

Журнал физической химии, 2019, T. 93, № 10, стр. 1500-1505
Строение и конформационная динамика молекулы 2,2-диметилпропаналя в низших возбужденных синглетном (S1) и триплетном (Т1) электронных состояниях
И. А. Годунов a, В. А. Батаев a, *, Н. Н. Яковлев a
a Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
* E-mail: v.a.bataev@gmail.com
Поступила в редакцию 15.03.2019
После доработки 15.03.2019
Принята к публикации 09.04.2019
Аннотация
Многоконфигурационным методом CASPT2/def2-TZVPP проведено исследование геометрического строения, формы ППЭ и конформационной динамики молекулы 2,2-диметилпропананя в низших возбужденных синглетном и триплетном электронных состояниях. Для каждого электронного состояния задача о ядерных движениях большой амплитуды (вращении трет-бутильной группы и инверсии неплоского карбонильного фрагмента) решена вариационным методом. Показано, что электронное возбуждение в оба состояния вызывает пирамидализацию карбонильного фрагмента ССHO и поворот трет-бутильной группы. Полученными результатами подтверждены качественные выводы проведенного ранее экспериментального исследования S1 ← S0 вибронного спектра молекулы 2,2-диметилпропаналя.
Для понимания, описания и прогнозирования молекулярных фотофизических и фотохимических процессов необходимы данные о строении и динамике молекул в возбужденных электронных состояниях. В то же время исследование многоатомных молекул в возбужденных электронных состояниях в настоящее время представляет задачу повышенной сложности. Теоретические исследования требуют расчетных методов высокого уровня, как правило, многоконфигурационных. Но и в этом случае заранее неясно, насколько надежны и точны получаемые результаты. Их сравнение с соответствующими экспериментальными данными далеко не всегда возможно. Необходимую детальную экспериментальную информацию можно получить, практически, только методом вибронной (ровибронной) спектроскопии. Анализ таких спектров для сложных многоатомных молекул часто не позволяет получить их однозначную интерпретацию без дополнительных данных, которые могут быть получены в результате расчетов.
Поэтому наиболее надежным и рациональным подходом является проведение совместных исследований сложных многоатомных молекул в возбужденных электронных состояниях теоретическими и экспериментальными методами. Используя такой подход, нами были проведены исследования ряда конформационно нежестких молекул карбонильных соединения (см., например, [1, 2]).
Молекулы карбонильных соединений широко используются в химии и фотохимии. Строение простейших из них было детально исследовано в основном (S0) и низших возбужденных электронных состояниях: формальдегида (H2CO) и его замещенных [3–5], ацетальдегида (CH3CHO) [3, 4], ацетона ((CH3)2CO) [3, 4].
Недавно мы исследовали строение более сложных алифатических альдегидов в низшем возбужденном синглетном (S1) электронном состоянии: пропаналя (CH3CH2CHO) [6, 7] и 2-метилпропаналя ((CH3)2CHCHO) [8]. В начале 1990-х годов, мы экспериментально исследовали S1 ← S0 вибронный спектр поглощения в газовой фазе молекулы 2,2-диметилпропаналя (ДМПА, (CH3)3CCHO) [9] (рис. 1). Анализ его колебательной структуры и распределения интенсивностей вибронных полос позволили сделать качественные выводы о смещении минимумов потенциальной энергии ДМПА в S1-состоянии относительно минимумов в S0-состоянии по координате внутреннего вращения (трет-бутильной группы, (CH3)3C) и о неплоском строении карбонильного фрагмента (CCHO) ДМПА в S1-состоянии. Были найдены: “начало” электронного перехода (${\text{0}}_{0}^{0}$) и ряд фундаментальных частот в S1-состоянии. Кроме того, для молекулы ДМПА в S1-состоянии были оценены (методом согласования рассчитанных и экспериментальных вращательных контуров вибронных полос) геометрические параметры, а также высоты потенциальных барьеров внутреннего вращения (группы (CH3)3C) и инверсии карбонильного фрагмента (CCHO), неплоского в S1-состоянии.

Главные выводы о строении молекулы ДМПА в S1-состоянии имели качественный характер, а оценки, возможно, были недостаточно точны. Поэтому было весьма желательно уточнить полученные в работе [9] результаты с помощью надежного расчета. Это было невозможно в 90-е годы прошлого века, но сейчас такая возможность появилась, и мы провели необходимую работу.
В данной работе представлены результаты расчетов строения молекулы в основном (S0) и в низших возбужденных триплетном (T1) и синглетном (S1) электронных состояниях и проведено сравнение с экспериментальными данными [9].
МЕТОДЫ РАСЧЕТА
При выборе квантово-химического метода изучения молекулы ДМПА в S1- и Т1-состояниях мы опирались на наш и литературный опыт теоретического исследования родственных молекул алифатических альдегидов H2CO [3–5, 10], СH3CHO [3, 4], СH3CH2CHO [6] и (СH3)2CHCHO [8]. При использовании для расчетов молекул карбонильных соединений в возбужденных электронных состояниях многоконфигурационных методов квантовой химии оптимален, как правило, выбор активного пространства из пяти МО, локализованных на карбонильном фрагменте (σCO, πCO, nO, $\pi _{{{\text{CO}}}}^{*}$, $\sigma _{{{\text{CO}}}}^{*}$) и заселенных шестью электронами. Основанный на этом активном пространстве метод CASPT2 успешно применялся для исследования свойств молекул в низколежащих возбужденных электронных состояниях (см., например, [11]).
С учетом этих результатов в данной работе строение поверхности потенциальной энергии (ППЭ) молекулы ДМПА анализировали методом CASPT2 в базисном наборе АО def2-TZVPP. Для S0-состояния также использовали метод MP2. Расчеты проводили с помощью программного комплекса Molpro [12].
Задачу о ядерных движениях большой амплитуды (вращении трет-бутильной группы и инверсии карбонильного фрагмента) решали вариационным методом. В общем случае гамильтониан колебательного движения в многомодовой системе можно представить в форме:
Внутреннее вращение и инверсия в молекуле ДМПА рассматривали в одномерном (1D, т.е. при n = 1) и двумерном (2D, n = 2) приближениях. В качестве переменных для внутреннего вращения брали среднее арифметическое значение трех двугранных углов СCCO (φ1), а для инверсии – угол выхода связи CHа из плоскости CCO (φ2). Значения Bij(φ1, φ2) и V(φ1, φ2) рассчитывали для различных геометрических конфигураций молекулы, определяемых изменением переменных φ1, φ2 в диапазонах 0 ≤ φ1 ≤ 60°, 0° ≤ φ2 ≤ 60° с шагом не более, чем 10 для φ1 и 5° для φ2 (остальные геометрические параметры оптимизировали в ходе квантово-механического расчета). По этим опорным значениям функции Bij и V аппроксимировали (методом наименьших квадратов) модельными функциями переменных φ1 и φ2, выбранными с учетом требований симметрии.
В 2D-приближении кинематические параметры В11, В12, В22 и потенциальную поверхность V совместного торсионно-инверсионного движения в прямоугольной области 0 ≤ φ1 ≤ 360°, –60° ≤ ≤ φ2 ≤ 60° аппроксимировали линейными комбинациями тригонометрических функций:
В этих выражениях смешанные произведения $\cos (3K{{\varphi }_{1}})\sin L{{\varphi }_{2}}$ и $\sin (3K{{\varphi }_{1}})\cos L{{\varphi }_{2}}$ отсутствуют в силу свойств симметрии рассматриваемой молекулы.
В 1D-приближении для внутреннего вращения и инверсии использовали следующие тригонометрические аппроксимации для кинематического параметра В и потенциальной функции V:
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Строение молекулы ДМПА в S0-, S1- и T1-состояниях
Строение молекулы ДМПА в S0-состоянии экспериментально исследовано методами микроволновой [16] и колебательной [17–19] спектроскопии. В результате анализа микроволнового спектра [16] определены вращательные постоянные и другие параметры для основного, низших двух возбужденных состояний, соответствующих торсионному колебанию трет-бутильной группы, и низших возбужденных состояний трех метильных групп. Вычислены высоты потенциальных барьеров внутреннего вращения трет-бутильной группы, 1186 и 1176 кал/моль (415 и 411 см–1), из наблюдаемых расщеплений в первом и втором возбужденных состояний и 1100 кал/моль (385 см–1) из соотношения интенсивностей первого и основного состояний, а также высоты потенциальных барьеров внутреннего вращения метильных групп 2600, 3500 и 3800 кал/моль (198, 232 и 242 см–1, соответственно) по соотношению интенсивностей возбужденных и основного состояний.
Различие высот потенциальных барьеров двух симметричных относительно плоскости симметрии молекулы метильных групп качественно объяснено их одновременным вращением в одной и противоположной фазах. Геометрические параметры молекулы ДМПА в [16] не были определены из-за недостатка экспериментальных данных.
Колебательные спектры молекулы ДМПА в работах [18, 19] исследованы в низкочастотной области ниже 600 см–1. В частности, из частоты торсионного колебания трет-бутильной группы 70 см–1 вычислена высота потенциального барьера внутреннего вращения этой группы, равная 337 см–1 [19]. В [17] колебательные спектры получены в широкой области (выше 200 см–1), однако сделано отнесение лишь части частот колебаний. Таким образом, данные о колебательных частотах молекулы ДМПА в S0-состоянии далеко не полны.
Наши расчеты методами MP2 и CASPT2 в базисном наборе АО def2-TZVPP согласованно определяют устойчивую (соответствующую минимуму энергии) заслоненную конформацию молекулы ДМПА в S0-состоянии и ее соответствие точечной группе симметрии CS. Рассчитанные методами МР2 и CASPT2 высоты потенциальных барьеров вращения трет-бутильной группы составляют 531 и 518 см–1 соответственно. Вычисленное гармоническое значение частоты торсионного колебания трет-бутильной группы равно 78 см–1 (MP2) и 79 см–1 (CASPT2). Энергия фундаментального перехода 1 ← 0 для этого торсионного колебания, найденная вариационным методом (см. выше) для ППЭ, построенных обоими методами, равна 85 см–1. Видно, что соответствующие результаты расчетов молекулы ДМПА в S0-состоянии, полученные двумя методами, близки и удовлетворительно согласуются с экспериментальными величинами.
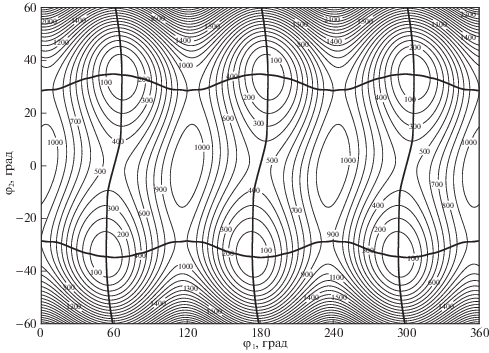
Наши расчеты показывают, что электронное возбуждение S1 ← S0 молекулы ДМПА вызывает значительные изменения геометрического строения молекулы: поворот трет-бутильной группы относительно карбонильной группы С=О, а также пирамидализацию карбонильного фрагмента CCHO, плоского в S0-состоянии (рис. 2 и табл. 1). В результате на сечении ППЭ молекулы в S1-состоянии по координатам внутреннего вращения и инверсии неплоского карбонильного фрагмента наблюдаются шесть симметричных потенциальных минимумов (рис. 3). Из табл. 1 видно, что в результате электронного возбуждения S1 ← S0 также увеличивается межъядерное расстояние R(C=O) и уменьшается валентный угол ∠CCO. Следует отметить, что электронное возбуждение мало влияет на геометрическое строение и относительную ориентацию всех трех метильных групп. Полученные данные расчета хорошо согласуются с выводами, сделанными в результате анализа экспериментального спектра [9]. В частности, для угла выхода связи СHa из плоскости ССО (φ2), описывающего степень пирамидализации карбонильного фрагмента, в [9] получена оценка φ2 ≈ 30°, расчет в настоящей работе дает 34.5° (табл. 1). Электронное возбуждение в T1-состояние вызывает похожие изменения геометрического строения и в настоящей работе детально не рассматривается.
Рис. 2.
Проекции Ньюмена устойчивых конформаций молекулы ДМПА в основном и возбужденных электронных состояниях.

Таблица 1.
Основные геометрические параметры и вращательные постоянные молекулы ДМПА в S0-, T1- и S1-состояниях, рассчитанные методом CASPT2/def2-TZVPP
| Параметр | S0 | S1 | T1 |
|---|---|---|---|
| R(C2O3) | 1.212 | 1.341 | 1.315 |
| R(C2C4) | 1.510 | 1.512 | 1.525 |
| R(C4C5) | 1.519 | 1.529 | 1.528 |
| R(C4C6) | 1.531 | 1.525 | 1.525 |
| R(C4C7) | 1.531 | 1.534 | 1.532 |
| R(C2H1) | 1.106 | 1.090 | 1.094 |
| ∠(O3C2H1) | 120.2 | 112.9 | 111.6 |
| ∠(O3C2C4) | 125.5 | 116.0 | 115.9 |
| ∠(C2C4C5) | 110.8 | 109.7 | 109.6 |
| ∠(C2C4C6) | 107.2 | 107.9 | 107.4 |
| ∠(C2C4C7) | 107.2 | 109.6 | 109.9 |
| ∠(C5C4C6) | 111.0 | 110.1 | 110.0 |
| ∠(C5C4C7) | 111.0 | 109.6 | 109.8 |
| ∠(C6C4C7) | 109.4 | 109.9 | 110.0 |
| d(O3C2C4C5) | 0.0 | 53.6 | 54.5 |
| d(O3C2C4C6) | 121.3 | 173.6 | 174.0 |
| d(O3C2C4C7) | –121.3 | –66.8 | –66.4 |
| d(O3C2C4H1) | 180.0 | –139.7 | –134.5 |
| φ1 | 0 | 66.5 | 65.9 |
| φ2 | 0 | 34.5 | 39.6 |
| A | 4501 | 4544 | 4535 |
| B | 2795 | 2775 | 2785 |
| C | 2754 | 2746 | 2755 |
| E | 0 | 28 845 | 26 010 |
Примечание. Межъядерные расстояния приведены в Å, углы – в град, вращательные постоянные – в МГц и адиабатические энергии Е возбужденных электронных состояний – в см–1. Экспериментальные значения вращательных постоянных для S0-состояния: А = 4496, B = 2771.56 и C = 2735.43 МГц. [16]. Экспериментальная величина “начала” электронного S1 ← S0-перехода составляет 29211 см–1 [9].
Рис. 3.
Карта двумерного торсионно-инверсионного сечения полной ППЭ молекулы ДМПА в S1-состоянии. Жирными линиями обозначены одномерные пути минимальной энергии.
В табл. 2 представлен ряд рассчитанных фундаментальных колебательных частот для молекулы ДМПА в S0-, S1- и T1-состояниях, а также имеющиеся соответствующие экспериментальные значения. Сравнение экспериментальных значений частот с рассчитанными показывает, что частоты 321 и 270 см–1 следует отнести к колебаниям ρ(СС3) и δ(CCO) соответственно (табл. 2), тогда как в работе [9] было принято обратное отнесение. Частота 279 см–1 соответствует согласованному колебанию симметричных метильных волчков. Из табл. 2 видно, что при электронном возбуждении S1 ← S0 частота валентного колебания ν(C=O) сильно уменьшается, понижается также частота плоского деформационного колебания δ(CCO). Аналогичные изменения наблюдаются и при электронном возбуждении T1 ← S0, они типичны для nπ*-электронного возбуждения молекул карбонильных соединений. Из табл. 2 видно, что соответствующие экспериментальные и расчетные результаты удовлетворительно согласуются.
Таблица 2.
Колебательные частоты (см–1) углеродного скелета молекулы ДМПА в S0-, T1- и S1-состояниях
| Отнесение | S0 | S0 (эксп) | T1 | S1 | S1 (эксп) |
|---|---|---|---|---|---|
| торс. τ(CHO) | 79 | – | 104 | 104 | 108 |
| ρ(CC3) | 344 | 322 | 317 | 338 | 321 |
| торс. τ(CH3) | 198 | 222 | 218 | 214 | |
| 257 | 264 | 276 | 272 | 279 | |
| 280 | 278 | 282 | 279 | ||
| δ(CCO) | 602 | 587 | 227 | 224 | 270 |
| ν(CC3) | 782 | – | 747 | 756 | 607 |
| ν(CO) | 1767 | – | 1207 | 1200 | 1150 |
Описание движений большой амплитуды молекулы ДМПА в S1- и T1-состояниях
В молекуле ДМПА в S0-состоянии вращения трет-бутильной и трех метильных групп относятся к движениям большой амплитуды. В молекуле в S1- и T1-состояниях к ним добавляется инверсионное колебание неплоского карбонильного фрагмента. Эти движения являются существенно ангармоническими (особенно инверсионное колебание, для которого в S1-состоянии наблюдается инверсионное расщепление нулевого колебательного уровня [9]). Для корректного описания таких движений энергии колебательных уровней и колебательные волновые функции рассчитываются в задачах разной размерности вариационным методом. В предыдущем разделе было указано, что вращение метильных волчков слабо связано с другими движениями большой амплитуды, поэтому с целью упрощения задачи (понижения ее размерности) вращения метильных волчков в настоящей работе не рассматривались. 2D-потенциальные функции молекулы ДМПА в S1- и T1-состояниях были построены как сечения полной ППЭ по координатам вращения трет-бутильного волчка (φ1) и инверсии неплоского карбонильного фрагмента (φ2). На рис. 3 приведена 2D-потенциальная функция ДМПА в S1-состоянии. Для T1-состояния потенциальная функция очень похожа и в данной работе не приводится.
При аппроксимации 1D-потенциальных функций молекулы ДМПА получены следующие высоты потенциальных барьеров внутреннего вращения (VВВ) и инверсии (Vинв): VВВ = 906 и 892 см–1, Vинв = = 485 и 741 см–1 для S1- и T1-состояний соответственно. Экспериментально полученная высота VВВ для S1-состояния составляет 910 см–1. Определить вид потенциальной функции инверсии в работе [9] не удалось из-за недостатка экспериментальных данных; была найдена лишь величина инверсионного расщепления нулевого колебательного уровня (40 см–1), на основе которой была получена достаточно грубая оценка Vинв ≈ 600 см–1.
В 1D-модели внутреннего вращения по мере изменения угла φ1 координата инверсии φ2 не остается фиксированной, а определяется в результате оптимизации геометрии. Поэтому путь минимальной энергии, отвечающий внутреннему вращению, на 2D-потенциальной поверхности оказывается несколько криволинейным. То же самое относится к пути минимальной энергии 1D-инверсионного движения. Из рис. 3 видно, что инверсия карбонильного фрагмента в молекуле ДМПА сопровождается поворотом трет-бутильной группы (приблизительно на 15°), т.е. эти два движения большой амплитуды не являются независимыми.
Отнесение нижних уровней энергии по симметрии и определение колебательных квантовых чисел для двух составляющих движений (внутреннего вращения и инверсии) проводили на основании анализа структуры узловых поверхностей и областей локализации рассчитанных волновых функций. Результаты расчетов для ряда низших колебательных состояний приведены в табл. 3. Из табл. 3 видно, что экспериментальная и рассчитанные энергии торсионного перехода в S1-состоянии вполне удовлетворительно согласуются между собой. Различие экспериментальной и рассчитанной величин инверсионного расщепления нулевого колебательного уровня для S1-состояния более существенно.
Таблица 3.
Рассчитанные энергии 1D-торсионных и инверсионных и 2D-торсионно-инверсионных уровней энергии молекулы ДМПА в S1- и T1-состоянии (см–1)
| Отнесение | S1 | T1 | ||
|---|---|---|---|---|
| 1D | 2D | 1D | 2D | |
| (0,0) a1 | 0.0 | 0.0 | 0.0 | 0.0 |
| (0,0) a2 | 23.1 | 14.5 | 4.6 | 3.1 |
| (1,0) a1 | 123.2 | 98.9 | 104.7 | 98.6 |
| (1,0) a2 | 100.1 | 112.4 | 101.6 | 101.8 |
| (2,0) a2 | 220.4 | 190.7 | 202.1 | 193.7 |
| (2,0) a1 | 197.3 | 203.1 | 199.0 | 196.2 |
| (3,0) a1 | 314.1 | 280.9 | 296.3 | 258.7 |
| (3,0) a2 | 291.0 | 287.7 | 293.2 | 286.4 |
| (0,1) a1 | 339.4 | 385.2 | 410.8 | 436.3 |
Примечание. Числа в скобках (${{{v}}_{{\text{t}}}}$, ${{{v}}_{{\text{i}}}}$) соответствуют значениям торсионного и инверсионного квантовых чисел в предположении полного разделения колебаний, символы после скобок обозначают неприводимое представление для колебательной волновой функции. Приведены только уровни симметрии А-типа. Экспериментальные энергии торсионно-инверсионных уровней: (0,0) a2, (1,0) a1 – равны 40 и 108 см–1 соответственно [9].
Следует отметить, что для родственной молекулы ацетальдегида (CH3CHO) в S1-состоянии экспериментальная величина инверсионного расщепления нулевого колебательного уровня составляет 33–34 см–1 [20–22], а высота барьера инверсии определена как 530 [20], 585 [21] и 639 см–1 [22]. Учитывая эти данные, высоту потенциального барьера инверсии молекулы ДМПА в S1-состоянии можно оценить в 500–50 см–1.
Ранее для молекулы ацетальдегида в T1-состоянии была найдена экспериментальная высота барьера инверсии 1110 см–1 [23], что примерно в 1.5 раза выше, чем для S1-состояния. Вычисленные для молекулы ДМПА в S1- и T1-состояниях барьеры инверсии находятся в таком же соотношении.
ЗАКЛЮЧЕНИЕ
Проведены квантово-химические расчеты методом CASPT2/def2-TZVPP параметров конформационно нежесткой молекулы ДМПА в низших возбужденных электронных S1- и T1-состояниях. Кроме того, для каждого электронного состояния задача о ядерных движениях большой амплитуды (вращении трет-бутильной группы и инверсии неплоского карбонильного фрагмента) решалась вариационным методом. Проведенные расчеты показали, что электронные возбуждения S1 ← S0 и T1 ← S0 вызывают повороты трет-бутильной группы относительно карбонильного фрагмента, который в отличие от основного S0-состояния становится пирамидальным.
Определены угол поворота трет-бутильной группы и степень пирамидализации при возбуждениях S1 ← S0 и T1 ← S0. Обнаружены взаимосвязь вращения трет-бутильной группы и инверсии и практически независимые от них ориентация и движение метильных групп. Определены геометрические параметры и колебательные частоты молекулы ДМПА в S1- и T1-состояниях. В соответствии с расчетом, для S1-состояния переотнесены колебательные частоты 321 и 270 см–1 к колебаниям ρ(СС3) и δ(ССО), соответственно.
В одно- (1D) и двумерном (2D) приближениях вычислены энергии торсионных (трет-бутильной группы) и инверсионных уровней энергии молекулы ДМПА в S1- и T1-состояниях. Вычислены высоты потенциальных барьеров внутреннего вращения и инверсии молекулы ДМПА в S1- и T1-состояниях. Высота потенциального барьера внутреннего вращения в S1-состоянии хорошо согласуется с экспериментальной. Соотношение вычисленных высот потенциальных барьеров инверсии для ДМПА согласуется с найденными ранее для родственной молекулы ацетальдегида.
Расчеты подтвердили качественные выводы проведенного ранее экспериментального исследования S1 ← S0 вибронного спектра молекулы ДМПА.
Работа выполнена с использованием оборудования Центра коллективного пользования сверхвысокопроизводительными вычислительными ресурсами МГУ имени М.В. Ломоносова.
Список литературы
Bataev V.A., Pupyshev V.I., Abramenkov A.V., Godunov I.A. // Russ. J. Phys. Chem. 2000. V. 74. Suppl. 2. S279.
Годунов И.А., Батаев В.А., Яковлев Н.Н., Пупышев В.И. // Журн. физ. химии. 2002. Т. 76. № 11. С. 1912.
Angeli C., Borini S., Ferrighi L., Cimiraglia R. // THEOCHEM. 2005. V. 718. P. 55 (см. и цитированную литературу).
Angeli C., Borini S., Ferrighi L., Cimiraglia R. // J. Chem. Phys. 2005. V. 122. P. 114304 (см. и цитированную литературу).
Bokarev S.I., Dolgov E.K., Bataev V.A., Godunov I.A. // Int. J. Quantum Chem. 2009. V. 109. P. 569 (см. и цитированную литературу).
Kudich A.V., Bataev V.A., Abramenkov A.V. et al. // THEOCHEM. 2003. V. 631. P. 39.
Godunov I.A., Yakovlev N.N., Maslov D.V. et al. // Phys. Chem. Chem. Phys. 2016. V. 18. P. 15244.
Godunov I.A., Terentiev R.V., Maslov D.V. et al. // Struct. Chem. 2019. V. 30. № 2. P. 529.https://doi.org/10.1007/s11224-018-1243-9
Яковлев Н.Н., Годунов И.А. // Журн. физ. химии. 1993. Т. 67. № 9. С. 1826.
Merchan M., Roos B.O. // Theor. Chim. Acta. 1995. V. 92. № 4. P. 227.
Roos B.O., Andersson K., Fülscher M.P. et al. // Adv. Chem. Phys. 1996. V. 93. P. 219.https://doi.org/10.1002/9780470141526.ch5
Werner H.-J., Knowles P.J., Knizia G. et al., see http://www.molpro.net;
Harthcock M.A., Laane J. // J. Mol. Spectrosc. 1982. V. 91. P. 300.
Harthcock M.A., Laane J. // J. Phys. Chem. 1985. V. 89. P. 4231.
Абраменков А.В. //Журн. физ. химии. 1995. Т. 69. № 6. С. 1048.
Ronn A.M., Woods R.C. // J. Chem. Phys. 1966. V. 45. № 10. P. 3831.
Crowder G.A. // J. Chem. Soc., Perkin Trans II. 1973. № 9. P. 1241.
Durig J.R., Craven S.M., Mulligan J.H. et al. // J. Chem. Phys. 1973. V. 58. № 4. P. 1281.
Durig J.R., Kenton R., Phan H.V., Little T.S. // J. Mol. Struct. 1991. V. 247. P. 237.
Weersink R.A., Cramb D.T., Wallace S.C., Gordon R.D. // J. Chem. Phys. 1995. V. 102. № 2. P. 623.
Liu H., Lim E.C., Muñoz-Caro C., Niño A. et al. // J. Mol. Spectrosc. 1996. V. 175. № 1. P. 172.
Liu H., Lim E.C., Niño A. et al. // J. Mol. Spectrosc. 1998. V. 190. № 1. P. 78.
Yakovlev N.N., Godunov I.A. // Can. J. Chem. 1992. V. 70. P. 931.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии