

Журнал физической химии, 2019, T. 93, № 10, стр. 1445-1454
Развитие расчетных методов в лаборатории химической термодинамики химического факультета МГУ
А. Л. Восков a, Н. А. Коваленко a, И. Б. Куценок a, И. А. Успенская a, *
a Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
* E-mail: ira@td.chem.msu.ru
Поступила в редакцию 15.03.2019
После доработки 15.03.2019
Принята к публикации 09.04.2019
Аннотация
Представлен краткий обзор работ по развитию расчетных методов, выполненных в лаборатории химической термодинамики Химического факультета МГУ имени М.В. Ломоносова. Перечислены основные этапы становления расчетных работ, имена ученых, которые внесли основной вклад в развитие этого направления, дано описание наиболее значимых результатов, полученных за последние 60 лет.
В 2019 г. кафедра физической химии и лаборатория химической термодинамики отмечают свой девяностолетний юбилей. Как известно, прошлое в значительной степени определяет настоящее и будущее, поэтому нам хотелось бы провести небольшой исторический экскурс и отразить этапы развития расчетных методов в нашей лаборатории, показать, как своевременный учет тенденций развития той или иной области знания может стать залогом последующего процветания научного коллектива.
Первым химиком, проводившим термодинамические исследования в Московском университете, был, по-видимому, профессор И.А. Каблуков, который вел у студентов занятия по физической химии, начиная с 1884 г. В 1915 г. лекции по химической термодинамике и приложениям математики к химии начал читать приват-доцент А.В. Раковский. С 01.10.1929 Химическое отделение Физико-математического факультета Московского университета было выделено в самостоятельный Химический факультет, в состав которого вошли восемь кафедр, в том числе, кафедра физической химии, включающая в себя шесть лабораторий. Профессор А.В. Раковский стал руководителем этой новой кафедры и входящей в ее состав лаборатории галургии – науки о физико-химических свойствах природных солей и технологии их переработки. Начало тридцатых годов ознаменовалось крупномасштабными и весьма неоднозначными экспериментами в области образования, которые благополучно закончились к середине 30-х, и к 1936 г. лаборатория галургии постепенно приобрела свое современное название лаборатории химической термодинамики. С момента основания до середины 60-х гг. XX века основное внимание сотрудников лаборатории уделялось экспериментальным исследованиям термодинамических свойств систем разной природы и компонентности, однако по мере развития вычислительных методов и техники, доля расчетно-теоретических работ неуклонно возрастала. На начальном этапе развития этого направления основное внимание уделялось методологии решения расчетных задач, в последние годы значительные успехи достигнуты в практической реализации расчетных методов (в том числе, в разработке программного обеспечения). Краткий обзор таких исследований и является целью настоящей работы.
Несомненно, заслугой А.В. Раковского является активное использование математического аппарата в практике термодинамических исследований, он первым в России стал применять гиббсовскую термодинамику, как в преподавании, так и в своих научных трудах. Его последователем можно считать и Я.И. Герасимова, долгие годы возглавлявшего кафедру и лабораторию, читавшего курс физической химии на химфаке МГУ. Тем не менее, становление именно расчетных методов в лаборатории химической термодинамики связано, в большей степени, с именами проф. А.М. Евсеева и его ученика проф. Г.Ф. Воронина, которые в середине XX-го столетия стали активно применять методы статистической термодинамики и термодинамические расчеты с использованием появившихся тогда больших ЭВМ. На определенном жизненном этапе пути этих двух неординарных ученых разошлись: Александр Михайлович возглавил межкафедральную лабораторию химической кибернетики, а Геннадий Федорович более сорока лет руководил лабораторией химической термодинамики. Наиболее значимые и интересные работы, связанные с развитием расчетных методов химической термодинамики, принадлежат проф. Г.Ф. Воронину и работавшим с ним сотрудникам. Прекрасное знание предмета, вычислительных методов, способность предвидеть перспективы развития компьютерной техники позволили правильно расставить приоритеты в деятельности лаборатории, что и определило, в конечном счете, вполне устойчивое положение, занимаемое ею в настоящее время.
Говоря о развитии расчетно-теоретических работ в лаборатории химической термодинамики, следует выделить несколько аспектов: во-первых, это развитие новых методов расчета термодинамических свойств фаз, фазовых и химических равновесий, во-вторых, создание новых термодинамических моделей и, наконец, разработка программного обеспечения для соответствующих расчетов. Далее каждое из этих направлений исследований рассмотрено отдельно, хотя и очевидно, что это взаимосвязанные работы.
РАЗВИТИЕ НОВЫХ МЕТОДОВ РАСЧЕТА ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ ФАЗ И РАВНОВЕСИЙ С ИХ УЧАСТИЕМ
Цель расчета равновесий в гетерогенной системе – определение состава и количества каждой из фаз, образующих эту систему, при заданном наборе внешних переменных. Если характеристические функции всех фаз известны (т.е. имеются их термодинамические модели) и параметры существования системы фиксированы, то такая задача, как правило, имеет единственное решение, т.е. является математически корректной. Однако в реальности эта единственность решения не гарантирована даже при математической корректности постановки задачи из-за ряда особенностей изучаемых объектов [1]. Это может быть множественность возможных моделей внутреннего строения системы, полнота набора составляющих, экстраполяционные возможности термодинамических моделей и т.п. В этой связи одной из актуальных проблем расчетных методов химической термодинамики является разработка способов однозначного определения энергии Гиббса всех фаз интересующей системы по минимальному набору исходных данных и связанная с нею проблема решения корректных и некорректных задач расчета фазовых равновесий.
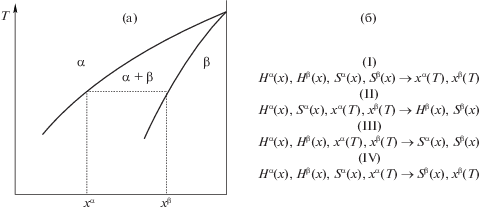
Эти проблемы подробно рассмотрены в ряде работ Воронина с соавторами в 80–90-е годы двадцатого столетия [2–7]. Их результаты кратко можно проиллюстрировать с помощью схемы, представленной на рис. 1. Примем, что для изучаемой двухкомпонентной системы, в которой образуются фазы α и β, известны следующие данные: Hα, Hβ, Sα, Sβ, xα(T), xβ(T), где Hi и Si – энтальпия и энтропия i-й фазы, а xi(T) – уравнение соответствующей кривой фазового равновесия. Как было показано в работах [2–4], комбинации четырех из этих функций достаточно для восстановления двух оставшихся, т.е. задача оптимизации системы может быть решена математически корректно. Для плохо обусловленных задач (когда не хватает данных для построения термодинамических моделей), целесообразно попытаться сначала оценить некоторые термодинамические свойства, чтобы перевести задачу в класс математически корректных. Например, в работе Дегтярева с соав. [8] было показано, что хорошие результаты удается получить, оценивая энтропию стехиометрической фазы или раствора.
Рис. 1.
Иллюстрация математически корректного решения задачи расчета равновесий: а – фрагмент фазовой диаграммы двухфазной системы А–В, б – возможные комбинации данных, обеспечивающих корректное решение задачи: (I) – расчет равновесий по известным значениям термодинамических функций, (II) – расчет функций образования фазы (β) по известным свойствам и составу фазы (α), (III) – расчет энтропий образования фаз (α) и (β) по энтальпиям образования и равновесным составам фаз, (IV) расчет одной из ветвей фазовой диаграммы по другой.
Если нет возможности доопределить задачу, поставив ее корректно, на помощь может прийти интуиция исследователя. Нам представляется весьма неординарным и, к сожалению, недостаточно оцененным профессиональным сообществом, способ восстановления недостающих участков фазовых T–x-диаграмм по их известным фрагментам. Методика расчета подробно описана в работе [9], а конкретные результаты ее применения представлены, например, в работах [10, 11].
При изучении фазовых диаграмм часто удается зафиксировать только температуру ликвидуса, поэтому возможность восстановления линии солидуса можно рассматривать как реальную практически значимую задачу. Для бинарной системы соответствующие расчеты основаны на минимизации целевой функции:
(1)
$f({{a}^{l}},{{b}^{l}},{{a}^{s}},{{b}^{s}}) = \sum\limits_{i = 1}^n {{{w}_{i}}{{{({{{\bar {T}}}_{i}} - {{T}_{i}}(\bar {x}_{{\text{B}}}^{{\text{l}}},x_{i}^{s}))}}^{2}}} $(2)
$\begin{gathered} - {{\Delta }_{{\text{m}}}}{{G}_{{\text{A}}}}(T) = RT\ln ((1 - \bar {x}_{{\text{B}}}^{{\text{l}}}){\text{/}}(1 - x_{{\text{B}}}^{{\text{s}}})) + \\ + \;(\mu _{{\text{A}}}^{{{\text{ex}}}}(T,\bar {x}_{{\text{B}}}^{{\text{l}}}) - \mu _{{\text{A}}}^{{{\text{ex}}}}(T,x_{{\text{B}}}^{{\text{s}}})), \\ - {{\Delta }_{{\text{m}}}}{{G}_{{\text{B}}}}(T) = RT\ln (\bar {x}_{{\text{B}}}^{{\text{l}}}{\text{/}}x_{{\text{B}}}^{{\text{s}}}) + (\mu _{{\text{B}}}^{{{\text{ex}}}}(T,\bar {x}_{{\text{B}}}^{{\text{l}}}) \\ - \;\mu _{{\text{B}}}^{{{\text{ex}}}}(T,x_{{\text{B}}}^{{\text{s}}})), \\ \end{gathered} $(3)
${{\Delta }_{{\text{m}}}}{{G}_{j}}(T) = {{\Delta }_{{\text{m}}}}{{H}_{j}}(T) - {{\Delta }_{{\text{m}}}}{{S}_{j}}(T).$Векторы ${{\vec {a}}^{\alpha }}\, = \,(a_{0}^{\alpha },a_{1}^{\alpha },......a_{{{{n}^{\alpha }} - 1}}^{\alpha })$ и ${{\vec {b}}^{\alpha }}\, = \,(b_{0}^{\alpha },b_{1}^{\alpha },......b_{{{{n}^{\alpha }} - 1}}^{\alpha })$ содержат неизвестные параметры термодинамических моделей сосуществующих фаз. Если воспользоваться простейшими полиномиальными зависимостями избыточной энергии Гиббса раствора α в виде
(4)
${{G}^{{{\text{ex}}}}}(T,{{x}^{\alpha }}) = {{x}^{\alpha }}(1 - {{x}^{\alpha }})\sum\limits_{i = 0}^{{{n}^{\alpha }} - 1} {(a_{i}^{\alpha } + Tb_{i}^{\alpha }){{{({{x}^{\alpha }})}}^{i}}} ,$Получаемые таким образом описания термодинамических свойств фаз являются, в некотором роде, фиктивными, наборы параметров могут не соответствовать действительным значениям термодинамических функций. Но в данном случае корреляция между параметрами, с которой приходится сталкиваться при обычной параметризации моделей, облегчает выбор адекватных моделей для расчета равновесий и восстановления участков фазовой диаграммы по известным ее фрагментам. На результат описания диаграммы существенно влияет число варьируемых параметров, но не их конкретный набор.
Анализируя вклад лаборатории в развитие расчетных методов химической термодинамики, нельзя не упомянуть о методе “выпуклых оболочек”. Этот метод является одним из наиболее эффективных и перспективных способов расчета фазовых диаграмм; он представляет собой прямой метод определения координат гиббсовской “поверхности рассеянной энергии” гетерогенной системы. Основное отличие его от других методов расчета фазовых равновесий связано с использованием геометрических способов построения огибающих термодинамических потенциалов и нахождения экстремумов характеристической функции системы. Этот метод позволяет непосредственно определять устойчивые равновесные состояния многофазной системы, соответствующие глобальному минимуму энергии Гиббса при фиксированных давлении и температуре. В отличие от других способов решения задачи расчета фазовых равновесий, при использовании метода выпуклых оболочек не требуется задания начальных приближений определяемых неизвестных. Существенное преимущество метода – возможность рассчитывать сразу серию равновесий при изменяющихся значениях количеств или концентраций компонентов, т.е. во всей области составов системы.
Автором идеи метода следует считать самого Дж.В. Гиббса, хотя формулировки понятия выпуклости и вогнутости [12] появились в математике спустя несколько десятилетий после публикации его работ. За прошедшие годы метод выпуклых оболочек применяли довольно часто и российские, и зарубежные авторы [13–17], однако следует подчеркнуть, что теоретические основы метода, предложенного в свое время Дж.В. Гиббсом, получили дальнейшее развитие именно в работах Г.Ф. Воронина [18–20].
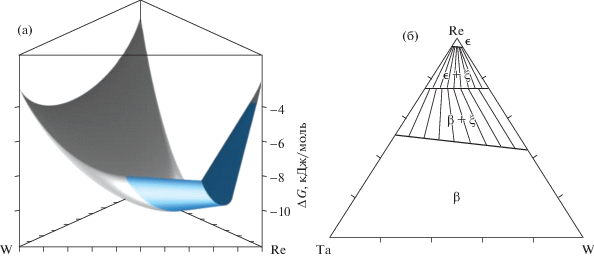
Одна из проблем при переходе от выпуклой оболочки к фазовой диаграмме заключается в идентификации особых точек оболочки и их проектировании на координатные оси, плоскости или объем диаграммы (в зависимости от размерности системы). На этом этапе возникает ряд проблем, решение которых нельзя отнести к разряду тривиальных. Чтобы представить фазовую диаграмму в привычном виде, надо сформулировать и запрограммировать правила сопряжений симплексов фазовой диаграммы в ее особых точках, решить проблему описания различий между двух- и трехфазными областями диаграммы и построения коннод в двухфазных областях. Оригинальное решение этой задачи было предложено А.Л. Восковым, более подробно этот материал изложен в работе [21]. На рис. 2, 3 в качестве иллюстрации получаемых результатов изображены выпуклые оболочки и изотермические сечения систем со стехиометрическими фазами (CaO–Al2O3–SiO2 , T = 800 K) и растворами (Ta–W–Re, T = 2293 K).

Совместная оптимизация данных по термодинамическим свойствам и фазовым диаграммам (CALPHAD method). В настоящее время метод выпуклых оболочек является активным инструментом для расчета фазовых диаграмм систем разной природы – оксидных, металлических, солевых, органических и пр. При термодинамическом моделировании сложных многокомпонентных систем (а именно они представляют практический интерес) принято использовать принцип последовательного возрастания размерности задачи (в англоязычной литературе его называют CALPHAD-method, CALculation of PHAse Diagrams) [22, 23]. Такой подход позволяет получать наборы параметров модели, адекватно описывающих не только интересующую систему, но и все составляющие ее подсистемы. Тем самым удается формировать не просто базы данных о термодинамических свойствах фаз разных систем, но и базы параметров моделей, с помощью которых могут быть рассчитаны фазовые и химические равновесия, интересующие практиков. Основоположниками метода принято считать Л. Кауфмана и Г. Бернстейна [24], которые использовали в своих первых расчетных работах самые простые модели фаз. Получающиеся при этом результаты во многих случаях заметно отличались от экспериментальных данных, однако в настоящее время уровень термодинамического моделирования таков, что удается воспроизводить экспериментальные значения в пределах погрешности измерения свойств.
Работы по согласованию термодинамических свойств и условий фазовых равновесий активно проводятся в лаборатории химической термодинамики с конца 70-х годов прошлого столетия. Результаты таких расчетов востребованы индустрией: собственно, реальный сектор экономики и определяет приоритеты в моделировании тех или иных объектов. Из работ последних лет, ориентированных на решение конкретных технологических задач, следует особо отметить публикации по оптимизации систем, включающих воду, аммиак и углекислый газ [25, 26]. Это не так часто встречающийся в нашей практике пример, когда четко просматривается логическая цепочка от фундаментальной задачи построения уравнений состояния до разработки новой технологии производства карбамида. Серия работ сотрудников лаборатории посвящена моделированию водно-солевых систем, представляющих интерес для разработки новых и оптимизации существующих условий производства удобрений [27, 28], созданию термодинамической модели процесса экстракционного разделения нитратов РЗЭ [29, 30], моделированию металлических систем [31, 32]. В целом, работы по согласованию термодинамических свойств и условий фазовых равновесий ведутся на уровне, не уступающем общемировому, но есть одна особенность, которая отличает публикации сотрудников лаборатории химической термодинамики от публикаций других научных коллективов, работающих в этом направлении. Это – оценка доверительных интервалов параметров предлагаемых аналитических зависимостей, что позволяет проводить более строгую дискриминацию моделей. Для определения доверительных интервалов параметров вычисляется матрица Якоби
(5)
$\begin{gathered} {{J}_{{ij}}} = \left( {\frac{{\partial (w_{i}^{{1/2}}{{z}^{{{\text{расч}}}}}({{X}_{1}},{{\beta }_{{0,1}}}, \ldots ,{{\beta }_{{0,k}}}))}}{{\partial {{\beta }_{j}}}}} \right), \\ 1 \leqslant i \leqslant n,\quad 1 \leqslant j \leqslant k, \\ \end{gathered} $(6)
$\begin{gathered} F({{Z}_{1}}, \ldots ,{{Z}_{n}},{{\beta }_{1}}, \ldots ,{{\beta }_{k}}) = \\ = \;{{\sum\limits_Z {\sum\limits_i {{{w}_{{Z,j}}}(Z_{i}^{{{\text{эксп}}}} - Z_{i}^{{{\text{расч}}}}({{\beta }_{1}}, \ldots ,{{\beta }_{k}}))} } }^{2}}, \\ \end{gathered} $Задача оценки статистической значимости параметров термодинамических моделей относится к классу нетривиальных и решается она в ограниченном числе научных групп.
Метод CALPHAD широко практикуется в настоящее время, но его использование предполагает довольно большой объем работы по согласованию термодинамических свойств и условий фазовых равновесий в системах малой размерности. Это объемная, кропотливая работа, в значительной степени ориентированная на решение задач, которые актуальны не только сегодня, но и будут востребованы в будущем. Если какой-либо объект содержит несколько компонентов и при этом является весьма перспективным с точки зрения практического использования, то моделируют непосредственно многокомпонентную систему, и только спустя время (или параллельно) оптимизируют системы меньшей размерности. Именно такая ситуация была в 90-е годы прошлого столетия, когда начался “бум высокотемпературной сверхпроводимости”. Более подробно вклад сотрудников лаборатории в моделирование оксидных сверхпроводников описан далее; это серьезная веха в истории развития расчетных методов, заслуживающая отдельного рассмотрения.
Аппроксимация термодинамических свойств фаз с использованием комбинаций функций Планка–Эйнштейна. Проблема аппроксимации температурной зависимости термодинамических свойств индивидуальных веществ была и остается актуальной задачей аналитического представления экспериментальных данных. Традиционно используемые полиномиальные зависимости не подходят для решения задач корректной экстраполяции данных; при использовании таких зависимостей в широком интервале температур возникает проблема “кусочечного” описания с необходимостью согласования набора полиномов в узловых точках. В работе Г.Ф. Воронина и И.Б. Куценка [33] для описания температурной зависимости теплоемкости (и получаемых при ее интегрировании функций) предложено использовать комбинацию функций Планка–Эйнштейна:
(8)
$\begin{gathered} {{c}_{p}}(T) = \mathop \sum \limits_{i = 1}^k \,{{{\alpha }}_{i}}{{C}_{E}}({{\theta }_{i}}{\text{/}}T), \\ {{C}_{E}}(x) = 3R{{x}^{2}}\frac{{\exp (x)}}{{{{{\left[ {\exp (x) - 1} \right]}}^{2}}}}, \\ \end{gathered} $(9)
$\begin{gathered} S(T) = \mathop \sum \limits_{i = 1}^k \,{{{\alpha }}_{i}}{{S}_{E}}({{\theta }_{i}}{\text{/}}T), \\ {{S}_{E}}(x) = 3R\left[ {\frac{x}{{(\exp (x) - 1)}} - \ln (1 - \exp ( - x))} \right]. \\ \end{gathered} $Для расчета варьируемых параметров αi (безразмерная величина) и θi(K) можно использовать результаты измерений теплоемкости от низких температур вплоть до точки плавления, хотя обычно ограничиваются комнатной температурой. Аппроксимирующие коэффициенты вычисляются при минимизации суммы квадратов отклонений между рассчитанными и измеренными значениями теплоемкости:
(10)
${{\chi }^{2}} = \mathop \sum \limits_{j = 1}^N {{\left[ {{{C}_{{pj}}}({{T}_{j}}) - \mathop \sum \limits_{i = 1}^k \,{{{\alpha }}_{i}}{{C}_{E}}\left( {\frac{{{{\theta }_{i}}}}{{{{T}_{j}}}}} \right)} \right]}^{2}},$Возможности функций Планка–Эйнштейна не ограничиваются удобным аналитическим описанием обычных температурных зависимостей свойств индивидуальных веществ. Сотрудниками лаборатории предложен оригинальный способ описания экспериментальных данных при наличии аномалий на кривых Cp(T) [37] и прогноза значений стандартных энтропий при 298.15 К при отсутствии результатов адиабатических измерений в “гелиевой” области [38]. Как показано в работе [39] на примере олова, использование комбинированных функций Планка–Эйнштейна позволяет оценить энтропию и энтальпию фазового перехода двух кристаллических модификаций при известном значении Ttr, если сам переход не реализуется в силу, например, кинетических факторов.
Совсем недавно на основе функций Планка–Эйнштейна разработана модель групповых функциональных вкладов для описания термодинамических свойств цеолитов; результаты представлены в работе [40]. Цеолиты – алюмосиликаты, состав которых задается формулой:
(11)
$\begin{gathered} {{С}_{p}}({{{\vec {\alpha }}}^{{\left( 1 \right)}}},...,{{{\vec {\alpha }}}^{{\left( m \right)}}},{{{\vec {\theta }}}^{{\left( 1 \right)}}},...,{{{\vec {\theta }}}^{{\left( m \right)}}},T) = \\ = \sum\limits_{i = m}^{i = 1} {{{f}_{i}}(\vec {n})} \sum\limits_{j = m}^{j = 1} {\alpha _{j}^{{\left( i \right)}}{{C}_{E}}\left( {\frac{{\theta _{j}^{{\left( i \right)}}}}{T}} \right)} , \\ \end{gathered} $Для оптимизации числа и значений параметров модели было использовано более 3000 экспериментальных точек – значений теплоемкости и теплосодержания. С помощью разработанной модели успешно описаны термодинамические свойства 46-и цеолитов, точность оценки теплоемкости и энтропии при 298.15 K составила, в среднем, около 5%. При более высоких температурах точность аппроксимации несколько падает и составляет приблизительно 5% для энтропии и 9% для теплоемкостей при 500 K. При понижении температуры точность также уменьшается и при 100 K составляет 7% для энтропии и 6% для теплоемкости, а при 50 K – 14% и 6.6% соответственно. При дальнейшем падении температуры погрешность превышает 20%. Эти цифры могут смутить термохимиков, однако следует помнить, что речь идет об оценках свойств очень сложного класса веществ, поэтому их следует сравнивать не с погрешностью измерений экспериментальных данных (которая для цеолитов заметно выше, чем для обычных веществ из-за проблем воспроизводимости состава), а с точностью других методов оценки.
В литературе описаны несколько различных, так называемых, аддитивных моделей термодинамических свойств цеолитов, среди которых особо следует отметить модель Вилларда (Р. Vieillard) [41]. Она включает свойства 35-и цеолитов разного состава (если рассматривать цеолиты близкого состава и одной структуры как одно соединение, то 26-и цеолитов), применима для T = = 298.15–500 K, точность оценки свойств составляет около 3%, что превосходит предлагаемую нами для модели групповых функциональных вкладов. Однако, модель Вилларда предполагает наличие сведений о мольных объемах цеолитов (которые не всегда доступны), неприменима при температурах ниже комнатной и не позволяет получать набор самосогласованных термодинамических данных (энтальпийный и энтропийный член оцениваются независимо), что принципиально важно для последующих расчетов устойчивости фаз.
Если говорить о развитии в лаборатории методов априорной оценки термодинамических свойств веществ, то следует подчеркнуть, что интерес к ним не пропадал никогда, хотя “пики активности” приходятся на разное время. Одной из первых работ этого направления является публикация [42], в которой Г.Ф. Ворониным был предложен оригинальный способ оценки энтропий химических соединений по известным значениям молярной массы, кратчайшего межатомного расстояния в кристаллах и характеристической температуры Дебая. В связи с публикацией большого количества данных о термодинамических свойствах смешанных оксидных фаз, в 90-е годы появилась возможность разработать методы прогноза теплоемкости в системах с высокотемпературной сверхпроводимостью [43]. Особенностью предложенного метода является построение системы инкрементов теплоемкости для некоррелированных между собой параметров температурной зависимости Cp(T), что отличает его от большинства аналогичных методов оценки.
Одной из проблем, связанной с разработкой методов оценки термодинамических свойств веществ, является невозможность (в подавляющем большинстве методов) получения самосогласованных данных. Теплоемкость, энтропия, энтальпия и энергия Гиббса образования оцениваются отдельно, в то время как эти функции связаны между собой строгими термодинамическими соотношениями. Невозможность учета такой взаимосвязи можно считать одним из недостатков многих существующих методов оценки. При использовании инкрементных методов оценки энергии Гиббса образования (см., например, [44]) значения функций рассчитываются как линейная комбинация вкладов определенных структурных фрагментов, в результате чего соседние по составу фазы оказываются в состоянии безразличного равновесия. Оригинальный способ оценки термодинамических свойств фаз в гомологических рядах, позволяющий решить проблему “безразличных” равновесий, был предложен в работе [45] и применен для оценки термодинамических свойств интерметаллических соединений [46] и фаз с низкой устойчивостью в системе U-O [47]. На основе анализа представительного массива данных для систем металл – кислород было показано, что для изоструктурных соединений зависимость стандартных термодинамических функций от состава (который выражается через n) можно аппроксимировать зависимостью:
(12)
$Z(n){\text{/}}n = {{\beta }_{Z}} + {{\alpha }_{Z}}{\text{/}}n + {{\gamma }_{Z}}{\text{/}}{{n}^{{\text{2}}}} + \ldots ,$Согласно справочным данным, стандартная энтропия и энтальпия образования диоксида титана при 1200 К составляет ΔfS°(TiO2) = −21.5R, ΔH/R(TiO2) = −113367R.
СОЗДАНИЕ НОВЫХ И МОДЕРНИЗАЦИЯ СУЩЕСТВУЮЩИХ ТЕРМОДИНАМИЧЕСКИХ МОДЕЛЕЙ
Моделирование систем с высокотемпературными сверхпроводящими фазами (ВТСП). Одной из серьезных вех в развитии расчетных методов химической термодинамики в лаборатории было моделирование свойств высокотемпературных сверхпроводников. Сотрудники лаборатории активно вели и экспериментальные исследования этих систем, но вся расчетная часть была сосредоточена в группе Г.Ф. Воронина.
За десятилетний период интенсивных исследований были разработаны термодинамические модели упорядочения дефектов в структурах YBa2Cu3O6 + z (Y123) и Y2Ba4Cu7Oz (Y247), адекватно описывающие экспериментальные данные, в том числе и параметры фазовых переходов с образованием сверхпроводящих состояний, рассчитаны равновесия в системе YBa2Cu3O6–YBa2Cu3O7 при разных температурах и парциальных давлениях кислорода, включая области метастабильных состояний фаз Y124 и Y247. При анализе множества опубликованных данных о термодинамических свойствах фаз Y123 и Y124 (это более 3500 точек в интервале от 250 до 1300 К и давлений от 1 бар до 100 кбар, полученных разными методами и опубликованных в 110-и статьях) применялась оригинальная статистическая модель, предложенная Е.Б. Рудным, позволяющая оценивать как случайные, так и систематические погрешности измерений [48]. Итогам термодинамических исследований фазы Y123 в прошлом столетии посвящен один из проектов ИЮПАК (2000 г.), в котором наряду с российскими участниками были представители США, Канады и Швейцарии. Наиболее полно результаты проведенных исследований представлены в работах [49–52].
Обобщенная модель локального состава. В начале XXI века одним из объектов исследований лаборатории стали водно-органические солевые системы, что потребовало расширения спектра используемых термодинамических моделей. При моделировании свойств растворов в таких системах сотрудниками лаборатории была предложена обобщенная модель локального состава (Generalized Local Composition Model, GLCM), см. например, работы [53, 54]. Эта модель сочетает в себе преимущества моделей UNIQUAC и Цубоки–Катаямы (Tsuboka-Katayama), первая из которых подходит для моделирования равновесий в системах с различающимися размерами частиц, но не всегда позволяет адекватно воспроизводить термохимические данные растворов (в отличие от второй модели).
Более подробно эта модель описана и проанализирована в статье Н.А. Коваленко “Обобщенная модель локального состава, ее возможности и ограничения”, представленной в этом же номере “Журнала физической химии”.
Модель ионной жидкости с подрешетками. В работах В.А. Лысенко была предложена оригинальная модель жидкой фазы для описания равновесий в керамических системах, в первую очередь, сверхпроводниковых [52, 55, 56]. В рамках этой модели жидкость рассматривается как смесь нескольких жидкостей, состав которых отвечает химическому элементу (выбирается в качестве компонента) или бинарному соединению, имеющему ионный характер (автор называет их ионными жидкостями). Однокомпонентные жидкости состоят из нейтральных атомов и имеют одну подрешетку фиксированной емкости 1 моль. Ионные жидкости образованы катионами и анионами, расположенными в соответствующих подрешетках, при этом суммарная емкость обеих подрешеток также составляет 1 моль. При образовании расплава происходит смешение анионов и нейтральных атомов в анионной подрешетке, а катионов – в катионной. Как обычно, дополнительно на такие системы накладывается условие электронейтральности и материального баланса.
РАЗРАБОТКА ПРОГРАММНОГО ОБЕСПЕЧЕНИЯ ДЛЯ РАСЧЕТОВ ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ ФАЗ, ФАЗОВЫХ И ХИМИЧЕСКИХ РАВНОВЕСИЙ
Большинство разработок сотрудников лаборатории представлено в открытом доступе на сайте http://td.chem.msu.ru/. Программные комплексы PhDi, TernAPI и CpFit используются не только в практике научных исследований, но и в образовательном процессе.
Программный комплекс PhDi. В основе расчета лежит метод выпуклых оболочек. Из выпуклой оболочки множества выделяется выпуклая функция, построенная на множестве соответствующих термодинамических потенциалов фаз, которая является характеристической функцией всей равновесной системы. Выпуклость такой функции гарантирует выполнение условий стабильности для соединений и растворов, термодинамический потенциал которых имеет с ней общие точки. Для расчета фазовой диаграммы в рабочем окне программы необходимо записать в явном виде термодинамические модели всех фаз интересующей системы. Источником данных о свойствах фаз могут быть термодинамические справочники, компьютерные базы данных, статьи, результаты эксперимента, компиляции других данных и т.п. Программный комплекс укомплектован базой данных. Результаты расчета фазовых диаграмм с помощью PhDi представлены, например, в работах [57–60].
Программный комплекс TernAPI. Программа TernAPI предназначена для расчета фазовых диаграмм тройных систем методом выпуклых оболочек. К основным особенностям программы следует отнести:
• высокую надежность автоматического нахождения равновесного состояния системы, т.е. устойчивость к “сваливанию” в локальный минимум энергии Гиббса,
• быстрый расчет изобарно-изотермических сечений фазовых диаграмм тройных систем (1–30 с),
• возможность задания термодинамических моделей фаз в свободной аналитической форме с использованием специального языка, основанного на YAML и Ruby,
• наличие встроенного набора термодинамических моделей растворов (идеальный раствор смешения, полиномы Редлиха–Кистера, NRTL, UNIQUAC),
• наличие встроенной база данных SGTE Unary с термодинамическими свойствами простых веществ.
Более подробное описание программы и примеры получаемых результатов представлены в работах [21, 28, 61].
В программе CpFit реализована термодинамическая модель теплоемкости и других термодинамических функций, основанная на использовании суммы функций Планка–Эйнштейна с эмпирическими параметрами αi и θi (см. (8), (9)), предложенная в работе [33]. Эта модель позволяет аппроксимировать температурные зависимости теплоемкости, энтропии и энтальпии в широком температурном диапазоне, используя единый набор параметров. В отличие от полиномиальных моделей она может обеспечить физически корректную экстраполяцию на более широкий температурный интервал. В настоящее время в программе предусмотрена возможность аналитического описания аномалий на кривых Cp(T).
Комплекс вычислительных программ AUTOEQUIL [62] ориентирован на математическое моделирование сложных физико-химических равновесных систем как известного, так и неопределенного молекулярного состава при исследовании свойств новых комплексонов и экстрагентов. Расчет химических равновесий проводится по модели идеально-ассоциированных растворов с учетом материального баланса системы. Разработанные эффективные алгоритмы решения “обратной задачи” обеспечивают ее безаварийное решение, автоматизированный выбор начальных оценок констант равновесий и сходимость к истинным оценкам параметров [63]. Комплекс AUTOEQUIL работает в нескольких режимах программного меню, задаваемых пользователем в карте ввода, написан на языке программирования FORTRAN. Примеры получаемых результатов представлены в работах [64–66].
Программа расчета равновесий AUTOEQUIL использована в режиме решения прямой задачи в программном комплексе BPEQUIL (Blood Plasma Equil) [67], который позволяет рассчитывать эндогенные равновесия плазмы крови, имитирующий “естественную” плазму системой из 120-и равновесий со значимыми по концентрациям продуктами реакций в термодинамическом базисе из 46-и эндогенных лигандов и шести катионов металлов, присутствующих в биосистемах. В программу встроен блок анализа результатов с выделением значимых комплексных форм математическими методами последовательного анализа многомерных совокупностей равновесий. На основании проводимых расчетов можно определить равновесный состав плазмы крови при введении в нее терапевтического лиганда заданной концентрации. База данных программного комплекса содержит информацию о базисных компонентах плазмы крови с заданными концентрациями и их комплексах, катионах, лигандах и константах комплексообразования. Программа написана на языке программирования FORTRAN, интерфейс пользователя разработан с использованием языка программирования Object Pascal в среде Delphi.
ЗАКЛЮЧЕНИЕ
Формат краткого обзора не позволяет в полной мере отразить все работы, проводимые за последние 60 лет сотрудниками лаборатории по развитию расчетных методов химической термодинамики. Мы надеемся, что это направление будет активно развиваться и далее, так что к 100-летнему юбилею Химического факультета, кафедры физической химии и лаборатории химической термодинамики мы сможем поделиться новыми интересными разработками наших сотрудников.
Расчетно-теоретические работы последнего десятилетия, представленные в обзоре, проводятся в рамках программы “Химическая термодинамика” (АААА-А16-116061750195-2), при поддержке РФФИ и ряда коммерческих организаций.
Список литературы
Воронин Г.Ф. Расчеты фазовых равновесий при неполном наборе термодинамических данных. В сб. Неформальные математические модели в химической термодинамике / Под ред. В.А. Титова, И.И. Яковлева. Новосибирск: Наука, 1991. С. 116.
Воронин Г.Ф., Дегтярев С.А. // Журн. физ. химии. 1981. Т. 55. № 3. С. 607.
Воронин Г.Ф. В сб. Математические проблемы фазовых равновесий. Новосибирск: Наука, 1983. С. 3.
Voronin G.F., Degterov S.A. // Physica C. 1991. V. 176. P. 387.
Дегтярев С.А., Воронин Г.Ф. // Журн. физ. химии. 1987. Т. 61. № 3. С. 611.
Дегтярев С.А., Воронин Г.Ф. // Там же. 1987. Т. 61. № 3. С. 617.
Degtyarev S.A., Voronin G.F. // CALPHAD. 1988. V. 12. № 1. P. 73.
Decterov S.A., Pelton A.D., Seifert H.-J. et al. // Z. Metallkd. 2001. V. 92. № 6. P. 533.
Воронин Г.Ф. // Журн. физ. химии. 1988. Т. 62. № 4. С. 1084.
Воронин Г.Ф. // Там же. 1988. Т. 62. № 5. С. 1362.
Voronine G.F. // J. Therm. Analysis. 1989. V. 35. № 7. P. 2379.
Wightman A.S. Introduction to: Israel R.B. Convexity in the Theory of Lattice Gases. Princeton, New Jersey, 1979.
Lee D.D., Choy J.H., Lee J.K. // J. Phase Equilib. 1992. V. 13. № 4. P. 365.
Hildebrandt D., Glasser D. // Chem. Eng. J. 1994. V. 54. № 3. P. 187.
Chen S.-L., Zhang J.-Y., Lu X.-G., Chou K.-C., Chang Y.A. // J. Phase Equlib. Diffus. 2006. V. 27. № 2. P. 121.
Cool T., Batrol A., Kasenga M., Modi K., García R.A. // CALPHAD. 2010. V. 34. № 4. P. 393.
Perevoshchikova N., Appolaire B., Teixeira J., Aeby-Gautier E., Denis S. // Computational Materials Science. 2012. V. 61. P. 54.
Воронин Г.Ф. // Журн. физ. химии. 2003. V. 77. № 10. С. 1874.
Воронин Г.Ф. // Там же. 2005. V. 79. № 12. С. 2126.
Воронин Г.Ф., Восков А.Л. // Вест. Моск. ун-та. Сер. 2. Химия. 2013. Т. 54. № 1. С. 3.
Восков А.Л., Воронин Г.Ф. // Журн. физ. химии. 2010. V. 84. № 4. С. 605.
Kattner U.R. // Tecnol. Metal. Mater. Min. 2016. V. 13. № 1. P. 3.https://doi.org/10.4322/2176-1523.1059
Kroupa A. // Comp. Mater. Sci. 2013. V. 66. № 1. P. 3. https://doi.org/10.1016/j.commatsci.2012.02.003
Кауфман Л., Бернстейн Г. Расчет диаграмм состояния с помощью ЭВМ. Пер. с англ. Удовского А. Л. Под ред. Аптекаря И. Л. и Шиняева А. Я. Москва: Мир, 1972. 326 с.
Воронин Г.Ф., Генкин М.В., Куценок И.Б. // Журн. физ. химии. 2015. Т. 89. № 11. С. 1958.
Voskov A.L., Voronin G.F. // J. Chem. Eng. Data. 2016. V. 61. № 12. P. 4110.https://doi.org/10.1021/acs.jced.6b00557
Kosova D.A., Voskov A.L., Kovalenko N.A., Uspenskaya I.A. // Fluid Phase Equilibria. 2016. V. 425. P. 312.https://doi.org/10.1016/j.fluid.2016.06.021
Dzuban A.V., Voskov A.L., Uspenskaya I.A. // J. Chem. Eng. Data. 2013. V. 58. № 9. P. 2440.https://doi.org/10.1021/je400255c
Maksimov A.I., Kovalenko N.A. // J. Chem. Eng. Data. 2016. V. 61. № 12. P. 4222.https://doi.org/10.1021/acs.jced.6b00582
Курдакова С.В., Коваленко Н.А., Успенская И.А. // Вестн. Московского ун-та. Серия 2: Химия. 2016. Т. 57. № 3. С. 131.https://doi.org/10.3103/S0027131416030068
Lysenko V.A. // J. Alloys & Comp. 2019. V. 776. P. 850.https://doi.org/10.1016/j.jallcom.2018.10.223
Vassiliev V.P., Lysenko V.A. // Ibid. 2015. V. 629. P. 326.https://doi.org/10.1016/j.jallcom.2014.12.217
Voronin G.F., Kutsenok I.B. // J. Chem. Eng. Data. 2013. V. 58. № 7. P. 2083.https://doi.org/10.1021/je400316m
Khvan A.V., Dinsdale A.T., Uspenskaya I.A. et al. // CALPHAD. 2018. V. 60. P.144.https://doi.org/10.1016/j.calphad.2017.12.008
Kosova D.A., Druzhinina A.I., Tiflova L.A. et al // J. Chem. Thermodyn. 2018. V. 118. P. 206.https://doi.org/10.1016/j.jct.2017.11.016
Kosova D.A., Druzhinina A.I., Tiflova L.A. et al // Ibid. 2019. V. 132. P. 432.https://doi.org/10.1016/j.jct.2019.01.021
Voskov A.L., Kutsenok I.B., Voronin G.F. // CALPHAD. 2018. V. 61. P. 50.https://doi.org/10.1016/j.calphad.2018.02.001
Uspenskaya I.A., Kulikov L.A. // J. Chem. Eng. Data. 2015. V. 60. № 8. P. 2320.https://doi.org/10.1021/acs.jced.5b00217
Khvan A.V., Babkina T.S., Dinsdale A.T. et al. // CALPAHAD. 2019. V. 65. № 6. P. 50.https://doi.org/10.1016/j.calphad.2019.02.003
Voskov A.L., Kutsenok I.B., Voronin G.F., Kozin N.Yu. // Ibid. In press.
Vieillard P. // European J. of Mineralogy. 2010. V. 22. № 6. P. 823.
Воронин Г.Ф. // Журн. физ. хим. 1970. Т. 44. № 12. С. 3013.
Воронин Г.Ф., Успенская И.А. // Журн. физ. химии. 1997. Т. 71. № 11. С. 1927.
Mostafa A.G., Eakman J.M., Yarbro S.L. // Ind. Eng. Chem. Res. 1995. V. 34. P. 4577.
Воронин Г.Ф., Зайцева И.А. // Журн. физ. химии. 1996. Т. 70. № 7. С. 1201.
Uspenskaya I.A., Goryacheva V.I., Kutsenok I.B. // Russ. J. Phys. Chem. 2001. Suppl. P. 135.
Успенская И.А., Быков М.А. // Журн. неорган. химии. 2013. Т. 48. № 10. С. 1715.
Rudnyi E.B. // Chemometrics and Intelligent Laboratory Systems. 1996. V. 34. № 1. P. 41.
Rudnyi E.B., Kuzmenko V.V., Voronin G.F. // J. Phys. Chem. Ref. Data. 1998. V. 27. № 5. P. 855.https://doi.org/10.1063/1.556023
Voronin G.F., Chase M., Conder K. et al. // Pure&Appl. Chem. 2000. V. 72. № 3. P. 463.https://doi.org/10.1351/pac200072030463
Decterov S.A., Rudnyi E.B., Voronin G.F. // Physica C. 2007. V. 454. P. 70.https://doi.org/10.1016/j.physc.2007.01.014
Лысенко В.А., Кузьменко В.В., Успенская И.А., Воронин Г.Ф. // Журн. рос. хим. об-ва им. Д.И. Менделеева. 2001. Т. 45. № 3. С. 86.
Коваленко Н.А. Термодинамические свойства и фазовые равновесия в системах, образованных 18-краун-6, водой, пропанолами и бутанолами: Дисс. на соискание степени канд. хим. наук. М.: МГУ, 2013. 126 с.
Коваленко Н.А. Термодинамические свойства и фазовые равновесия в системах, образованных 18-краун-6, водой, пропанолами и бутанолами: Автореф. дис. … канд. хим. наук. М.: МГУ, 2013. 24 с.
Лысенко В.А. // Изв. РАН. Сер. неорган. материалы. 1998. Т. 34. № 9. С. 1108.
Лысенко В.А. // Журн. физ. химии. 2007. Т. 81. № 8. С. 1192.
Простакова В.А., Ломако М.О., Восков А.Л. и др. // Вестн. Московского ун-та. Сер. 2. Химия. 2010. Т. 51. № 2. С. 81.
Belov G.V., Emelina A.L., Goriacheva V.I. et al. // J. Alloys&Compounds. 2008. V. 452. № 1. P. 133.https://doi.org/10.1016/j.jallcom.2007.01.180
Белов Г.В., Воронин Г.Ф., Горячева В.И. и др. // Математическое моделирование. 2006. Т. 18. № 1. С. 67.
Kosova D.A., Voskov A.L., Kovalenko N.A., Uspenskaya I.A. // Fluid Phase Equilib. 2016. V. 425. P. 312.https://doi.org/10.1016/j.fluid.2016.06.021
Voskov A.L., Dzuban A.V., Maksimov A.V. // Fluid Phase Equilib. 2015. V. 388. P. 50.https://doi.org/10.1016/j.fluid.2014.12.028
Кирьянов Ю.А., Николаева Л.С. AUTOEQUIL // Свидетельство о государственной регистрации программы на ЭВМ, 2008 г. 200861226.
Евсеев А.М., Николаева Л.С., Кирьянов Ю.А. // Журн. физ. химии. 1988. Т. 62. № 5. С. 1153.
Николаева Л.С., Семенов А.Н. // Там же. 2018. Т. 92. № 2. С. 335.
Николаева Л.С., Белов Г.В., Рулев Ю.А., Семенов А.Н. // Журн. физ. химии. 2013. Т. 87. № 3. С. 457.
Николаева Л.С., Семенов А.Н., Бурова Л.И. // Там же. 2010. Т. 84. № 12. С. 2039.
Белов Г.В., Кирьянов Ю.А., Николаева Л.С. BPEQUIL, Свидетельство о государственной регистрации программы на ЭВМ. 2011. № 2011616552.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии