянием 2.36–2.41 Å, характерным для PdCoO2 (ICCD 27-1324), что свидетельствует о большой вероятности контакта двух металлов в этом образце. По-видимому, таких частиц немного, так как по данным РФЭС содержание окисленного палладия на поверхности катализаторов не превышает 11%, а методом ТПВ окисленный палладий не обнаружен.
Таким образом, из данных РФЭС, ТПВ и ПЭМ следует, что палладий в композитах присутствует преимущественно в восстановленном состоянии, а кобальт в окисленном (в основном в форме CoO, и некоторое количество смешанного оксида PdCoO2). Стоит отметить, что взаимодействие металлов друг с другом обнаружено для образца PdCo(0.7)/C с относительно более высоким содержанием кобальта, но оно не исключается и для другого биметаллического композита.
Суммируя полученные данные, можно описать структуру биметаллических образцов как включающую Pd0, CoO и небольшое количество PdCoO2 в виде мелкодисперсных частиц, отчасти запрессованных в матрицу из углеродного материала низкой степени активации, содержащего преимущественно ультрамикропоры с узкими устьями, а отчасти покрытых углеродными оболочками графеновой или аморфной природы. Судя по данным ТПВ, восстановление кобальта из оксида протекает при температуре выше 300°C.
Каталитические испытания в ГДХ хлорбензола
Результаты тестирования катализаторов в реакции ГДХ хлорбензола в интервале температур от 150 до 300°C представлены на рис. 7.
Рис. 7.
Зависимости конверсии хлорбензола и селективности образования продуктов от температуры в реакции газофазного ГДХ хлорбензола в присутствии катализаторов Pd/C, Co/C и PdCo/C.
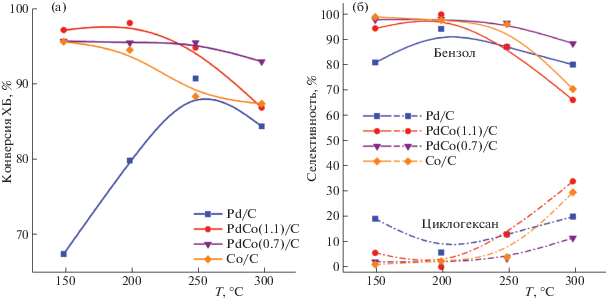
Все катализаторы проявили высокую активность в реакции парофазного ГДХ хлорбензола. Удивительно, что при относительно низких температурах реакции (150–200°С) конверсия ХБ выше на Co/C, содержащем преимущественно СоО, чем в присутствии Pd/C, в составе которого преобладает металлический палладий; при более высоких температурах они обеспечивают примерно одинаковую конверсию ХБ. Биметаллические катализаторы сходны по свойствам с катализатором Со/С при низких температурах, при 300°С PdCo(0.7)/C обеспечивает наибольшую из всех катализаторов конверсию ХБ.
Конверсия ХБ в присутствии катализатора Pd/C с ростом температуры проходит через максимум, а на остальных катализаторах в большей (Co/C, PdCo(1.1)/C) или меньшей (PdCo(0.7)/C) степени снижается с ростом температуры. Можно предположить ряд причин, вызывающих такое снижение, например, спекание частиц металлов и хлорирование металлов выделяющимся при ГДХ хлороводородом, которое должно нарастать с увеличением продолжительности опыта.
Содержание металлов в катализаторах различается. Чтобы нивелировать это различие, мы рассчитали значения TOF, отнесенные к общему содержанию металлов, определенному методом ААС (табл. 5). Полученные результаты являются лишь оценочными: в использованных условиях конверсия ХБ очень высокая, следовательно, максимально возможная скорость реакции при этом не достигается.
Таблица 5.
Значения TOF в расчете на молярное содержание металлов в образцах
| Т, °C | Образец | TOF, 1/ч | TOF(Pd), 1/ч | TOF(Co), 1/ч |
|---|---|---|---|---|
| 150 | Pd/C | 959 | 959 | – |
| PdCo(1.1)/C | 346 | 910 | 559 | |
| PdCo(0.7)/C | 204 | 712 | 287 | |
| Co/C | 327 | – | 327 | |
| 200 | Pd/C | 1135 | 1135 | – |
| PdCo(1.1)/C | 350 | 919 | 564 | |
| PdCo(0.7)/C | 204 | 710 | 286 | |
| Co/C | 323 | – | 323 | |
| 250 | Pd/C | 1290 | 1290 | – |
| PdCo(1.1)/C | 338 | 888 | 546 | |
| PdCo(0.7)/C | 204 | 710 | 286 | |
| Co/C | 300 | – | 300 | |
| 300 | Pd/C | 1200 | 1200 | – |
| PdCo(1.1)/C | 310 | 814 | 500 | |
| PdCo(0.7)/C | 198 | 692 | 278 | |
| Co/C | 300 | – | 300 |
Как видно из табл. 5, наиболее активным катализатором при всех температурах является Pd/C, причем его активность несколько возрастает с повышением температуры реакции. Добавление кобальта приводит к сильному снижению активности, с ростом его содержания падение активности более заметно. Система PdCo(0.7)/C работает значительно хуже, чем Pd/C, хотя содержание палладия в этом катализаторе, по данным ААС, примерно в два раза выше, и обладает даже меньшей активностью, чем Со/С. При увеличении содержания палладия в образцах в ряду Pd/C < PdCo(1.1)/C < PdCo(0.7)/C значения TOF при всех исследованных температурах снижаются. Учитывая незначительные различия в размере частиц металлов в этих образцах, причинами снижения эффективности могут быть: (1) вовлечение части палладия в образование двойного оксида PdCoO2, что согласуется с наиболее высокой долей окисленного палладия на поверхности (11%, табл. 4) и наименее выраженным обогащением поверхности палладием по сравнению с другими образцами (табл. 1); (2) снижение доступной поверхности палладия в связи с его капсулированием преобладающим количеством объемного оксида кобальта, или (3) капсулирование палладия углеродом, вызванное образованием карбидной или углеродной оболочки, или попаданием в закрытые или ультрамалые поры углеродного носителя. Поскольку величины удельной площади поверхности и размеры пор катализаторов Pd/C и PdCo(0.7)/C практически не различаются, более весомыми представляются первые две причины. Однако вид изотермы адсорбции азота на биметаллическом образце PdCo(0.7)/C иной по сравнению с остальными образцами, т.е. структура носителя имеет определенные особенности, которые могут воздействовать на доступность палладия, оксида кобальта или биметаллических активных центров для адсорбции и активации реагентов. Катализатор с меньшим содержанием кобальта PdCo(1.1)/C несколько более активен по сравнению с другими кобальтсодержащими образцами.
Как отмечено в нашей работе [13], значительная активность в ГДХ катализатора Со/С, не содержащего, по данным РФЭС, металлического кобальта, свидетельствует о возможности протекания реакции ГДХ на активных центрах СоО.
В составе продуктов, полученных в присутствии всех катализаторов, при относительно низких температурах преобладает бензол, однако при повышении температуры реакции селективность по бензолу снижается, и в реакционной смеси обнаруживается до 35% циклогексана, в результате смещения равновесия реакции С6Н6 + Н2 = С6Н12 вправо при повышении температуры и сильной адсорбции бензола на каталитических центрах. Селективность образования циклогексана при 300°С уменьшается в ряду PdCo(0.7)/C < Pd/C < < Co/C < PdCo(1.1)/C.
Для катализаторов ГДХ важнейшей характеристикой является стабильность в реакционных условиях, где они подвергаются спеканию, а также хлорированию под действием НСl, неизбежного продукта реакции. Отметим, что методика проведения каталитических испытаний подразумевала длительный эксперимент (табл. 4): зависимости конверсии ХБ от температуры строили на основании анализа изменения величины конверсии при определенной температуре от времени, выбирая стационарное значение конверсии, не меняющееся на протяжении как минимум 1 ч эксперимента. При относительно невысоких температурах реакции (150–250°С) после перехода к следующей температуре происходило постепенное повышение конверсии ХБ со временем, пока она не достигала стационарных значений. При более высокой температуре (250–300°С для Со-содержащих катализаторов, 300°С для Pd/C) катализаторы практически сразу обеспечивали стационарные величины конверсии ХБ. Так, в присутствии Pd/C при 250°С в течение первых 60 мин конверсия ХБ возрастала от 55 до 92% (стационарное значение); при 300°С уже в первой пробе для ГЖХ анализа обнаружена конверсия 84%, далее в течение 120 мин она колебалась на уровне 84–87%. В присутствии Co/С конверсия ХБ вырастала за первые 100 мин опыта от 20 до 99% и от 35 до 94% при 100 и 150°С, соответственно, а затем сохраняла достигнутое значение в течение следующих 100 мин опыта. При 250 и 300°С на этом катализаторе конверсия ХБ сразу достигала стационарных значений, приведенных на рис. 7а. В случае биметаллических катализаторов начальное значение конверсии после достижения каждой из температур было выше, чем для Co/C, это особенно заметно для катализатора PdCo(1.1)/C с более высоким отношением Pd/Co. При 100 и 250°С начальная величина конверсии ХБ составляла 75–77% и возрастала до стационарного значения 98–95% за 100 мин. При 300°С конверсия ХБ составляла около 90% на всем протяжении эксперимента (150 мин). Во всех случаях в изотермических условиях селективность по бензолу возрастала симбатно с увеличением конверсии.
Одно из положительных действий неблагородного компонента может состоять в предотвращении хлорирования благородного металла [6]. Чтобы выявить изменение степени окисления металлов в ходе ГДХ, мы провели исследование образцов после каталитических испытаний методом РФЭС.
Как показал метод РФЭС (табл. 1), после каталитических испытаний содержание Pd на поверхности образцов Pd/C и PdCo(1.1)/C снижается в 1.5–2 раза, а содержание Co на поверхности всех Co-содержащих образцов, наоборот, возрастает в 1.5–3 раза. Повышение концентрации Co может быть связано с выгоранием углеродной оболочки вокруг частиц CoO во время каталитического эксперимента, или выходом кобальта на поверхность из объема частиц катализатора за счет взаимодействия с НСl.
Спектры Pd3d-электронов (рис. 4в) образцов после каталитических испытаний несколько изменяются: наряду с дублетом асимметричных линий с энергией связи компоненты Pd3d5/2-электронов около 335.3 эВ, характерной для Pd0, как в исходных образцах, в них увеличивается вклад от атомов палладия в окисленном состоянии. Доля атомов палладия в окисленном и металлическом состоянии приведена в табл. 4.
Энергия связи Pd3d5/2- и Pd3d3/2-электронов для атомов палладия в окисленном состоянии составляет около 337.6 и 343.0 эВ соответственно, что примерно совпадает с величинами, характерными для оксида (337 и 342.2–342.5 эВ) или хлорида палладия (338 и 343.3–343.7 эВ). Относительно высокое содержание кислорода в носителе, выявленное методом РФЭС и СЭМ-ЭДС, делает вероятным образование не только хлорида, но и оксида палладия в ходе каталитического эксперимента.
Спектры Co2p3/2-электронов после каталитических испытаний также несколько изменяются. Положение пика, характеризующего энергию связи Co2p3/2-электронов, в спектрах РФЭС образцов PdCo/C после каталитических испытаний не меняется (781.0 эВ) (рис. 4г). Однако на основании положения этого пика трудно определить, происходит ли частичное превращение оксида в хлорид, поскольку энергии связи Co в оксиде и хлориде отличаются незначительно [40, 41]. В то же время относительная интенсивность сателлитов, характерных для Co2+, после каталитических испытаний возрастает, что особенно заметно для катализаторов 2Со/С и PdCo(0.7)/C, содержащих большее количество кобальта. Наблюдаемое изменение соотношения интенсивностей этих сателлитов может указывать на присутствие в них вклада трехвалентного кобальта. Таким образом, есть основания полагать, что под действием реакционной среды происходит более глубокое окисление кобальта, а палладий присутствует в образцах в нескольких формах, например, в виде частиц, не покрытых углеродом, включающих аморфную или кристаллическую углеродную оболочку, и запрессованных в углеродную матрицу, причем эти формы в разной степени устойчивы к хлорированию.
После каталитических испытаний во всех катализаторах присутствует значительное количество хлора. В спектрах Cl2p-электронов исследованных образцов (рис. 4д) можно выделить вклад от двух состояний атомов хлора, отвечающих их ковалентным связям с углеродом (Cl–C) и с металлом (палладий или кобальт). Результаты такой обработки приведены в табл. 4. Видно, что для Pd/C эти вклады примерно одинаковы, но для кобальтсодержащих катализаторов преобладает состояние Сl–Ме. Можно предположить, что различие связано с тем, что кобальт в большей степени подвергается хлорированию, чем палладий. Подтверждением служит тот факт, что по данным РФЭС в биметаллических катализаторах окисленного палладия после опыта больше всего на 4%, чем для Pd/C, независимо от времени опыта.
Повышение содержания кобальта на поверхности катализаторов после длительных каталитических испытаний, отмеченное при исследовании методом РФЭС, может быть вызвано удалением углеродных оболочек с кобальтсодержащих частиц при продолжительном воздействии реакционной среды, например, за счет хлорирования углеродной поверхности с последующим ее разрушением. Известно, что под действием HCl возможна функционализация поверхности активированного угля, хотя она протекает в меньшей степени, чем под действием кислот-окислителей, например, HNO3 [42, 43].
В табл. 4 приведены результаты расчета массового содержания хлора в форме Ме–Сl (на основании спектров РФЭС), отнесенного к общему времени каталитических испытаний и суммарному содержанию металлов на поверхности после каталитических испытаний, определенному методом РФЭС. Сопоставление результатов для Pd/C и Co/С показывает, что кобальт подвергается хлорированию в существенно большей степени. В то же время на поверхности образца PdCo(0.7)/C наблюдается минимальное количество хлора, связанного с металлами. Таким образом, присутствие двух металлов при определенных отношениях может способствовать стабилизации каталитической системы в реакционной среде.
Согласно данным РФЭС и СЭМ-ЭДС, в приготовленных катализаторах присутствуют примеси щелочных и щелочноземельных металлов, причем их содержание выше в образцах Pd/C и PdCo(0.7)/C. Такие примеси также могут связывать хлор и способствовать уменьшению степени дезактивации, однако выделить их вклад на основании данных РФЭС невозможно.
ЗАКЛЮЧЕНИЕ
Полученные в работе результаты комплексного физико-химического исследования свидетельствуют, что пиролиз опилок, пропитанных растворами солей двух металлов, палладия и кобальта, позволяет синтезировать биметаллические композитные материалы, включающие ультрамикропористый углерод с большим содержанием кислородсодержащих функциональных групп на поверхности, а также наночастицы Pd0, CoO и, возможно, PdCoO2. Таким образом, показано, что в условиях пиролиза происходит практически полное восстановление палладия из соли до металла, но восстановления кобальта не происходит, либо после вынесения образцов на воздух он окисляется до СоО. По данным ПЭМ, распределение металлсодержащих частиц по размеру в биметаллических катализаторах несколько шире, чем в монометаллических, а средний размер больше. Многие из металлсодержащих частиц расположены не на поверхности носителя, а запрессованы в его матрицу или покрыты углеродной оболочкой. Образующийся при пиролизе углеродный материал сходен по структуре с активированным углем, однако его природа, судя по данным КР-спектроскопии и низкотемпературной адсорбции азота, зависит от природы металлов, присутствующих в исходном материале. Так, степень графитизации углеродного носителя в Co-содержащих образцах в некоторой степени снижается по сравнению с Pd/C. Текстура всех полученных композитов характеризуется наличием ультрамикропор и пор с узким горлом. Доля микропор выше для Co-содержащих образцов.
Полученные биметаллические композиты проявили высокую стабильность и активность в реакции газофазного ГДХ хлорбензола, причем конверсия хлорбензола для всех Co-содержащих катализаторов была выше 85% во всем исследованном температурном интервале (150–300°С), однако их активность ниже по сравнению с Pd/C, что может объясняться снижением доступности палладия для адсорбции реагентов при введении кобальта. С увеличением времени каталитических испытаний в изотермическом режиме при низких температурах конверсия хлорбензола повышается до достижения стационарных высоких значений, а при высоких температурах (300°С) стационарное значение конверсии достигается сразу после начала опыта. Повышение конверсии с ростом времени эксперимента в изотермических условиях сопровождается существенным ростом селективности образования бензола; при увеличении температуры опыта эта величина проходит через максимум. Другим углеводородным продуктом ГДХ был циклогексан. Судя по данным РФЭС, в ходе продолжительных каталитических испытаний протекает хлорирование металлов и разрушение углеродной структуры под действием хлороводорода, сопровождающееся выходом частиц активных металлов на поверхность катализатора. Анализ элементного состава и электронного состояния элементов в образцах до и после каталитических испытаний показал, что в биметаллических образцах протекает преимущественное окисление кобальта по сравнению с палладием. По-видимому, в композитах присутствуют несколько форм палладия, которые подвергаются хлорированию в большей и меньшей степени. Так, в ходе продолжительных каталитических испытаний после достижения некоторого содержания хлорированного палладия далее оно не увеличивается. Таким образом, введение кобальта способствует предотвращению дезактивации Pd-содержащих центров катализатора, повышая тем самым эффективность использования благородного металла. Присутствующие в композитах примеси щелочных и щелочноземельных металлов также способны связывать НСl, замедляя окисление активных металлов. В биметаллических катализаторах возможно присутствие биметаллических частиц оксида PdCoO2, восстановление которых присутствующим в реакционной среде водородом может обеспечивать новые биметаллические активные центры гидродехлорирования, обеспечивающие повышенную конверсию ХБ при относительно малых температурах реакции по сравнению с Pd/C.
Работа выполнена при поддержке Российского фонда фундаментальных исследований (код проекта № 18-33-00785). Авторы выражают благодарность Программе развития МГУ имени М.В. Ломоносова за доступ к оборудованию для РФЭС. В работе были использованы аналитические приборы Омского регионального центра коллективного пользования СО РАН.
Список литературы
Bedia J., Arevalo-Bastante A., Grau J.M. et al. // J. Catal. 2017. V. 352. P. 562.
Wu W., Xu J., Ohnishi R. // Appl. Catal. B. 2005. V. 60. P. 129.
Arevalo-Bastante A., Alvarez-Montero M.A., Bedia J. et al. // Ibid. 2015. V. 179. P. 551.
Coq B., Hub S., Figueras F., Tournigant D. // Appl. Catal. A. 1993. V. 101. P. 41.
Bonarowska M., Kaszkur Z., Lomot D. et al. // Appl. Catal. B. 2015, V. 162. P. 45.
Golubina E.V., Lokteva E.S., Lunin V.V. et al. // Appl. Catal. A. 2006. V. 302. P. 32.
Seshu Babu N., Lingaiah N., Vinod Kumar J., Sai Prasad P.S. // Ibid. 2009. V. 367. P. 70.
Seshu Babu N., Lingaiah N., Sai Prasad P.S. // Appl. Catal. B. 2012. V. 111–112. P. 309.
Han Y., Gu G., Sun J. et al. // Appl. Surf. Sci. 2015. V. 355. P. 183.
Martin-Martinez M., Gómez-Sainero L.M., Bedia J. et al. // Appl. Catal. B. 2016. V. 184. P. 55.
Klokov S.V., Lokteva E.S., Golubina E.V. et al. // Catal. Commun. 2016. V. 77. P. 37.
Локтева Е.С., Клоков С.В., Голубина Е.В. и др. // Изв. АН. 2016. Т. 65. С. 2618.
Klokov S.V., Lokteva E.S., Golubina E.V. et al. //Appl. Surf. Sci. 2019 V. 463. P. 395.
Thommes M., Kaneko K., Neimark A.V. et al. // Pure Appl. Chem. 2015. V. 87. P. 1051.
Landers J., Gor G.Yu., Neimark A.V. // Colloids Surf. A. 2013. V. 437. P. 3.
CarrottI P.J.M., Cansado I.P.P., Ribeiro Carrott M.M.L. // Appl. Surf. Sci. 2006. V. 252. P. 5948.
Sulman E.M., Kosivtsov Yu.Yu., Sidorov A.I. et al. // Int. J. Energy Env. 2016. V. 10. P. 100.
Xiang Z., Liang J., Morgan H.M., Jr. et al. // Bioresour. Technol. 2018. V. 247. P. 804.
Chattopadhyay J., Pathak T.S., Srivastava R., Singh. A.C. // Energy. 2016. V. 103. P. 513.
Persson H., Duman I., Wanga S. et al. // J. Anal. Appl. Pyrolysis. 2019. V. 138. P. 54.
Wang Y., Alsmeyer D.C., McCreery R.L. // Chem. Mater. 1990. V. 2. P. 557.
Adamopoulos G., Robertson J., Morrison N.A., Godet C. // J. Appl. Phys. 2004. V. 96. P. 6348.
McDonald-Wharry J., Manley-Harris M., Pickering K. // Carbon. 2013. V. 59. P. 383.
Pawlyta M., Rouzaud J.-N., Duber S. // Ibid. 2015. V. 84. P. 479.
Knight D.S., White W.B. // J. Mater. Res. 1989. V. 4. P. 385.
Tang C.-W., Wang C.-B., Chien S.-H. // Thermochim. Acta. 2008. V. 473. P. 68.
Chiriu D., Ricci P.C., Polcaro A. et al. // J. Spectrosc. 2014. V. 12.
Nie R., Miao M., Du W. et al. // Appl. Catal. B. 2016. V. 180. P. 607.
Weber K., Quicker P. // Fuel. 2018. V. 217. P. 240.
Hronec M., Fulajtarova K., Vavra I. et al. // Appl. Catal. B. 2016. V. 181. P. 210.
Huang W., Zuo Z., Han P. // J. Electron. Spectrosc. Relat. Phenom. 2009. V. 173. P. 88.
Karski S., Rogowski J., Goluchowska J. // J. Mol. Catal. A: Chem. 2005. V. 240. P. 155.
Bischoff S., Weigt A., Fujimoto K., Lucke B. // J. Mol. Catal. A: Chem. 1995. V. 95. P. 259.
Sepulveda J.H., Fígoli N.S. // Appl. Surf. Sci. 1993. V. 68. P. 257.
Boudart M., Hwang H.S. // J. Catal. 1975. V. 39. P. 44.
Borg Ø., Rønning M., Storsæter S. et al. // Stud. Surf. Sci. Catal. 2007. V. 163. P. 255.
Kim J.K., Lee J.K., Kang K.H. et al. // J. Mol. Catal. A: Chem. 2015. V. 410. P. 184
Zhang H., Lancelot C., Chu W. et al. // J. Mater. Chem. 2009. V. 19. P. 9241.
Martin-Martinez M., Alvarez-Montero A., Gomez-Sainero L.M. et al. // Appl. Catal., B. 2015. V. 162. P. 532.
Hasik M., Kurkowska I., Bernasik A. // React. Funct. Polym. 2006. V. 66. P. 1703.
Carrara C., Irusta S., Lombardo E., Cornaglia L. // Appl. Catal. A. 2001. V. 217. P. 27.
Chen J.P., Wu S. // Langmuir. 2004. V. 20. P. 2233
Singha S., Sarkar U., Luharuka P. // Sci. Total Environ. 2013. V. 447. P. 472.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии