

Журнал физической химии, 2019, T. 93, № 10, стр. 1494-1499
Предсказание энтальпий сублимации циклических производных мочевины с использованием модели молекулярного электростатического потенциала
О. Н. Рыжова a, *, О. В. Дорофеева a
a Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
* E-mail: ron@phys.chem.msu.ru
Поступила в редакцию 15.03.2019
После доработки 15.03.2019
Принята к публикации 09.04.2019
Аннотация
Для оценки величин энтальпий сублимации циклических производных мочевины предложено уравнение, рассматривающее корреляцию между энтальпией сублимации и рассчитанными величинами плотности кристалла, молекулярной поверхности и четырьмя параметрами, характеризующими распределение электростатического потенциала на поверхности молекулы.
В настоящее время энтальпия образования (${{\Delta }_{{\text{f}}}}H_{{298}}^{^\circ }$) газообразных органических соединений может быть предсказана с высокой точностью с использованием квантово-химических расчетов, и это позволяет оценить энтальпию образования кристаллического соединения из известного соотношения
(1)
${{\Delta }_{{{\text{sub}}}}}H_{{298}}^{^\circ } = {{\Delta }_{{\text{f}}}}H_{{298}}^{^\circ }({\text{газ}}) - {{\Delta }_{{\text{f}}}}H_{{298}}^{^\circ }({\text{крист}}),$Наибольшее распространение для предсказания энтальпии сублимации получили методы QSPR (“Quantitative Structure – Property Relationship”, или “количественное соотношение структура–свойство”) [6]. QSPR-моделирование заключается в поиске корреляции между свойством органического соединения и его структурой, которая характеризуется дескрипторами. В качестве дескриптора может выступать любое число, которое можно рассчитать, исходя из структурной формулы. В литературе можно найти более десятка работ, в которых подход QSPR был применен для оценки энтальпии сублимации [7–12]. В этих работах использовались наборы экспериментальных данных, содержащие от 30 до 1800 значений энтальпии сублимации, а QSPR-уравнение содержало от 3 до 18 дескрипторов; среднее квадратичное отклонение для различных моделей составляло от 5 до 20 кДж моль–1, а максимальное расхождение достигало 10–75 кДж моль–1.
Особое место среди QSPR-моделей занимают модели, использующие в качестве дескрипторов параметры электростатического потенциала на поверхности молекулы. В работах Политцера и соавторов [13–15] было показано, что различные макроскопические свойства, зависящие от молекулярных взаимодействий, могут быть предсказаны на основе их корреляции с параметрами электростатического потенциала. В частности, для энтальпии сублимации было предложено уравнение [14]
(2)
${{\Delta }_{{{\text{sub}}}}}H_{{298}}^{^\circ } = a{{({{A}_{{\text{S}}}})}^{2}} + b\sqrt {\sigma _{{{\text{tot}}}}^{2}{v}} + c,$Анализ опубликованных в литературе моделей для оценки энтальпий сублимации показывает, что при их разработке не уделяется достаточного внимания достоверности экспериментальных величин, используемых для параметризации модели, тогда как от этого в первую очередь зависит ее предсказательная точность. Поэтому при исследовании энергетических соединений с высоким содержанием азота [20] при параметризации модифицированного уравнения (2) мы использовали только те соединения, экспериментальные значения энтальпии сублимации которых были подтверждены квантово-химическими расчетами. Однако и в этом случае не удалось достичь высокой точности оцененных величин (среднее квадратичное отклонение составляло 8.7 кДж моль–1, а максимальное отклонение достигало 23 кДж моль–1).
Поскольку в работе [20] рассмотрено 185 молекул, относящихся к различным классам соединений, мы предположили, что точность модели можно повысить за счет включения в уравнение (2) дополнительных дескрипторов. Было предложено новое уравнение [21, 22]
(3)
${{\Delta }_{{{\text{sub}}}}}H_{{298}}^{^\circ } = a{\rho } + b{{A}_{{\text{S}}}} + c{{\bar {V}}_{{\text{S}}}} + d(\sigma _{{{\text{tot}}}}^{2}{v}) + e{\Pi } + f,$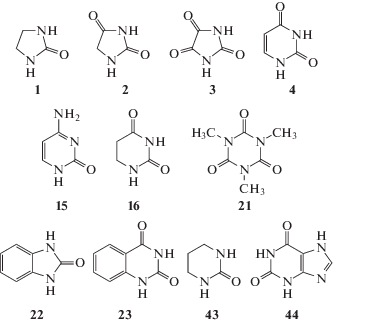
МЕТОДЫ РАСЧЕТА
Для определения коэффициентов a, b, c, d, e и f в уравнении (3) были отобраны 23 соединения, экспериментальные значения энтальпий сублимации которых были предварительно подтверждены квантово-химическими расчетами. Энтальпии образования 44 соединений в газовой фазе были рассчитаны методом Gaussian-4 (G4) [23] с использованием программного комплекса Gaussian 16 [24]. Для 23 из этих соединений (табл. 1) расхождение между рассчитанным значением ${{\Delta }_{{\text{f}}}}H_{{298}}^{^\circ }$(газ) и экспериментальным, определенным из соотношения (1), не превышало 5 кДж моль–1, и на этом основании экспериментальные данные были признаны надежными. Для остального 21 соединения (табл. 2) это расхождение превышало 5 кДж моль–1 или отсутствовали данные по ${{\Delta }_{{\text{f}}}}H_{{298}}^{^\circ }$(крист) и поэтому нельзя было определить значение ${{\Delta }_{{\text{f}}}}H_{{298}}^{^\circ }$(газ) из эксперимента.
Таблица 1.
Энтальпии сублимации (в кДж моль–1) циклических производных мочевины, отобранных для определения коэффициентов a, b, c, d, e и f в уравнении (3)
| № | Соединение | Эксперимент | Расчет | Δ* |
|---|---|---|---|---|
| 1 | Этиленмочевина | 96.6 ± 0.8 [25] | 92.8 | 3.8 |
| 83.7 ± 1.9 [26] | ||||
| 2 | Гидантоин | 116.3 ± 0.7 [27] | 118.2 | –1.9 |
| 114.2 ± 1.0 [28] | ||||
| 3 | Парабановая кислота | 119.4 ± 0.6 [29] | 122.1 | –2.7 |
| 4 | Урацил | 131.4 ± 0.4 [30] | 129.0 | 2.4 |
| 5 | 5,6-Дигидроурацил | 115.4 ± 1.0 [31] | 117.5 | –2.1 |
| 6 | 5-Метилурацил (тимин) | 127.2 ± 0.3 [30] | 125.1 | 2.1 |
| 134.1 ± 4.2 [32] | ||||
| 7 | 6-Метилурацил | 131 ± 7.0 [32] | 132.3 | –1.3 |
| 8 | 1,3-Диметилурацил | 96.9 ± 1.2 [33] | 107.4 | –10.5 |
| 96.8 [34] | ||||
| 96.4 ± 1.4 [35] | ||||
| 9 | 5,6-Диметилурацил | 132.4 ± 1.4 [36] | 128.9 | 3.5 |
| 10 | 1,3,5-Триметилурацил | 94.9 ± 0.6 [36] | 100.6 | –5.7 |
| 103.5 ± 1.5 (326 К) [37] | ||||
| 11 | 1,3,5,6-Тетраметилурацил | 101.7 ± 0.9 [36] | 103.8 | –2.1 |
| 12 | 6-Амино-1-метилурацил | 158.2 ± 4.4 [38] | 156.4 | 1.8 |
| 13 | 6-Азаурацил | 120.7 ± 1.7 [39] | 115.3 | 5.4 |
| 118.1 ± 2.0 [39] | ||||
| 14 | 6-Азатимин | 109.1 ± 2.3 [39] | 109.7 | –0.6 |
| 15 | Цитозин | 156.4 ± 2.0 [40] | 153.7 | 2.7 |
| 16 | Барбитуровая кислота | 115.1 ± 0.7 [41] | 121.0 | –5.9 |
| 120.3 ± 1.2 [41, 42] | ||||
| 17 | 1,3-Диметилбарбитуровая кислота | 92.3 ± 0.6 [43] | 96.7 | –4.4 |
| 18 | 5,5'-Диметилбарбитуровая кислота | 115.8 ± 0.5 [44] | 112.3 | 3.5 |
| 19 | 1,5,5-Триметилбарбитуровая кислота | 106.2 ± 0.4 [45] | 100.5 | 5.7 |
| 20 | 1,3-Диэтилбарбитуровая кислота | 99.5 ± 0.6 [46] | 98.4 | 1.1 |
| 21 | Триметил изоцианурат | 88.2 ± 1.2 [47] | 81.0 | 7.2 |
| 22 | 2-Бензимидазолинон | 126.4 ± 2.4 [48] | 128.6 | –2.2 |
| 23 | Бензоиленмочевина | 128.3 ± 2.2 [49] | 128.1 | 0.2 |
Таблица 2.
Энтальпии сублимации (в кДж моль–1) циклических производных мочевины, рассчитанных по уравнению (3)
| № | Соединение | Эксперимент | Расчет | Δ* |
|---|---|---|---|---|
| 24 | 1-Метилурацил | 121.7 ± 4.0 (386 К) [50] | 117.5 | 4.2 |
| 112.5 ± 2.6 (398 К) [51] | –5.0 | |||
| 25 | 3-Метилурацил | 118.8 ± 3.0 (382 К) [50] | 120.8 | –2.0 |
| 26 | 5,6-Дигидро-5-метилурацил | 124 ± 0.7 [52] | 117.7 | 6.3 |
| 27 | 5,6-Дигидро-6-метилурацил | 118.8 ± 0.8 [52] | 116.3 | 2.5 |
| 28 | 1-Метилтимин | 124.4 ± 1.3 (398 К) [51] | 109.7 | 14.7 |
| 29 | 1,3,6-Триметилурацил | 106.7 ± 2.5 (320 К) [51] | 107.8 | –1.1 |
| 30 | 1,3-Диметил-5-этилурацил | 98.7 ± 1.7 (316 К) [37] | 104.2 | –5.5 |
| 99.3 ± 0.2 (308 К) [53] | –4.9 | |||
| 110 ± 1.2 (330 К) [53] | 5.8 | |||
| 31 | 1,3-Диметил-5-пропилурацил | 111 ± 1.6 (322 К) [37] | 107.8 | 3.2 |
| 32 | 1,3-Диметил-5-изопропилурацил | 102.9 ± 1.6 (322 К) [37] | 103.5 | –0.6 |
| 33 | 1,3-Диэтилтимин | 89.8 ± 0.4 [54] | 100.8 | –11.0 |
| 95.0 ± 2.1 [51] | –5.8 | |||
| 34 | 5-Аминоурацил | 138.1 [55] | 128.0 | 10.0 |
| 35 | 6-Аминоурацил | 150.4 [55] | 171.1 | –20.7 |
| 36 | 6-Амино-1,3-диметилурацил | 148.7 [55] | 146.7 | 2.0 |
| 37 | 5-Нитроурацил | 161.4 ± 3.5 [32] | 145.0 | 16.4 |
| 38 | 1-Метилцитозин | 141.2 ± 0.6 (471 К) [56] | 129.3 | 11.9 |
| 149.1 ± 9.0 [57] | 19.8 | |||
| 39 | 1,3,5-Триметилбарбитуровая кислота | 96.1 ± 0.8 [45] | 94.2 | 1.9 |
| 40 | 5-Изопропилбарбитуровая кислота | 123.6 ± 0.6 [58] | 115.7 | 7.9 |
| 41 | 5,5-Диэтилбарбитуровая кислота | 117.3 ± 0.6 [59] | 107.6 | 9.7 |
| 42 | Тетраметилбарбитуровая кислота | 87.7 ± 0.5 [45] | 87.1 | 0.6 |
| 43 | Пропиленмочевина | 113.4 ± 0.7 [25] | 85.6 | 27.8 |
| 89.3 ± 2.5 [26] | 3.7 | |||
| 44 | Ксантин | 175.5 ± 1.3 [60] | 131.8 | 43.7 |
Волновая функция, необходимая для определения электронной плотности и расчета электростатического потенциала, была получена методом DFT/B3LYP/6-311++G(3df,2p). Все дескрипторы из уравнения (3) рассчитывались с помощью программы Multiwfn [61]. В результате минимизации среднего квадратичного отклонения между рассчитанными и экспериментальными значениями энтальпии сублимации 23 соединений были определены a, b, c, d, e и f в уравнении (3).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Уравнение (3) ранее использовалось для оценки энтальпий сублимации алифатических производных мочевины [22] и, как видно из табл. 3, привело к хорошему согласию рассчитанных и экспериментальных значений. Однако это уравнение, примененное без перепараметризации к, казалось бы, родственному классу циклических производных мочевины, привело к увеличению среднего квадратичного отклонения с 2.3 кДж моль–1 до 13.6 кДж моль–1. Значительно лучший результат был достигнут, когда коэффициенты в уравнении (3) были получены на основе экспериментальных данных для циклических производных мочевины (табл. 3). Из 23 соединений только для шести (соединения 8, 10, 13, 16, 19 и 21 в табл. 1) расхождения между рассчитанной и экспериментальной энтальпией сублимации превышает 5 кДж моль–1. Наибольшее расхождение наблюдается для 1,3-диметилурацила (8), для которого экспериментальная величина подтверждена тремя исследованиями. Возможно, это расхождение связано с тем, что по сравнению с другими метилзамещенными урацила у 1,3-диметилурацила обнаружено существование не полностью кристаллизованной фазы при фазовых переходах “кристалл-кристалл” [62]. В таком случае теоретические методы могут давать завышенные оценки, как это наблюдалось в случае 1-аминоадамантана [21], который при комнатной температуре образует фазу, только на 50% являющуюся кристаллической. Что касается расхождений для 10, 13, 16, 19 и 21, их не следует считать значимыми, поскольку они могут быть обусловлены небольшой точностью экспериментальных данных.
Таблица 3.
Точность предсказания энтальпий сублимации производных мочевины
| Параметр | Алифатические производные мочевины [22] | Циклические производные мочевины | |
|---|---|---|---|
| Коэффициенты a, b, c, d, e и f для алифатических соединений [22] | Коэффициенты a, b, c, d, e и f для циклических соединений | ||
| Количество соединений | 13 | 23 | 23 |
| Среднее квадратичное отклонение, кДж моль–1 | 2.3 | 13.6 | 4.2 |
| Максимальное отклонение, кДж моль–1 | 4.7 | 20.4 | 7.2 |
| Минимальное отклонение, кДж моль–1 | –3.0 | –25.3 | –10.5 |
В табл. 2 показаны результаты использования уравнения (3) для другого 21 циклического производного мочевины, которое не использовалось в параметризации модели из-за недостаточной уверенности в надежности их экспериментальных данных. Для восьми соединений (25, 27, 29, 31, 32, 36, 39 и 42) уравнение (3) хорошо воспроизводит экспериментальные данные. Относительно небольшие расхождения, не превышающие 6 кДж моль–1, наблюдаются для соединений 24, 26, 30 и 33, экспериментальные данные которых несколько противоречивы. Для группы соединений (28, 34, 35, 37, 38, 40 и 41) рассчитанные значения ${{\Delta }_{{{\text{sub}}}}}H_{{298}}^{^\circ }$ отличаются от экспериментальных на 8–20 кДж моль–1, и эти расхождения, скорее всего, связаны с неточностью экспериментальных измерений. Рассчитанные в настоящей работе значения ${{\Delta }_{{\text{f}}}}H_{{298}}^{^\circ }$(газ) отличаются от экспериментальных на 14–20 кДж моль–1 для 34, 35, 37, 40 и 41, что значительно превышает неточность метода G4, а потому указывает на неточность экспериментальных данных для этих соединений.
Особое внимание следует обратить на соединения 43 и 44 с самыми большими расхождениями между экспериментальными и рассчитанными энтальпиями сублимации. Расхождение в 43.7 кДж моль–1, полученное для ксантина (44), объяснятся тем, что это соединение в кристалле присутствует в виде цвиттер-ионного таутомера, и в этом случае неправильно рассматривать неполярную структуру 44, отвечающую газообразному состоянию. По этой же причине модель электростатического потенциала дает по сравнению с экспериментом заниженные на 50−60 кДж моль–1 значения энтальпий сублимации аминокислот, которые являются самыми известными цвиттер-ионными соединениями. Столь же большое занижение значений ${{\Delta }_{{{\text{sub}}}}}H_{{298}}^{^\circ }$ аминокислот дают методы групповых вкладов [2, 3]. Для пропиленмочевины (43) наблюдается хорошее согласие с экспериментальной величиной [26], однако нет основания сомневаться в точности экспериментальных данных [25], поскольку величина ${{\Delta }_{{{\text{sub}}}}}H_{{298}}^{^\circ }$(газ), определенная из соотношения (1) с использованием экспериментальных значений ${{\Delta }_{{\text{f}}}}H_{{298}}^{^\circ }$(крист) и ${{\Delta }_{{{\text{sub}}}}}H_{{298}}^{^\circ }$ из [25], хорошо согласуется со значением, рассчитанным в настоящей работе методом G4. Причину столь большого расхождения, возможно, прояснят новые структурные и термохимические исследования пропиленмочевины.
Суммируя результаты, полученные в настоящей и предыдущих [21, 22] работах, можно сделать вывод, что при рассмотрении классов структурно близких соединений модель электростатического потенциала может давать хорошую точность предсказаний энтальпий сублимации, приближенную к точности экспериментальных измерений. Предложенное нами уравнение (3) дает для трех изученных классов соединений (адамантаны, алифатические и циклические производные мочевины) средние квадратичные отклонения 3.7, 2.3 и 4.2 кДж моль–1 соответственно, что намного лучше, чем для часто использующегося уравнения (2). Из рассмотренных в настоящей работе 44 циклических производных мочевины, уравнение (3) воспроизводит экспериментальные значения энтальпии сублимации в пределах 6 кДж моль–1 для 32 соединений. Этот достаточно неплохой результат обусловлен также и выполненным с помощью квантово-химических расчетов анализом точности экспериментальных данных, который позволил исключить из параметризации модели ненадежные величины энтальпий сублимации. Выполненные расчеты энтальпий образования газообразных соединений позволяют предположить неточность в экспериментальных данных, по меньшей мере, пяти соединений (34, 35, 37, 40 и 41).
Особую трудность при разработке и применении методов оценки энтальпии сублимации представляют полиморфизм, твердофазные превращения, образование пластических и жидких кристаллов, которые мало предсказуемы и создают значительные трудности при разработке успешного метода оценки энтальпии сублимации [63]. Твердые соединения, которые образуют пластические и жидкие кристаллы или не являются полностью кристаллическими, а также существующие в виде цвиттер-ионов, не должны рассматриваться как при разработке, так и применении методов, предсказывающих энтальпии сублимации. К сожалению, из опубликованных результатов не всегда можно понять природу фазовых переходов, а также является ли исследуемое вещество полностью кристаллическим.
Работа выполнена при поддержке Российского фонда фундаментальных исследований (код проекта № 17-03-00449).
Список литературы
Yang J., Hu W., Usvyat D., Matthews D., Schütz M., Chan G.K. // Science. 2014. V. 345. P. 640.
Gharagheizi F., Sattari M., Tirandazi B. // Ind. Eng. Chem. Res. 2011. V. 50. P. 2482.
Mathieu D. // Ind. Eng. Chem. Res. 2012. V. 51. P. 2814.
Keshavarz M.H., Bashavard B., Goshadro A. et al. // Therm. Anal. Calorim. 2015. V. 120. P. 1941.
Naef R., Acree W.E. Jr. // Molecules. 2017. V. 22. P. 1059.
Roy K., Kar S., Das R.N. A Primer on QSAR/QSPR Modeling. Fundamental Concepts. Springer, 2015.
Ouvrard C., Mitchell J.B.O. // Acta Cryst. 2003. V. B59. P. 676.
Жохова Н.И., Баскин И.И., Палюлин В.А. и др. // Журн. прикл. химии. 2003. Т. 76. С. 1966.
Gharagheizi F. // Thermochim. Acta. 2008. V. 469. P. 8.
Perlovich G.L., Raevsky O.A. // Cryst. Growth Des. 2010. V. 10. P. 2707.
Bagheria M., Bagheria M., Gandomi A.H., Golbraikh A. // Thermochim. Acta. 2012. V. 543. P. 96.
Salahinejad M., Le T.C., Winkler D.A. // J. Chem. Inf. Model. 2013. V. 53. P. 223.
Politzer P., Murray J.S. // Theor. Chem. Acc. 2002. V. 108. P. 134.
Politzer P., Murray J.S., Grice M.E. et al. // Mol. Phys. 1997. V. 91. P. 923.
Murray J.S., Politzer P. // WIREs Comput. Mol. Sci. 2011. V. 1. P. 153.
Byrd E.F.C., Rice B.M. // J. Phys. Chem. A. 2006. V. 110. P. 1005.
Singh H.J., Upadhyay M.K., Sengupta S.K. // J. Mol. Model. 2014. V. 20. P. 2205.
Tan B., Li Z., Guo X. et al. // Ibid. 2017. V. 23. P. 10.
Jaidann M., Roy S., Abou-Rachid H., Lussier L.-S. // J. Hazard. Mater. 2010. V. 176. P. 165.
Suntsova M.A., Dorofeeva O.V. // J. Mol. Graph. Model. 2017. V. 72. P. 220.
Dorofeeva O.V., Filimonova M.A. // Chem. Phys. Lett. 2018. V. 711. 231.
Dorofeeva O.V., Suchkova T.A. // J. Chem. Thermodyn. 2019. V. 131. P. 254.
Curtiss L.A., Redfern P.C., Raghavachari K. // J. Chem. Phys. 2007. V. 126. P. 084108.
Frisch M.J., Trucks G.W., Schlegel H.B. et al. Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford, CT, USA, 2016.
Ribeiro da Silva M.D.M.C., Ribeiro da Silva M.A.V., Freitas V.L.S. et al. // J. Chem. Thermodyn. 2008. V. 40. P. 386.
Farias R.F., Oliveira O.A., Medeiros J.V., Airoldi C. // Thermochim. Acta. 1999. V. 328. P. 241.
Silva A.L.R., Cimas Á., Vale N. et al. // J. Chem. Thermodyn. 2013. V. 58. P. 158.
Ramos F., Ledo J.M., Flores H. et al. // Thermochim. Acta. 2017. V. 655. P. 181.
Ribeiro da Silva M.D.M.C., Ribeiro da Silva M.A.V., Freitas V.L.S. et al. // J. Chem. Thermodyn. 2008. V. 40. P. 1378.
Emel’yanenko V.N., Verevkin S.P., Notario R. // Ibid. 2015. V. 87. P. 129.
Galvão T.L.P., Rocha I.M., Ribeiro da Silva M.D.M.C., Ribeiro da Silva M.A.V. // J. Phys. Chem. A. 2013. V. 117. P. 5826.
Ribeiro da Silva M.A.V., Amaral L.M.P.F., Szterner P. // J. Chem. Thermodyn. 2011. V. 43. P. 1924.
Imamura A., Takahashi K., Murata S., Sakiyama M. // Ibid. 1989. V. 21. P. 237.
Brunetti B., Irrera S., Portalone G. // J. Chem. Eng. Data. 2015. V. 60. P. 74.
Murata S., Sakiyama M., Seki S. // Thermochim. Acta. 1985. V. 88. P. 121.
Notario R., Emel’yanenko V.N., Roux M.V. et al. // J. Phys. Chem. A. 2013. V. 117. P. 244.
Kaminski M., Zielenkiewicz W. // J. Chem. Thermodyn. 1996. V. 28. P. 153.
Ribeiro da Silva M.A.V., Amaral L.M.P.F., Szterner P. // Ibid. 2011. V. 43. P. 1763.
Amaral L.M.P.F., Szterner P., Morais V.M.F. et al. // Ibid. 2016. V. 96. P. 93.
Emel’yanenko V.N., Zaitsau D.H., Shoifet E. et al. // J. Phys. Chem. A. 2015. V. 119. P. 9680.
Roux M.V., Temprado M., Notario R. et al. // J. Phys. Chem. A. 2008. V. 112. P. 7455.
Солдатова Т.В., Кабо Г.Я., Козыро А.А., Френкель М.Л. // Журн. физ. химии. 1990. Т. 64. С. 336.
Roux M.V., Notario R., Foces-Foces C. et al. // J. Phys. Chem. A. 2011. V. 115. P. 3167.
Roux M.V., Notario R., Foces-Foces C. et al. // Ibid. 2010. V. 114. P. 3583.
Notario R., Roux M.V., Ros F. et al. // J. Chem. Thermodyn. 2014. V. 74. P. 144.
Notario R., Roux M.V., Ros F. et al. // Ibid. 2014. V. 77. P. 151.
Imamura A., Murata S., Sakiyama M. // Ibid.1988. V. 20. P. 389.
Morais V.M.F., Miranda M.S., Matos M.A.R., Liebman J.F. // Mol. Phys. 2006. V. 104. P. 325.
Miranda M.S., Matos M.A.R., Morais V.M.F., Liebman J.F. // J. Chem. Eng. Data. 2011. V. 56. P. 4516.
Brunetti B., Piacente V., Portalone G. // J. Chem. Eng. Data. 2000. V. 45. P. 242.
Teplitsky A.B., Yanson I.K., Glukhova O.T. et al. // Biophys. Chem. 1980. V. 11. P. 17.
Amaral L.M.P.F., Szterner P., Ribeiro da Silva M.A.V. // J. Chem. Thermodyn. 2013. V. 64. P. 187.
Colomina V., Jimenez P., Turrion C. et al. // Thermochim. Acta. 1983. V. 68. P. 371.
Sabbah R., Komorowski S.J. // Ibid.1980. V. 41. P. 379.
Zielenkiewicz W., Szterner P., Kaminski M. // J. Chem. Eng. Data. 2003. V. 48. P. 1132.
Zielenkiewicz A., Wszelaka-Rylik M., Poznański J., Zielenkiewicz W. // J. Solution Chem. 1998. V. 27. P. 235.
Burkinshaw P.M., Mortimer C.T. // J. Chem. Soc., Dalton Trans. 1984. P. 75.
Szterner P., Galvão T.L.P., Amaral L.M.P.F. et al. // Thermochim. Acta. 2016. V. 625. P. 36.
Ribeiro da Silva M.D.M.C., Ribeiro da Silva M.A.V., Freitas V.L.S., Roux M.V. et al. // J. Chem. Thermodyn. 2009. V. 41. P. 1400.
Emel’yanenko V.N., Zaitsau D.H., Verevkin S.P. // J. Chem. Eng. Data. 2017. V. 62. P. 2606.
Lu T., Chen F. // J. Comp. Chem. 2012. V. 33. P. 580.
Sakiyama M., Imamura A. // Thermochim. Acta. 1989. V. 142. P. 365.
Acree, W., Jr., Chickos J.S. // J. Phys. Chem. Ref. Data. 2017. V. 46. P. 013104.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии