

Журнал физической химии, 2019, T. 93, № 12, стр. 1790-1797
Каталитический метатезис N-метилформамида с диметилкарбонатом ассоциатами спиртов
А. Я. Самуилов a, Д. Р. Алекбаев a, Я. Д. Самуилов a, *
a Казанский национальный исследовательский технологический университет
Казань, Россия
* E-mail: ysamuilov@yandex.ru
Поступила в редакцию 08.02.2019
После доработки 08.02.2019
Принята к публикации 12.03.2019
Аннотация
Квантово-химическим методом B3LYP/6-311++G (df, p) изучен механизм метатезиса N-метилформамида с диметилкарбонатом, приводящий к образованию N,O-диметилкарбамата. Реакция включает три стадии: превращение формамида в иминол; присоединение диметилкарбоната по азометиновой связи иминола; распада образующегося продукта на карбамат и метилформиат. Вторая стадия лимитирует скорость взаимодействия. Все стадии протекают через согласованные циклические переходные состояния, и все они эффективно катализируются мономером и димером метанола.
Органические карбонаты вступают с карбонильными соединениями различного строения в реакции метатезиса, которые приводят к новым функционализированным соединениям:

Катализаторами этих процессов выступают карбонат калия [1, 8, 9], силикагель [2], фенол [6], окислы лантана [4, 10], кальция, магния, алюминия, циркония [10], метилат натрия [11] и др. Эти взаимодействия могут протекать и в отсутствии каких-либо катализаторов [12–15].
Особое внимание привлекает взаимодействие формамидов с органическими карбонатами, приводящими к образованию карбаматов. Эта реакция позволяет легко получать из первичных аминов и органических карбонатов в присутствии эфиров муравьиной кислоты карбаматы бесфосгенным способом. В этом случае превращения приобретают автокаталитический характер, например:
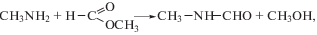
Доступность регентов делает этот способ привлекательным для получения карбаматов. Карбаматы являются мономерами для получения бесфосгенных полиуретанов [16, 17]. Ряд карбаматов проявляет высокую биологическую активность [18–20]. На их термическом разложении основан бесфосгенный метод получения изоцианатов [21, 22].

Формамиды являются простейшей моделью пептидов. Поэтому понимание механизмов протекания реакций с их участием представляет интерес для биохимии, химии окружающей среды.
Рассматриваемая реакция является частным случаем взаимообмена между сложными эфирами и амидами карбоновых кислот:
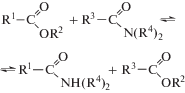
Эти превращения широко используются в химии полимеров [23–27].
До настоящего времени остаются неизвестными механизм и термодинамические параметры реакций органических карбонатов с амидами карбоновых кислот. С целью выяснения механизма этих реакций, нами проведено квантово-химическое изучение взаимодействия N-метилформамида с диметилкарбонатом.
Формамид, N-монозамещенные амиды карбоновых кислот существует в виде двух таутомеров [28–32]:

Принципиально возможно рассматривать реакции диметилкарбоната с N-метилформамидом как с участием его амидной формы, так и иминольной. Однако, как свидетельствуют экспериментальные данные, диметилкарбонат не взаимодействует с N,N-дизамещенными амидами карбоновых кислот, в которых невозможна амид-иминольная таутомерия. Они могут быть использованы как растворители при проведении реакций с участием органических карбонатов [33–37]. Поэтому наиболее вероятным является путь превращения с участием иминольной формы N-метилформамида. Экспериментальные данные указывают на то, что по азометиновой связи действительно легко присоединяются соединения со связями углерод – гетероатом с неподеленной парой электронов [38–43].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Квантово-химические расчеты проводились методом функционала плотности B3LYP/6-311++G (df, p) [44–48]. Был использован пакет прикладных программ Gaussian 09 [49]. В вычислениях проводилась полная оптимизация геометрии реагентов, промежуточных продуктов, переходных состояний и конечных соединений.
Поиск переходных состояний производили по первой отрицательной частоте колебания в матрице Гесса. Истинность переходных состояний определяли спуском от точек переходных состояний в обе стороны с помощью процедуры IRC c последующей оптимизацией геометрии полученных пред - и послереакционных комплексов.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
1. Стадии взаимодействия N-метилформамида с диметилкарбонатом и их термодинамические параметры
Проведенные вычисления показали, что взаимодействие N-метилформамида с диметилкарбонатом протекает в три стадии. Первая стадия реакции заключается в превращении цис-формы N-метилформамида в иминол:

На второй стадии происходит присоединение цис,цис-диметилкарбоната (наиболее устойчивого конформера [50, 51]) по π-связи иминола с образованием N,O-диметил-N-[(гидрокси)(метокси)метил]карбамата:
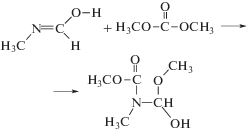
На третьей стадии происходит отщепление молекулы метилформиата от карбамата:
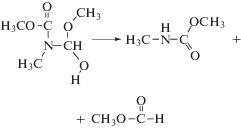
В табл. 1 приведены термодинамические параметры рассматриваемых превращений.
Таблица 1.
Изменения свободной энергии (∆G), энтальпии (∆Н) и энтропии (∆S) в реакциях (1)–(3); газовая фаза; 298 К
| Реакция | ∆G, кДж/моль | ∆Н, кДж/моль | ∆S, Дж/(моль K) |
|---|---|---|---|
| (1) | 53.0 | 51.0 | –6.7 |
| (2) | 64.9 | 13.7 | –171.4 |
| (3) | –128.2 | –71.2 | 191.4 |
Первые две стадии протекают эндотермично. Они характеризуются отрицательными величинами энтропий. Третья стадия протекает экзотермично. Она имеет большую положительную величину энтропии превращения. Суммарно весь цикл превращений имеет отрицательную энтальпию (–6.5 кДж/моль) и положительную энтропию (13.3 Дж/(К моль)).
2. Механизм превращения N-метилформамида в иминол
Некаталитическое мономолекулярное превращение N-метилформамида в иминол протекает через четырехчленное переходное состояние (ПС1):
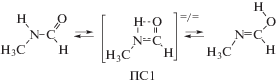
Образование иминола из формамида ускоряется водой [52], муравьиной кислотой [53]. Ассоциаты спиртов являются эффективными катализаторами таутомерных превращений [54]. В этих реакциях они выступают как бифункциональные кислотно-основные катализаторы. Поэтому мы рассмотрели возможность катализа мономером и димером метанола амид-иминольного превращения N-метилформамида. Каталитические превращения сопровождаются образованием пред- и послереакционных интермедиатов (ИМ), которые представляют собой комплексы с водородными связями. Взаимодействия протекают в соответствии со схемами:

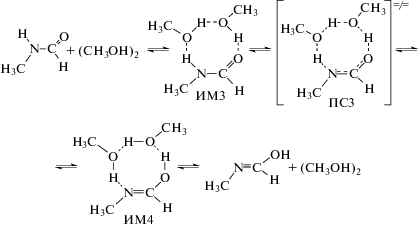
В табл. 2 приведены термодинамические параметры активации реакций (4)–(6).
Таблица 2.
Термодинамические параметры активации реакций (4)–(6) в газовой фазе при 298 К
| Реакция | ∆G≠, кДж/моль | ∆Н≠, кДж/моль | –∆S≠, Дж/(моль K) |
|---|---|---|---|
| 4 | 183.8 | 182.8 | 3.5 |
| 5 | 88.3 | 37.9 | 169.1 |
| 6 | 68.0 | 4.5 | 212.8 |
Из данных табл. 2 следует, что некаталитическая реакция (4) имеет большой энтальпийный барьер на пути превращения. В катализируемых мономером, димером метанола этот барьер резко снижается. Каталитические реакции (5), (6) имеют малые величины энтропий активации. Тем не менее, барьер свободной энергии в каталитических превращениях значительно меньший, чем в некаталитической реакции (4). Эти данные указывают на то, что спирты являются эффективными катализаторами амид-иминольной таутомерии.
3. Механизм взаимодействия диметилкарбоната с иминолом с образованием N,O-диметил-N-[(гидрокси)(метокси)метил]карбамата
Некаталитичекое взаимодействие диметилкарбоната с иминолом протекает через согласованное четырехчленное переходное состояние:
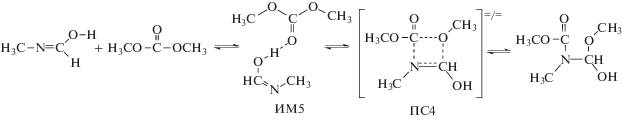
Как оказалось, эта стадия реакции также может катализироваться ассоциатами спиртов:
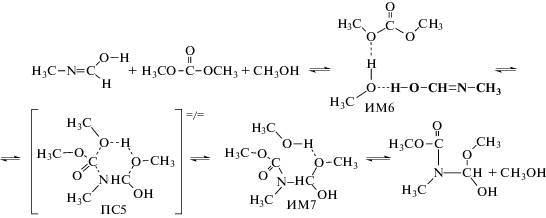
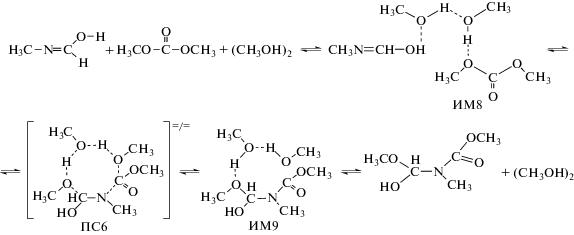
На рис. 1 приведены шаростержневые модели переходных состояний ПС4–ПС6.
Рис. 1.
Шаростержневые модели переходных состояний ПС4 – ПС6. Данные расчета методом B3LYP/6-311++(df, p). Указаны длины связей в Å.
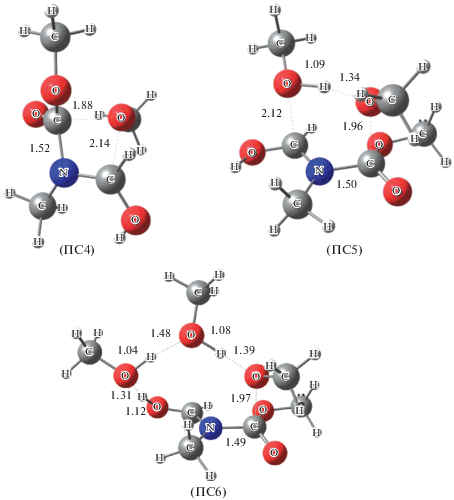
Превращения (7)–(9) протекают через согласованные циклические переходные состояния. В табл. 3 приведены термодинамические параметры активации реакций (7)–(9).
Таблица 3.
Свободные энергии, энтальпии и энтропии активации реакций (7)–(9) в газовой фазе при 298 К
| Реакция | ∆G≠, кДж/моль | ∆Н≠, кДж/моль | –∆S≠, Дж/(моль K) |
|---|---|---|---|
| (7) | 264.6 | 209.3 | 185.8 |
| (8) | 248.3 | 145.9 | 343.5 |
| (9) | 179.8 | 66.3 | 380.9 |
Катализ мономером, димером метанола и в этом случае приводит к резкому снижению энтальпийного барьера на пути реакций. Изменения свободных энергий активации контролируются энтальпийным членом.
Рассматриваемая реакция протекает как реакция метатезиса с участием σ-связи диметилкарбоната и π-связи иминола. Известно большое количество реакций метатезиса с участием σ-связей [55–57]. Однако описанные превращения, как правило, протекают при каталитическом содействии металлов. Уникальность рассматриваемой реакции метатезиса заключается в том, что она не требует участия соединений металлов.
4. Механизм распада N,O-диметил-N-[(гидрокси)(метокси)метил]карбамата на N,O-диметилкарбамат и метилформиат
Некатализируемый распад N,O-диметил-N-[(гидрокси)(метокси)метил]карбамата с образованием N,O-диметилкарбамата и метилформиата протекает через четырехчленное согласованное переходное состояние:
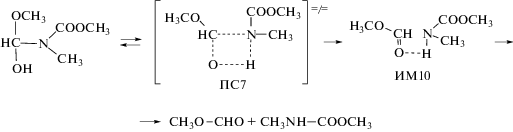
Эта стадия реакции также катализируется мономером и димером метанолом:
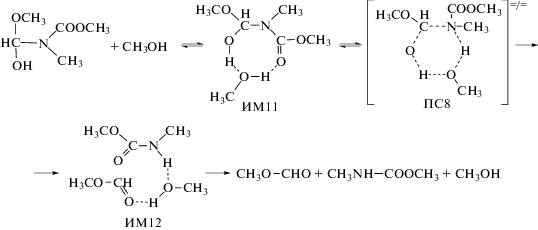
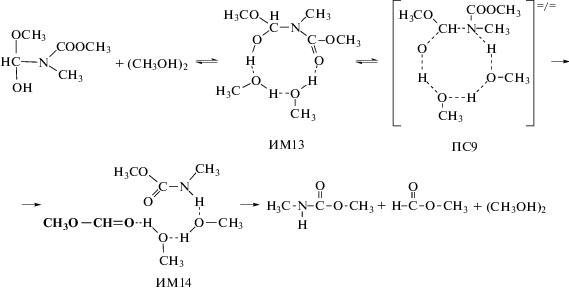
Реакции (10)–(12) протекают через циклические согласованные переходные состояния. Они сопровождаются образованием пред- и послереакционных комплексов (ИМ11-ИМ14), которые представляют собой комплексы с водородными связями.
В табл. 4 приведены термодинамические параметры активации реакций (10)–(12).
Таблица 4.
Свободные энергии, энтальпии, энтропии активации распада N,O-диметил-N-[(гидрокси)(метокси)метил]карбамата с образованием N,O-диметилкарбамата и метилформиата
| Реакция | ∆G≠, кДж/моль | ∆Н≠, кДж/моль | ∆S≠, Дж/(мольK) |
|---|---|---|---|
| (10) | 131.9 | 132.9 | 3.2 |
| (11) | 92.3 | 44.0 | –161.9 |
| (12) | 84.0 | 20.9 | –211.5 |
Как следует из данных табл. 4, катализируемые метанолом превращения имеют существенно более низкий энтальпийный и свободноэнергетический барьер на пути взаимодействий. Как и в других случаях, каталитический эффект димера метанола более значительный, чем мономера.
Таким образом, реакция метатезиса N-метилформамида с диметилкарбонатом протекает в три стадии: таутомеризации N-метилформамида; присоединения диметилкарбоната по азометиновой связи иминола с образованием N,O-диметил-N-[(гидрокси)(метокси)метил]карбамата; распада карбамата с образованием N,O-диметилкарбамата и метилформиата. Вторая стадия является лимитирующей. Все стадии катализируются мономерами и ассоциатами спиртов, и они протекают через согласованные циклические переходные состояния.
Список литературы
Selva M., Marque C.A., P. Tundo P. // Cazz. Chim. Ital. 1993. V. 123. P. 515.
Gupte S.P., Shivarkar A.B., Chaudhari R.V. // Chem. Commun. 2001. Iss. 24. P. 2620.
Shivarkar A.B., Gupte S.P., Chaudhari R.V. // J. Mol. Catal. A. 2004. V. 223. P. 85.
Guo X., Shang J., Li J. et al. // Synthet. Commun. 2011. V. 41. P. 1102.
Duong T., Prager R.H., Ward A.D., Kerr D.I.B. // Aust. J. Chem. 1976. V. 29. P. 2651.
Mason R.W. Non-phosgene route to the manufacture of organic isocyanates. US Pat. N 6781010.
Romano U., Tesei R. Method for preparing aromatic urethans. US Pat. N 4100351.
Kébir N., Benoit M., Legrand C., Burel F. // Europ. Polym. J. 2017. V. 96. P. 87.
Kébir N., Benoit M., Burel F. // Ibid. 2018. V. 107. P. 155.
Wu D., Fu X., Xiao F., Li J., Zhao N., Wei W. Sun Y. // Catal. Commun. 2008. V. 9. P. 680.
Gao J.J., Li H.Q., Zhang Y. // Chin. Chem. Lett. 2007. V. 18. P. 149.
Gattiglia E., Turturro F., Lamantia F.P., Valenza A. // J. Appl. Polym. Sci. 1992. V. 46. P. 1887.
Transreactionsin Condensation Polymers // Ed. S. Fakirov. Weimheim: Wiley-VCH. 1999. 485 p.
Costa D. A., Marize C., Oliveira F. // Int. J. Polym. Mater. 2002. V. 51. P. 393.
Reactive Extrusion. Principles and Applications // Eds. G. Beyer, Ch. Hopmann. Weimheim: Wiley-VCH. 2018. 414 p.
Sardon H., Pascual A., Mecerreyes D. et al. // Macromolecules. 2015. V. 48. P. 3153.
Maisonneuve L., Lamarzelle O., Rix E. et al. // Chem. Rev. 2015. V. 115. P. 12407.
Simon J.Y. The Toxicology and Biochemistry of Insecticides. Boca Raton: CRC Press. 2015. 380 p.
Ghosh A.K., Brindisi M. // J. Med. Chem. 2015. V. 58. P. 2895.
Cheong J.E., Zaffagni M., Chung I. et al. // Europ. J. Med. Chem. 2018. V. 144. P. 372.
Арико Ф., Тундо П. // Успехи. химии. 2010. Т. 79. С. 532
Wang P., Liu Sh., Deng Y. // Chin. J. Chem. 2017. V. 35. P. 821.
Pillon L.Z., Utracki L.A. // Polym. Eng. Sci. 1984. V. 24. P. 1300.
Ho J.-Ch., Wei K.-H. // J. Polym. Sci., Part B. 2000. V. 38. P. 2124.
Pai F.-Ch., Lai S.-M., Chu H.H. // J. Appl. Polym. Sci. 2013. V. 130. P. 2563.
Gug J., Sobkowicz M.J. // Ibid. 2016. V. 133. P. 43350.
Fakirov S. // Prog. Polym. Sci. 2018. https://doi.org/10.1016/j.progpolymsci.2018.09.003
Guo J.-X., Ho J.-J. // J. Phys. Chem. A. 1999. V. 103. P. 6433.
Allegrettia P.E., Milazzoa C.B., Castrob E.A. et al. // J. Mol. Struct. (Theochem) 2002. V. 589–590. P. 161.
Hazra M.K., Chakraborty T. // J. Phys. Chem. A. 2005. V. 109. P. 7621.
Guzmán-Angel D., Inostroza‑Rivera R., Gutiérrez‑Oliva S. et al. // Theor. Chem. Acc. 2016. V. 135. P. 37.
Vichietti R.M., da Silva A.B.F., Haiduke R.L.A. // Molec. Astrophys. 2018. V. 10. P. 1.
Gadge S.T., Mishra A., Gajengi A.L. et al. // RSC Adv. 2014. V. 4. P. 50271.
Bouhadir K.H., Abramian L., Ezzeddine A. et al. // Molecules. 2012. V. 17. P. 13290.
Marsault E., Hoveyda H.R., Peterson M.L. et al // J. Med. Chem. 2006. V. 49. P. 7190.
Wen-Chung Shieh W.-Ch., Dell S., Bach A. et al. // J. Org. Chem. 2003. V. 68. P. 1954.
James A. Cella J.A., Bacon S.W. // J. Org. Chem. 1984. V. 49. P. 1122.
Burgstahler A.W. // J. Am. Chem. Soc. 1951. V. 73. P. 3021.
Bredereck H., Simchen G., Goknel E. // Angew. Chem. Int. Ed. 1964. V. 3. P. 704.
Ulrich H., Sayigh A.A.R. // Angew. Chem. Int. Ed. 1966. V. 6. P. 844.
Yeap G.-Y., Mohammad A.-T., Osman H. // Mol. Cryst. Lig. Cryst. 2012. V. 552. P. 177.
Mohammad A.-T., Yeap G.-Y., Osman H. // J. Mol. Struct. 2015. V. 1087. P. 88.
Mohammed I.A., Ahmed M., Ikram R. et al. // Lat. Am. J. Pharm. 2018. V. 37. P. 540.
Tsuneda T. Density Functional Theory in Quantum Chemistry. Tokyo, Heidelberg, New York, Dordrecht, London: Springer, 2014. 200 p.
Sahni V. Quantal Density Functional Theory. Berlin, Heidelberg: Springer-Verlag, 2016. 413 p.
Becke A.D. // J. Chem. Phys. 1992. V. 96. P. 2155.
Becke A.D. // Ibid. 1992. V. 97. P. 9173.
Becke A.D. // Ibid. 1993. V. 98. P. 5648.
Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., Nakatsuji H., Caricato M., Li X., Hratchian H.P., Izmaylov A.F., Bloino J., Zheng G., Sonnenberg J.L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J.A., Peralta J.E., Ogliaro F., Bearpark M., Heyd J.J., Brothers E., Kudin K.N., Staroverov V.N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Rega N., Millam J.M., Klene M., Knox J.E., Cross J.B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R.E., Yazyev O., Austin A.J., Cammi R., Pomelli C., Ochterski J.W., Martin R.L., Morokuma K., Zakrzewski V.G., Voth G.A., Salvador P., Dannenberg J.J., Dapprich S., Daniels A.D., Farkas O., Foresman J.B., Ortiz J.V., Cioslowski J., Fox D.J. Gaussian 09, Revision A.1, Gaussian Inc., Wallingford CT, 2009.
Reddy S.K., Balasubramanian S. // J. Phys. Chem., B. 2012. V. 116. P. 14892.
Gontrani L., Russina O., Marincola F.C. // J. Chem. Phys. 2009. V. 131. P. 244503.
Guzmán-Angel D., Inostroza‑Rivera R., Gutiérrez-Oliva S. et al. // Theor. Chem. Acc. 2006. V. 135. P. 37.
Hazra M.K., Chakraborty T. // J. Phys. Chem. A. 2005. V. 109. P. 7621.
Samuilov A.Ya., Balabanova F.B., Samuilov Ya.D. et al. // Russ. J. Gen. Chem. 2015. V. 85. P. 1806.
Rory Waterman R. Organometallics. 2013. V. 32. P. 7249.
Bauer H., Alonso M., Färber Ch. et al. // Nature Catal. 2018. V. 1. P. 40.
Yadav S., Dixit R., Vanka K. et al. // Chem. Europ. J. 2018. V. 26. P. 1269.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии