

Журнал физической химии, 2019, T. 93, № 11, стр. 1628-1637
Структура и физико-химические свойства растворов ацетонитрил–о-дихлорбензол
А. И. Абрамович a, *, Е. С. Алексеев a, Т. В. Богдан a
a Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
* E-mail: a-abramovich@yandex.ru
Поступила в редакцию 22.04.2019
После доработки 22.04.2019
Принята к публикации 14.05.2019
Аннотация
На основе анализа концентрационных зависимостей степени деполяризации и коэффициентов молекулярного рассеяния света, плотности, скорости ультразвука, адиабатической сжимаемости, избыточных молярного объема и адиабатической сжимаемости в растворах ацетонитрил–о-дихлорбензол выделены три различающиеся по структуре интервала 0–0.2, 0.2–0.9, 0.9–1.0. Для детального описания структуры растворов в каждом интервале проведено молекулярно-динамическое моделирование. Обнаружено, что в первом интервале сольватация молекул ацетонитрила сопровождается локальной переориентацией молекул о-дихлорбензола от преимущественно параллельных к перпендикулярным взаимным расположениям; молекулы ацетонитрила попадают в менее плотные области структуры о-дихлорбензола и встраиваются в систему его невалентных контактов. Второй интервал – область максимальной неоднородности растворов, в которой существенную роль играют как гомо-, так и гетеромолекулярные взаимодействия и присутствуют агломераты и конгломераты из молекул обоих компонентов. В третьем интервале происходит сольватация молекул о-дихлорбензола, механизм которой отличается от механизма сольватации ацетонитрила: о-дихлорбензол слабо влияет на структуру ацетонитрила и его структура практически не изменяется.
Информация о структуре и физико-химических свойствах индивидуальных жидкостей и их растворов важна не только для фундаментальной науки, но и для многих экологических и технологических процессов. Ацетонитрил (АЦН) и орто-дихлорбензол (о-ДХБ) являются широко используемыми полярными растворителями; дипольные моменты – 3.92 и 2.25 D соответственно. В некоторых реакциях, где АЦН используют как растворитель, он не остается нейтральным, а способен влиять на ход превращения и приводить к новым продуктам реакции [1]. о-ДХБ является растворителем для многих ароматических соединений, в частности для фуллеренов. Так, при переходе от монохлорбензола к о-ДХБ растворимость фуллерена С60 увеличивается в 4 раза [2]. Кроме того, о-ДХБ используют в качестве промежуточного продукта в производстве красителей и сельскохозяйственных химикатов. Чтобы правильно прогнозировать поведение растворителя в реакционной смеси, необходимо понимать взаимное влияние компонентов раствора друг на друга и их структуру.
Благодаря наличию электрофильных и нуклеофильных центров молекулы АЦН могут ассоциироваться за счет различных функциональных групп. Теоретически были изучены кластеры, включающие от 2 до 13 молекул АЦН [3–6]. Было показано, что с увеличением количества молекул в кластере, он становится более стабильным. В [7] обнаружено, что в жидком АЦН возможно образование циклических ассоциатов из шести молекул.
Изучение структуры кристаллического АЦН при низких температурах методами нейтронографии показывает, что в одной из кристаллических модификаций реализуется антипараллельное расположение молекул [8], в другой – параллельное [9]. Отметим, что и в одной и в другой модификации, благодаря водородным связям N···H(Me), молекулы располагаются почти перпендикулярно друг к другу.
Структура жидкого о-ДХБ в области температур 293–368 K изучалась в [10]. На основе классической теории молекулярного рассеяния света (МРС) были рассчитаны изотермическая сжимаемость, внутреннее давление, радиальная и ориентационная функции корреляций молекул в указанном температурном интервале и показано, что локальная структура о-ДХБ определяется наличием в нем ортогональных (Т-конфигураций) и стопочных (S-конфигураций) контактов бензольных колец, причем – последние являются преобладающими.
Моделирование структуры жидкого о-ДХБ методом классической молекулярной динамики [11] показало, что его структура определяется тенденцией к образованию динамического хлор-агрегата, включающего почти все молекулы модельной системы. Анализ взаимного расположения бензольных колец показал, что в ближайшем окружении молекул преобладают параллельные контакты.
Особенности строения индивидуальных жидкостей сохраняются и в растворах, но структура растворов усложняется. Это связано с тем, что, во-первых, флуктуации плотности и ориентации молекул в растворах имеют более сложный характер, чем в индивидуальных жидкостях, во-вторых – возникают флуктуации концентрации. Кроме того, с изменением концентрации компонентов структура раствора во многих случаях существенно изменяется.
Ранее нами был обнаружен ряд особенностей на концентрационных зависимостях физико-химических параметров в системах бинарных растворов хлорбензол–о-ДХБ и хлорбензол–о-хлортолуол, прослежена эволюция структуры этих растворов при изменении концентрации [12, 13]. Для корректной интерпретации экспериментальных данных было предпринято молекулярно-динамическое моделирование растворов и показано, что в структурировании растворов существенную роль играет хлор-агрегация [11, 13].
В данной работе в качестве объекта исследования выбрана система растворов (х)АЦН–(1 – х)о-ДХБ (х – мольная доля), в которой существенную роль могут играть донорно-акцепторные и диполь-дипольные взаимодействия. Изучены оптические, объемноупругие свойства и структура этих растворов. Проведен анализ упаковок АЦН и о-ДХБ в кристалле и выявлены структурообразующие контакты между молекулами, реализующиеся в конденсированном состоянии. Методом молекулярной динамики проведено моделирование структуры растворов и выявлена роль межмолекулярных взаимодействий разного типа при изменении концентрации растворов. Полученная информация важна для прогнозирования поведения изученных жидкостей в растворах.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Экспериментальные методы. Параметры МРС (общий коэффициент светорассеяния R0 и степень деполяризации ∆U) измерялась при 298 K на приборе ФПС-3М с усовершенствованной системой регистрации и контроля интенсивности падающего и рассеянного излучений. Источником падающего излучения служил гелий-неоновый лазер ЛГ-75 (λ = 632.8 нм). Измерения проводились относительно бензола, для которого абсолютное значение общего коэффициента МРС R0 = 11.84 × 10–6 см–1 на длине волны 632.8 нм. Подробно методика измерений описана в [14]. Погрешность измерения R0 и степени деполяризации ∆U была 2.5 и 3% соответственно. Зная R0 и ∆U, были рассчитаны анизотропный Rα (рассеяние света на флуктуациях анизотропии поляризуемости молекул) и изотропной Ris (рассеяние света на флуктуациях плотности и концентрации) вклады в рассеяние. Для расчетов использовались формулы термодинамической теории светорассеяния:
Плотность ρ измерялась пикнометрическим методом с точностью ±4 × 10–4 г/см3. Молярный объем VM и его избыточная часть $V_{{\text{M}}}^{{\text{E}}}$ рассчитывались по формулам:Скорость ультразвука измерялась импульсным методом на частоте 5.5 МГц с точностью 0.1 м/с. Адиабатическая сжимаемость рассчитывалась по формуле Лапласа
а избыточная адиабатическая сжимаемость – по формулеСоздание модельных систем для расчетов. Были выполнены расчеты для чистых компонентов и бинарных систем тех же концентраций, что и в эксперименте. Для создания исходной модельной ячейки для растворов сначала была создана ячейка из 100 молекул: для этого помещали в ячейку необходимые количество молекул растворенного компонента и далее заполняли свободное пространство молекулами второго компонента до общего количества 100 молекул. После оптимизации геометрии малой ячейки, ее размножали до размеров 3 × 3 × 3 (2700 молекул) и оптимизировали размещение молекул при заданной экспериментальной плотности. Минимизацию энергии системы проводили с помощью программного пакета GROMACS методом скорейшего спуска до значений 10 кДж/(моль нм) с начальным шагом 0.01 нм. Ячейка для чистых компонентов состояла из 1000 молекул.
Получение молекулярно-динамических траекторий. Молекулярно-динамическое моделирование созданных модельных систем проводилось с использованием программного пакета GROMACS [15], параметры внутримолекулярных и ван-дер-ваальсовых взаимодействий задавались потенциалом OPLS-AA [16]. Для учета кулоновского взаимодействия использовались значения зарядов на атомах из базы данных NIST http://cccbdb.nist.gov/ [17] для АЦН – рассчитанные неэмпирически методом CCSD(T)/cc-pV(T + d)Z, для о-ДХБ – методом MP2/6-31G*. Моделирование проводили при наложении периодических граничных условий в NPТ-ансамбле при температуре 298.0 K и давлении 1 атм. Для интегрирования уравнений движения использовали алгоритм leap-frog с уточнением значений после применения ограничений (sd) с шагом интегрирования 1 фс. Для поддержания постоянных температуры и давления использовали термостат Нозе–Гувера и баростат Парринелло–Рамана. Для вычисления кулоновских взаимодействий использовался алгоритм PME (Particle-Mesh Ewald) с радиусом отсечения 10 Å. Радиус отсечения ван-дер-ваальсовых взаимодействий был 10 Å.
Для анализа были записаны молекулярно-динамические траектории длиной 500 пс с записью точек траектории каждые 0.5 пс.
Обработка траекторий. Для характеристики структуры о-ДХБ в чистом виде и в растворах были рассчитаны функции радиально-углового распределения G(r, θ) (ФРУР) расстояний между центрами масс бензольных колец молекул и углов между ними для каждой концентрации:
ФРУР дает сведения о радиальных и угловых корреляциях в окружении молекул. Максимумы ФРУР отвечают наиболее вероятному взаимному расположению молекул. Анализ положения максимумов позволяет сделать выводы о локальной структуре жидкости (о ближайшем окружении молекул) и об агломерации молекул посредством специфических взаимодействий бензольных колец. ФРУР обычно используется для характеристики взаимного расположения анизотропных молекул: дискообразных или стержнеобразных.
Для характеристики гомо- и гетеромолекулярных взаимодействий в растворах были получены функции радиального распределения расстояний (ФРР) между отдельными атомами, принадлежащими разным молекулам:
Анализ кристаллических упаковок. Из Кембриджского банка данных были выбраны данные по структуре кристаллов, в которых в качестве сокристаллизованного растворителя содержались молекулы о-ДХБ, или молекулы АЦН и соединения с атомами хлора в орто-положении в ароматическом кольце. Последний класс соединений особенно важен, поскольку дает прямую информацию о взаимном расположении молекул о-ДХБ и АЦН и реализованных контактах между ними; позволяет выявить характерные мотивы взаимного расположения молекул и виды межмолекулярных контактов между молекулами АЦН и о-ДХБ. Поскольку реализуемые в кристалле взаимные расположения молекул всегда соответствуют одному из минимумов потенциальной энергии системы, то полученные данные о кристаллических упаковках и межмолекулярных контактах использованы нами для верификации результатов моделирования и создания модели структурной организации растворов.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Концентрационные зависимости плотности ρ, скорости звука v и адиабатической сжимаемости βs представлены на рис. 1а, 1б и в соответственно, а концентрационные зависимости избыточных молярного объема $V_{{\text{m}}}^{{\text{E}}}$ и адиабатической сжимаемости $\beta _{{\text{s}}}^{{\text{E}}}$ – на рис. 2а и 2б. Видно, что плотность плавно уменьшается с возрастанием мольной доли АЦН; скорость звука проходит через минимум при х = 0.7, а зависимость βs(х) меняет наклон при этой же концентрации. Во всей области концентраций $V_{{\text{m}}}^{{\text{E}}}$ и $\beta _{{\text{s}}}^{{\text{E}}}$ имеют большие отрицательные значения и проходят через минимум: $V_{{\text{m}}}^{{\text{E}}}$ – в области концентраций 0.4–0.7, а $\beta _{{\text{s}}}^{{\text{E}}}$ – при х = 0.7. Это указывает на наличие сильных гетеромолекулярных взаимодействий в этой системе растворов.
Рис. 1.
Концентрационные зависимости плотности (а), скорости звука (б), адиабатической сжимаемости (в) для системы растворов ацетонитрил–о-дихлорбензол (х – мольная доля ацетонитрила).

Рис. 2.
Концентрационные зависимости избыточного молярного объема (а), избыточной адиабатической сжимаемости (б).

На рис. 3а представлена концентрационная зависимость степени деполяризации (∆U) изученных растворов. Наблюдается сложная немонотонная зависимость ∆U(х): в области малых концентраций 0 < х < 0.05 происходит резкое уменьшение ∆U, перегиб на зависимости при х = = 0.05, плавное уменьшение ∆U в области концентраций 0.1 < х < 0.9 и снова резкое уменьшение при х > 0.9. То есть, видны аномалии в области малых концентраций со стороны одного и другого компонентов. На рис. 3б представлены концентрационные зависимости общего коэффициента МРС (R0), коэффициентов изотропной (Ris) и анизотропной (Rα) части рассеяния. Из этих графиков видно, что МРС в области концентраций х = 0–0.55 определяется анизотропным рассеянием, т.е. рассеянием на флуктуациях анизотропии поляризуемости молекул, которое обусловлено взаимной ориентацией молекул. Это короткоживущие неоднородности (пикосекундный диапазон), включающие две–три молекулы. При х > 0.55 МРС определяется изотропным рассеянием – рассеянием на флуктуациях плотности и концентрации (на агломератах и конгломератах). В области средних концентраций наблюдается максимум на анизотропном рассеянии при х = 0.45, а на изотропном – при х = 0.7.
Рис. 3.
Концентрационные зависимости степени деполяризации (а) и коэффициентов молекулярного рассеяния света (б).
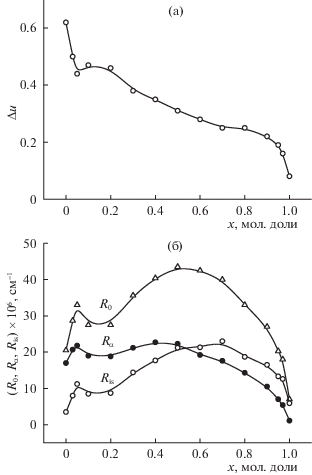
Обнаруженное не монотонное поведение концентрационных зависимостей изученных физико-химических параметров указывает на существенную перестройку структуры растворов при изменении концентрации. Можно выделить три различающиеся по структуре интервала: 0–0.2, 0.2–0.9, 0.9–1.0. Для корректного описания структуры растворов в каждом интервале было выполнено молекулярно-динамическое моделирование растворов.
Ранее в [11] была исследована система контактов между атомами хлора в жидком о-ДХБ (галоген-агрегация). Используя геометрический критерий 4.35 Å для расстояний между атомами хлора было получено, что при данном критерии 99% молекул в о-ДХБ объединены в единый хлор-агрегат. Ниже, при анализе строения о-ДХБ в кристаллической фазе, будет показано, что контакты между атомами хлора дополнительно стабилизируются взаимодействиями между атомами хлора и π-системой бензольного кольца.
На рис. 4а приведена ФРУР для жидкого о‑ДХБ. В области углов, близких к 0°, наблюдаются максимумы при 4–5, 7–8 и 10–12 Å. В области углов, близких к 90°, наблюдаются максимумы при 5–7 и 9–11 Å. Таким образом, в первой координационной сфере (4–7 Å) реализуются как параллельные, так и ортогональные контакты между молекулами, причем параллельные ориентации являются преобладающими. К такому же выводу пришли авторы [10], анализируя температурные зависимости ряда физико-химических параметров. Во второй координационной сфере и следующих – все конфигурации молекул равновероятны.
Рис. 4.
Функция радиально-углового распределения G(r, θ) молекул о-дихлорбензола: в индивидуальном о-дихлорбензоле (а), в растворе при х = 0.5 (б).

При образовании растворов наблюдается существенное изменение ориентации молекул о‑ДХБ. Например, при х = 0.5 во всей области близких расстояний наблюдаются преимущественно перпендикулярные ориентации во взаимном расположении молекул о-ДХБ (рис. 4б). То есть, в растворах при добавлении АЦН в о-ДХБ происходит переориентация молекул о-ДХБ от преимущественно параллельного расположения к перпендикулярному, что и является основной причиной преобладания анизотропного рассеяния и увеличения коэффициентов МРС в области концентраций 0 < х < 0.5 (см. рис. 3б).
На рис. 5а представлены ФРР для расстояний между атомами хлора в растворах. В области концентраций 0 < х < 0.5 вид ФРР меняется незначительно: при 4–5 Å функция имеет первый максимум, отражающий первую координационную сферу, и менее выраженный широкий максимум в области 7.5 Å. Однако при х = 0.5 можно отметить уменьшение высоты первого максимума, который с дальнейшим увеличением доли АЦН в системе уменьшается с одновременным увеличением высоты второго максимума, что соответствует агрегации сольватированных молекул о‑ДХБ в разбавленных растворах.
Рис. 5.
Функция радиального распределения расстояний: между атомами хлора в о-дихлорбензоле (а), между атомами азота в ацетонитриле (б) (кривые для разных концентраций сдвинуты относительно друг друга по оси ординат на 1 Å). Концентрационная зависимость гомо- и гетеромолекулярных взаимодействий (в).

На рис. 5б представлены ФРР для расстояний между атомами азота цианогруппы АЦН. Видно, что для индивидуального АЦН функция имеет “плечо” в области 4–5 Å и максимум при 6 Å. Существование на ФРР различных областей расстояний N···N в АЦН отражает разные варианты расположения молекул при разных межмолекулярных взаимодействиях: 1) линейное положение молекул (типа “голова-хвост”) с реализацией водородной связи Ме–С≡N···Me–С≡N, где расстояние N···N порядка 6 Å; 2) расположение молекул при образовании водородной связи под углом друг к другу с расстоянием N···N порядка 5 Å, 3) антипараллельное расположение молекул при донорно-акцепторном взаимодействии цианогрупп с расстоянием между атомами азота порядка 4 Å.
В растворах вид ФРР изменяется и при х = 0.5 вместо “плеча” при 4–5 Å появляется максимум – такой же высоты, что и максимум при 6 Å. При дальнейшем уменьшении концентрации АЦН высота первого максимума (при 4–5 Å) увеличивается, а второго (при 6–7 Å) уменьшается. При х = 0.03 вместо второго максимума появляется “плечо”, которое связано как с наличием линейных водородных связей, так и с существованием пространственного разделения молекул АЦН. Поскольку расстояние между атомами азота отражает разную ориентацию молекул, то можно сделать вывод об изменении характера взаимного расположения молекул АЦН в индивидуальной жидкости и в растворах. В интервале 0.03 < x < 0.5 на ФРР N···N проявляется область корреляции при 10 Å, которая наиболее заметна при х = 0.03. Наличие таких корреляций может быть связано с образованием кластеров из сольватированных молекул АЦН.
При х = 0.5 происходит существенное изменение локальной структуры о-ДХБ по сравнению с индивидуальной жидкостью: на ФРУР наблюдается преобладание перпендикулярных ориентаций над параллельными (см. рис. 4б и 4а). Таким образом, добавление АЦН в о-ДХБ приводит к переориентации молекул о-ДХБ. Мы отмечали появление на ФРУР корреляций в области 10 Å. Сопоставляя с ФРР (см. рис. 5б), мы можем предположить, что корреляции в области 10 Å при х = 0.5 обусловлены взаимным расположением молекул о‑ДХБ и АЦН.
Следует отметить, что в ранее исследованных нами бинарных системах, в которых оба компонента были ароматическими жидкостями (бензол–толуол, бензол–хлорбензол, хлорбензол–о-хлортолуол), при х = 0.5 каждый из компонентов сохранял свои индивидуальные особенности в растворе. ФРУР и ФРР для отдельного компонента в растворе выглядела как ФРУР для индивидуальной жидкости. Однако в исследуемой нами системе АЦН–о-ДХБ компоненты отличаются по своим молекулярным свойствам, и при х = = 0.5 корреляционные функции компонентов раствора отличаются от их вида для индивидуальных жидкостей. Такие отличия свидетельствуют о сильном влиянии компонентов друг на друга, что может происходить только вследствие гетеромолекулярных взаимодействий, наиболее сильно проявляющихся в области эквимолярных концентраций. На наличие сильных гетеромолекулярных взаимодействий указывает отрицательное отклонение от идеальности изученных растворов и большие величины избыточных молярного объема и адиабатической сжимаемости (см. рис. 2а и 2б).
Был проведен анализ гомо- и гетеромолекулярных взаимодействий в растворе (рис. 5в). Гетеромолекулярные взаимодействия Cl···Me между молекулами о-ДХБ и АЦН проявляются в области концентраций 0.2 < x < 0.9. При 0.2 < x < < 0.45 гетеромолекулярные контакты Cl···Me превалируют над контактами N···Me в АЦН. При 0.55 < x < 0.9 количество гетеромолекулярных контактов Cl..Me превышает количество контактов Cl…Cl в о-ДХБ. В области концентраций 0.45 < x < 0.55 в растворах присутствуют гомо- и гетеромолекулярные агломераты.
Результаты моделирования растворов мы дополнили анализом кристаллических упаковок соединений из Кембриджского банка данных https://www.ccdc.cam.ac.uk/] [18], которые: 1) содержали молекулы о-ДХБ в качестве сокристаллизованного растворителя, 2) содержали сокристаллизованные: молекулы АЦН и о-ДХБ, или АЦН и соединения с атомами хлора в орто-положении в ароматическом кольце. Последний класс соединений был особенно важен, поскольку давал прямую информацию о наиболее вероятном положении молекул о-ДХБ и АЦН, так как реализуемые в кристалле взаимные расположения молекул всегда соответствуют одному из минимумов потенциальной энергии системы.
Анализ упаковок кристаллов, содержащих молекулы о-ДХБ без примесей других веществ или в качестве сокристаллизованного растворителя, показывает, что, как правило, молекулы о-ДХБ в кристалле объединены с образованием бесконечных хлор-агрегатов разного строения (рис. 6а, 6б). В кристаллах можно выделить два основных вида контактов между атомами хлора при хлор-агрегации: 1) атомы хлора направлены “навстречу” друг другу, что приводит к образованию центросимметричного димера (рис. 6в); 2) короткие контакты между атомами хлора стабилизированы взаимодействиями между атомами хлора и атомами углерода π-системы бензольного кольца (рис. 6а, 6б, 6г). При этом атом хлора может выступать как в качестве нуклеофильного агента, координируя атом водорода связи С–Н, так и проявлять электрофильные свойства, взаимодействуя с π‑системой бензольного кольца. Контакты первого вида приводят к образованию “рыхлых” (неплотных) областей в кристалле, а контакты второго вида – к образованию плотных областей в кристалле, стабилизированных большим количеством невалентных контактов.
Рис. 6.
Расположение молекул о-дихлорбензола в кристаллических структурах: бесконечный хлор-агрегат в кристаллах о-дихлорбензола (а) (фрагмент кристаллической упаковки ABUMIT); цепочечный хлор-агрегат из молекул о-дихлорбензола в кристаллической структуре VIZPOK (б); образование центросимметричного димера из молекул о-дихлорбензола, приводящего к образованию менее плотных (рыхлых) областей в структуре (в); взаимодействия хлор···π‑система бензольного кольца, приводящие к образованию более плотных областей в кристалле (г); фрагмент упаковки кристаллической структуры UHUVEZ (д), где реализуются два вида расположения молекул – более плотное (г) и менее плотное (д).

Анализ кристаллических упаковок сокристаллизованных молекул АЦН и о-ДХБ, или молекул, содержащих о-дихлорбензольный фрагмент, показывает, что в кристалле молекулы АЦН и о-дихлорбензольные фрагменты других молекул находятся рядом, при этом атомы хлора координируют метильную группу АЦН и/или атом углерода цианогруппы (рис. 7). Анализ также показал, что при возникновении невалентных контактов между АЦН и орто-дихлорбензольными группами последние в кристалле взаимно расположены менее плотно (см. рис. 6в). В кристаллах гомомолекулярная ассоциация молекул АЦН, как правило, отсутствует. Как исключение можно привести пример кристаллической структуры RIZWIH (см. рис. 7в), в которой молекулы АЦН и о-ДХБ присутствуют в качестве сокристаллизованных растворителей, где обнаружена водородная связь N···Me между двумя молекулами АЦН (c расстоянием N···N 4.5 Å). Однако и в этом случае протяженные агломераты из молекул АЦН не образуются.
Рис. 7.
Взаимное расположение в кристалле молекул ацетонитрила и молекул, имеющих атомы хлора в орто-положении в бензольном кольце. Кристаллические структуры MAGLOW (а), KUNBIF (б), RIZWIH (в) [18].

Согласно проведенному нами анализу кристаллических структур в структуре о-ДХБ можно выделить области меньшей плотности, образованные направленными контактами Cl···Cl, и области большей плотности, в которых контакты Cl…Cl стабилизированы взаимодействием Cl···π-система бензольного кольца. Сокристаллизованные молекулы АЦН попадают в области меньшей плотности и встраиваются в систему невалентных контактов посредством взаимодействий Cl···Me, N···H(Cаром). Можно предположить, что характерные мотивы взаимного расположения молекул о‑ДХБ и АЦН сохраняются и в жидкой фазе. При образовании растворов молекулы АЦН также могут попадать в области меньшей плотности и встраиваться в систему невалентных контактов. Это приводит к переориентации молекул о-ДХБ, что мы и наблюдаем по данным моделирования и МРС.
Малый размер, малая поляризуемость и большой дипольный момент молекулы АЦН способствует тому, что в конденсированном состоянии при сближении молекул сближаются и одноименные заряды, поэтому силы притяжения между молекулами слабые, о чем свидетельствуют низкие температуры плавления и кипения АЦН (–44 и 80°С, соответственно). С другой стороны, наличие слабых водородных связей N···H(Cаром) (согласно данным ИК-спектроскопии) обеспечивает достаточно большой интервал существования жидкой фазы (120°, что сравнимо с жидкой водой). Существование таких направленных взаимодействий приводит к тому, что структура жидкого АЦН достаточно рыхлая и в ней нет плотных областей. Попадание о-ДХБ в АЦН приводит к уменьшению взаимного отталкивания молекул АЦН и образованию более стабильных плотных областей в растворах.
Таким образом, полученные экспериментальные данные и результаты моделирования позволили в системе растворов АН–о-ДХБ выделить три различающиеся по структуре интервала 0–0.2, 0.2–0.9, 0.9–1.0 и описать особенности структуры в каждом интервале.
В интервале 0 < x < 0.2 происходит сольватация молекул АЦН, которая сопровождается локальной переориентацией молекул о-ДХБ от преимущественно параллельных к перпендикулярным ориентациям. Сольватация молекул АЦН сопровождается процессами попадания молекул АЦН в менее плотные области структуры о-ДХБ и встраивания молекул АЦН с систему невалентных контактов в о-ДХБ. При 0 ≤ х ≤ 0.05 существует протяженный агломерат из молекул о-ДХБ с встроенными в него молекулами АЦН. При х = 0.05, соответствующей максимуму коэффициентов МРС, происходит распад этого протяженного агломерата на более мелкие, что в области 0.05 < x < 0.2 приводит к уменьшению коэффициентов МРС.
В интервале 0.2 < x < 0.9 проявляется максимальная неоднородность растворов. В этом интервале существенную роль играют как взаимодействия между молекулами одного сорта, так и гетеромолекулярные взаимодействия. При x < 0.5 происходит переориентация молекул о-ДХБ, при 0.25 < x < 0.45 гетеромолекулярные взаимодействия преобладают над взаимодействиями N···Ме в АЦН, а при 0.55 < x < 0.9 доля гетеромолекулярных взаимодействий выше, чем контактов Cl···Cl в о-ДХБ. Таким образом, в концентрационном интервале 0.2 < x < 0.9 в растворах присутствуют гомомолекулярные и гетеромолекулярные агломераты из молекул о-ДХБ и АЦН.
В интервале 0.9 < x < 1 происходит сольватация молекул о-ДХБ. Структура АЦН сохраняется (см. рис. 5б, ФРР для расстояний азот–азот). Механизм сольватации другой, о-ДХБ слабо влияет на структуру АЦН.
Авторы выражают благодарность Л.В. Ланшиной за критическое обсуждение результатов работы.
Список литературы
Suh S.-E., Chenoweth D.M. // Org. Lett. 2016. V. 18. № 16. P. 4080.
Безмельницын В.Н., Елецкий А.В., Окунь М.В. // Успехи физ. наук. 1998. Т. 168. № 11. С. 1197.
Defranceschi M., Peetersb D., Mathieua D. et al. // J. Mol. Struct. (Theochem). 1993. V. 287. P. 153.
Siebers J.G., Buck U., Beu T.A. // Chem. Phys. 1998. V. 239. P. 549.
Alberti M., Amat A., De Angelis F., Pirani F. // J. Phys. Chem. B. 2013. V. 117. № 23. P. 7065.
Nigam S., Majumder C. // J. Chem. Phys. 2008. V. 128. P. 214307.
Грибов Л.А., Новаков И.Л., Павлюченко А.И. // Журн. структур. химии. 2004. Т. 45. № 5. С. 816.
Barrow M.J. //Acta Crystallogr. 1981. V. B37. P. 2239.
Torrie B.H., Powell B.M. // Mol. Phys. 1992. V. 75. № 3. P. 613.
Каргин И.Д., Ланшина Л.В., Абрамович А.И. // Журн. физ. химии. 2017. Т. 91. С. 1737.
Алексеев Е.С., Богдан Т.В. // Журн. структур. химии. 2016. Т. 57. № 8. С. 1664.
Абрамович А.И., Ланшина Л.В., Каргин И.Д. // Russ. Chem. Bull. V. 66. № 5. P. 828.
Абрамович А.И., Алексеев Е.С., Богдан Т.В., Ланшина Л.В. // Журн. структур. химии. 2014. Т. 55. № 4. С. 686.
Ланшина Л.В., Абрамович А.И. // Журн. физ. химии. 2007. Т. 81. № 2. С. 187.
Berendsen H.J.C., van der Spoel D., van Drunen R. // Comp. Phys. Comm. 1995. V. 91. P. 43.
Jorgensen W.L., Maxwell D.S., Tirado-Rives J. // J. Am. Chem. Soc. 1996. V. 118. P. 11225.
Computational Chemistry Comparison and Benchmark DataBase National Institute of Standards and Technology. http://cccbdb.nist.gov/
The Cambridge Crystallographic Data Centre (CCDC). https://www.ccdc.cam.ac.uk/
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии