

Журнал физической химии, 2019, T. 93, № 7, стр. 1060-1070
Наноразмерные частицы Fe и Ni в процессе углекислотного риформинга лигнина: исследование методами ИК-спектроскопии
О. В. Арапова a, *, Г. Н. Бондаренко a, М. В. Цодиков a
a Российская академия наук, Институт нефтехимического синтеза им. А.В. Топчиева
Москва, Россия
* E-mail: arapova@ips.ac.ru
Поступила в редакцию 25.10.2018
После доработки 25.10.2018
Принята к публикации 20.11.2018
Аннотация
Методом колебательной спектроскопии изучены структурные изменения лигнина, поверхность которого была модифицирована наноразмерными никель- и железосодержащими частицами, в процессе углекислотного риформинга (УР), стимулированного микроволновым излучением. Показано, что в условиях конвективного нагрева, ванилиновая кислота превращается в синтез-газ, если созданы условия ее существования в мономерной форме. Нанесенные металлсодержащие компоненты оказывают существенные изменения на структурные характеристики лигнина. С использованием внутреннего стандарта найдено, что никель, нанесенный на поверхность лигнина из водного раствора ацетатной соли, взаимодействует с поверхностными альдегидными группами, окисляя их в карбоксилатные. In situ в каталитической ячейке ИК-спектрометра, показано, что в присутствии наноразмерных никельсодержащих частиц в среде СО2 при конвективном нагреве разложение лигнина приводит к образованию ванилиновых производных (кислота и спирт), которые прочно удерживаются на поверхности за счет водородного связывания. Наноразмерные частицы железа, нанесенные на поверхность из ацетилацетонатного комплекса, координируются прочно на алкокси-группах лигнина. При конвективном нагреве в среде СО2 органическая масса лигнина подвергается разложению с образованием легких продуктов (углеводородов и оксигенатов).
Эффективные методы переработки лигнина являются в настоящее время актуальной задачей, поскольку лигнин остается основным и пока мало перерабатываемым отходом целлюлозно-бумажной промышленности [1, 2]. Трудность промышленной переработки лигнина обусловлена сложностью его природы, многовариантностью структурных звеньев и связей между ними, а также нестойкостью этого природного полимера, необратимо меняющего свойства в результате химического или термического воздействия [3–6].
С другой стороны, лигнин является возобновляемым природным источником ароматических углеводородов [7], которые могут быть использованы в качестве компонентов моторных топлив или продуктов нефтехимии. Интерес к получению ценных продуктов из лигнина не ослабевает, вследствие чего появляется множество работ по использованию для разложения лигнина реакций каталитического гидролиза и крекинга, восстановления и окисления [3–6, 8]. В работе [9], посвященной изучению реакций окисления лигнина с использованием модельных соединений было показано, что полиоксометаллаты обладают высокой избирательностью по отношению к лигнину. В работе установлено, что степень превращения фенольных соединений в 10 раз выше, чем нефенольных, независимо от типа применяемой окислительной системы. Это дает основание полагать, что основную роль в реакциях окисления играют соединения со свободным фенольным гидроксилом.
Настоящая работа является логическим продолжением наших работ по переработке лигнина, содержащего высокодисперсные частицы никеля, обладающие способностью к поглощению микроволнового излучения (МВИ) и, как следствие, генерированию плазмы непосредственно в порах субстрата, что приводит к существенной интенсификации процесса углекислотного риформинга (УР) лигнина с образованием синтез-газа [10–13]. Методом высокотемпературной ИК-спектроскопии in situ было показано, что при нагревании лигнина в высокотемпературной ячейке ИК-фурье спектрометра на его поверхности образуется смесь ванилиновой кислоты и ванилинового спирта, которые прочно удерживаются на поверхности лигнина вплоть до 450°C. Было высказано предположение о том, что образующиеся ванилиновые производные являются продуктами первой стадии риформинга лигнина с последующим образованием синтез-газа [14]. Кроме того, представляло интерес исследовать методами ИК-спектроскопии влияние природы металла, введенного в лигнин, на эффективность и селективность превращения лигнина в условиях микроволнового излучения и конвективного нагрева. Поэтому в отличие от работы [14], в которой исследование ИК-спектров проводили для лигнина с введенными в него высокодисперсными частицами никеля, в настоящей работе представлены результаты по сравнительному изучению взаимодействия нанесенных наноразмерных частиц железа и никеля с функциональными группами лигнина и формирования промежуточных соединений при термическом воздействии in situ в ячейке ИК-Фурье спектрометра.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Использованные реагенты. Лигнин (смешанный древесного происхождения Кировской области). Состав, мас. %: С – 58.1, H –5.4, Al – 1.2, Si – 3.1, Ca – 0.6, Fe – 0.8, Mg – 0.04, S – 1.2, N – 0.2, O – 28.9, остальные – 0.5 (содержание каждого компонента менее 0.05%). Реагенты, растворители и газы: толуол (Sigma-Aldrich, 99%); Ni(OAc)2 · · 4H2O (Aldrich, 98%); фольга Ni (Sigma-Aldrich, 99.99%); Fe(acac)3 (Aldrich); ванилиновая кислота (Sigma-Aldrich, 97%); Ar и He (ОСЧ, 99.99%).
Железосодержащие компоненты на поверхности лигнина формировали путем нанесения солей металлов из растворов и дисперсных частиц никеля в растворе толуола, полученных методом металлопарового синтеза (МПС), описанного в [15].
При нанесении железосодержащих частиц использовали раствор ацетилацетоната железа Fe(acac)3 в этиловом спирте. Методика нанесения подробно описана в [11, 12]. Таким образом, для исследования были взяты: исходный лигнин (образец 1), железосодержащие образцы, полученные при нанесении Fe(acac)3 и содержащие 0.1; 0.5; 1.5; и 2.0% Fe (образцы 2, 3, 4 и 5 соответственно). Образец 6, содержал 2.78% Fe, нанесенного из коллоидного раствора металлических частиц в толуоле, полученных методом МПС.
Методика проведения углекислотного риформинга лигнина, стимулированного МВИ. Эксперименты по углекислотному риформингу образцов железосодержащего лигнина проводили на лабораторной установке, состоящей из магнетрона, волновода, кварцевого реактора и камеры поглощения остаточного излучения [11, 12]. В ходе проведения экспериментов через реактор пропускали углекислый газ или аргон со скоростью 60 мл/мин, время облучения составляло 10 мин при инициированной микроволновым излучением средней температуре реактора 700–800°С. Газообразные продукты реакции анализировали on-line методом газовой хроматографии на хроматографе Кристаллюкс-4000М. Анализ углеводородной части проводили с использованием насадочной колонки 1.5 м, заполненной гранулами (0.5 мм) α-Al2O3 с 15% нанесенной фазы сквалан с использованием пламенно-ионизационного детектора (ПИД) и He в качестве элюента. Содержание Н2, СН4, СО и СО2 определяли на насадочной колонке, заполненной углеродной фазой марки СКТ, в качестве элюента использовали Ar и детектор по теплопроводности.
ИК-спектральные исследования. ИК-спектры образцов лигнина получали в режиме пропускания для образцов в виде таблеток, прессованных с KBr (IFS-66v/sBruker, разрешение 2 см–1, скан. 30, диапазон 400–4000 см–1), в режиме отражения с поверхности (ATR) (кристалл Ge, скан. 100 разрешение 2 см–1, диапазон 600–4000 см–1) на ИК-микроскопе HYPERION 2000. Высокотемпературные ИК-спектры диффузного отражения in situ получали с использованием высокотемпературной ячейки PIKE Diffus IR, сопряженной с ИК-фурье-спектрометром VERTEX-70. Спектры регистрировали в непрерывном режиме в температурном интервале 20–450°C, (194 сканирования/спектр) с разрешением 2 см–1 в диапазоне 600–4000 см–1. Регистрацию спектров проводили для образца порошка ванилиновой кислоты, перетертого с бромистым калием, порошка лигнина в токе аргона или СО2, т.е. в условиях приближенных к эксперименту риформинга.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Исследование термического превращения ванилиновой кислоты методом высокотемпературной ИК-спектроскопии in situ
Ванилиновые производные (кислота и спирт) были идентифицированы методом высокотемпературной ИК-спектроскопии диффузного отражения in situ (ИКДО) на поверхности лигнина при нагревании его в высокотемпературной ячейке спектрометра [14]. Количество образующихся на поверхности лигнина ванилиновых производных и их состав зависели от условий проведения эксперимента (температура, присутствие в составе лигнина частиц никеля и способ его нанесения, атмосфера при нагреве: аргон или диоксид углерода). Было показано, что ванилиновые производные, образующиеся из лигнина, легко сорбируются на стенках ячейки, на специально внесенный в кюветное отделение спектрометра угольный сорбент, а на поверхности термически обработанного лигнина остаются даже при температуре 450°С. При этом никаких признаков присутствия синтез-газа в ИКДО спектрах поверхности лигнина не наблюдалось, хотя основным продуктом превращения лигнина под влиянием МВИ является именно синтез-газ [11–13]. Представляло интерес исследовать термическое разложение ванилиновой кислоты методом ИКДО.
На рис. 1 представлены ИКДО спектры ванилиновой кислоты (ВК), зарегистрированные при различной температуре в токе аргона. Спектр при комнатной температуре отражает все функциональные группы ВК: интенсивная полоса 3474 см–1 относится к валентным колебаниям фенольной ОН-группы. Широкая расщепленная полоса в области 3350–2750 см–1 указывает на наложение широкой полосы от ассоциированных карбоксилатной и фенольной –ОН-групп и полос валентных колебаний связей С–Н в группах ‒СН2–, –ОСН3 и СН в ароматическом кольце. Широкие полосы в области 2525–2644 см–1 вместе с длинноволновым сдвигом полосы от валентных колебаний связи C=O до 1690 см–1 подчеркивают существование ВК в виде димера через карбоксильную группу [16]. Малая интенсивность полос от связей С–О в области 1300–1100 см–1, сильное расщепление этих полос и полос валентных колебаний связи С–С в ароматическом кольце (1500–1600 см–1) также являются следствием проявления достаточно сильной межмолекулярной ассоциации между заместителями в ароматическом кольце ВК.
Рис. 1.
ИКДО-спектры ванилиновой кислоты в аргоне: 1 – 20°C, 2 – 200°C, 3 – 340°C, 4 – 25°C после остывания.
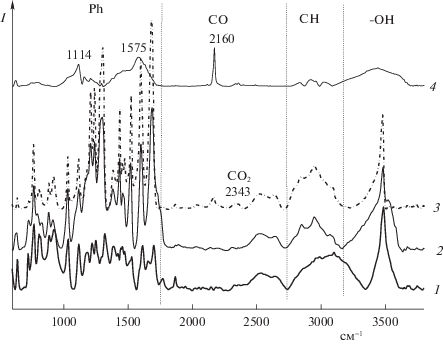
При нагревании ВК (рис. 1, спектры 2, 3) происходит существенное увеличение интенсивности и снятие расщепления полос в области 1100–1600 см–1, что вызвано разрушением значительного количества наиболее слабых межмолекулярных водородных связей при нагревании. В частности, ассоциация между ОН-фенольными и карбоксилатными группами снимается и в области валентных колебаний связей СН появляются три четкие полосы 2843, 2946 см–1 от метильных и метиленовых групп и 3070 см–1 от связей СН в три-замещенном ароматическом кольце. Димерная структура ВК через карбоксилатную группу сохраняется , поскольку в спектре хорошо проявляются две четкие полосы 2525 и 2644 см–1, при этом структура димера меняется, т.к. полоса от связи C=O смещается до 1683 см–1. При 340°C в спектре ВК появляются две новые слабые полосы 2160 и 2343 см–1, которые можно отнести к валентным колебаниям связей C=O, соответственно, в оксиде и диоксиде углерода. Дальнейшее повышение температуры приводит к росту интенсивности полосы 2160 см–1, все другие полосы от ВК и СО2 постепенно падают по интенсивности. В спектре образца, прокаленного при 450°C в течение 10 мин и затем охлажденного до комнатной температуры в токе аргона (спектр 4, рис. 1) отсутствуют все полосы, характеризующие ВК, наиболее интенсивно проявляется полоса 2160 см–1 от СО, адсорбированного на поверхности бромистого калия с твердыми остатками продукта термического разложения ВК. Можно полагать, что в составе продукта термического разложения ВК присутствуют в очень небольшом количестве ароматические кольца (полосы 1575 и 3031 см–1), метокси-группы (полосы 1114, 2843, 2946 см–1), широкая полоса с максимумом при 3435 см–1 скорее всего связана с адсорбированной на поверхности водой. Данный эксперимент однозначно показывает, что ВК в условиях термического нагрева может разлагаться с образованием синтез-газа. Следует отметить, что выделившийся при этом водород нельзя зафиксировать методом ИКС. Разложение ВК до синтез-газа происходит только в мономерных молекулах и затруднено высокой склонностью к межмолекулярной ассоциации. Поэтому в спектрах ИКДО лигнина фиксировались интенсивные полосы ванилиновых производных, прочно адсорбированных на поверхности лигнина даже при температуре 450°C, а полосы монооксида углерода не проявлялись.
Аналогично выглядят ИКДО спектры ВК, зарегистрированные при различных температурах в токе СО2 (рис. 2). Также при температуре выше 350°C начинают появляться полосы от монооксида углерода (2160 см–1). Полосы от димера ВК через карбоксилатную группу (2525 и 2644 см–1) сохраняются в спектре до 420°C, а затем при более высоких температурах интенсивность этих полос падает вместе с падением интенсивностей всех остальных полос, характеризующих ИК-спектр ВК.
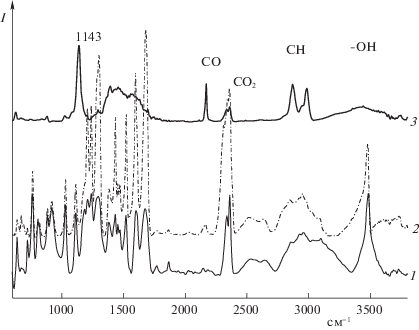
На рис. 3 представлено сравнение спектров продуктов термического разложения ВК в токе аргона и диоксида углерода. Видно, что полосы, характеризующие ВК в спектрах, отсутствуют, а полоса от монооксида углерода 2160 см–1, адсорбированного на поверхности остывших до комнатной температуры образцов имеет примерно равную интенсивность. В задачу данного исследования не входила идентификация твердых продуктов термического разложения ВК, однако можно констатировать, что кроме оксидов углерода и, по всей видимости, водорода в твердых продуктах термического разложения ВК присутствуют ароматические скорее всего конденсированные кольца (полосы 1575 см–1 от сопряженных связей С=С, полосы в области 720–850 см–1 от деформационных и в области 3000–3040 см–1 от валентных колебаний связей С–Н в ароматических кольцах) и алифатические группы –СН2–, –СН3 (полосы в области 2970–2890 и 1360–1470 см–1), связанные между собой простыми эфирными связями (интенсивные полосы в области 1110–1150 см–1). Можно отметить, что относительное содержание ароматики и алифатики в этих образцах существенно зависит от условий термической обработки: в токе аргона продукт термообработки содержит больше ароматики, а в продукте, полученном при нагревании в токе СО2 преобладают алифатические простые эфиры.
Углекислотный риформинг лигнина, содержащего высокодисперсные частицы железа
Сводные данные по составу и выходу газообразных продуктов, конверсии лигнина, потере его массы и селективности в образовании синтез-газа при МВИ и конвективном нагреве для различных лигнин содержащих материалов приведены в табл. 1.
Таблица 1.
Конверсия и удельный выход продуктов УР лигнина (№ 1), Fe-содержащих образцов лигнина (№ 2–5), полученных нанесением из ацетилацетоната железа, и Fe-содержащего образца № 6, полученного нанесением из коллоидного раствора железа
| № | Fe, мас. % | w(Н2), см3/(г мин) | w(СО), см3/(г мин) | w(СН4), см3/(г мин) | w(С2–С4), см3/(г мин) | α(Н2), % | α(RН), % | ${{S}_{{{{{\text{Н }}}_{2}} + {\text{С О }}}}}$, % |
|---|---|---|---|---|---|---|---|---|
| Микроволновое излучение | ||||||||
| 1 | – | 9.2 | 16.2 | 8.4 | 1.2 | 75.5 | 48.6 | 72.5 |
| 2 | 0.1 | 31.3 | 40.3 | 13.5 | 3.3 | 87.7 | 61.6 | 80.9 |
| 3 | 0.5 | 36.4 | 35.0 | 11.4 | 2.4 | 78.5 | 60.5 | 83.9 |
| 4 | 1.5 | 40.5 | 52.0 | 9.8 | 1.6 | 78.2 | 60.0 | 89.0 |
| 5 | 2.0 | 55.6 | 63.0 | 7.3 | 1.0 | 88.7 | 63.8 | 93.4 |
| 6 | 2.78 (МПС) | 47.2 | 53.7 | 8.2 | 1.2 | 83.2 | 62.6 | 91.4 |
| Конвективный нагрев | ||||||||
| 1 | – | 1.6 | 7.5 | 3.8 | 0.4 | 83.5 | 62.1 | 38.0 |
| 4 | 1.5 | 2.6 | 4.0 | 3.2 | 0.2 | 84.3 | 51.7 | 35.0 |
Сравнение данных табл. 1 с данными по УР в условиях МВИ для лигнина, содержащего Ni [13], показывают очень близкие результаты (конверсия органической массы лигнина α(RН), селективность по синтез-газу ${{S}_{{{{{\text{Н }}}_{2}} + {\text{С О }}}}}$) для экспериментов с 1.5 мас. % металла в составе лигнина. С другой стороны содержание Ni менее 1% в составе лигнина, практически не повышала конверсию лигнина по сравнению с конверсией лигнина, не содержащего металл. Содержание Fe 0.1 и 0.5% (эксперименты 2 и 3 в табл. 1) показывают достаточно эффективное образование продуктов в ходе УР лигнина при МВИ. Исследование ИК-спектров образцов лигнина после внесения в него ацетилацетоната железа и после УР позволяет выяснить влияние природы металла и предыстории образца на структурные изменения в ходе превращения лигнина в синтез-газ при его риформинге в ходе МВИ [10–12].
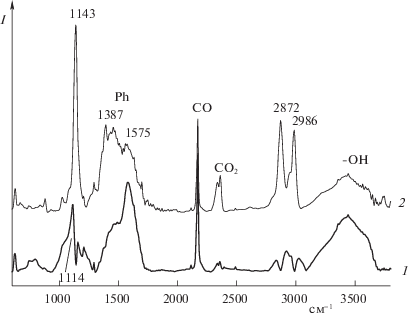
На рис. 4 представлены ИК-спектры поглощения образцов лигнина с содержанием (1.5% и 2.0 мас. %) металла солей никеля (ацетат) и железа (ацетилацетонат) в сравнении со спектром чистого лигнина. Все спектры приведены к одинаковым базовым линиям и отнормированы по полосе 2120 см–1 от нитрильных групп внутреннего стандарта – феррицианида калия K3[Fe(CN)6], аналогично тому, как это было выполнено в работе [14]. При такой обработке спектров интенсивность отдельных полос ИК-спектра пропорциональна содержанию функциональных групп в составе лигнина, к которым относится данная полоса.

Качественно все спектры рис. 1 идентичны и хорошо описывают все функциональные группы в составе лигнина, спектральные области поглощения которых отмечены на рис. 4. Но относительные интенсивности отдельных полос сильно меняются в зависимости от природы металла и его содержания в образце. Рост интенсивности полос валентных колебаний связей обычно происходит при увеличении полярности этих связей [17]. Сравнивая спектры лигнина с 1.5% содержанием соли металлов (рис. 4), можно видеть, что введение в состав лигнина никелевых и железных солей приводит к поляризации связей в разных функциональных группах лигнина: соль никеля приводит к повышению интенсивности полос от связей О–Ph–OH (полосы в области 1230–1400, 1604 и 3500 см–1), в то время как соль железа в большей мере поляризует связи О–Alk–OH (полосы в области 1030–1100, 2840–2970 и 3500 см–1). Причем под влиянием никелевых солей происходит значительно больший рост интенсивностей полос особенно от связей –ОН. Кроме того, как обсуждалось в [14], введение никеля в лигнин приводит к окислению альдегидных групп в составе лигнина и превращению их в кислотные группы, что сопровождается смещением и падением интенсивности полосы 1713 см–1. В спектрах лигнина, содержащего 1.5% железа, полоса от C=O в альдегидной группе лигнина совпадает и по положению и по интенсивности с такой полосой в спектре чистого лигнина. Введение 2.0% никеля в лигнин сопровождается незначительным понижением интенсивности, а 2.0% железа в лигнине приводит к очень значительному уменьшению интенсивности практически всех полос поглощения лигнина. Из этих данных можно заключить, что в образцы лигнина с 1.5% содержанием металлов оказываются наиболее подготовленными к риформингу, поскольку именно в этих образцах связи ОН, СН и С–С сильно поляризованы, причем присутствие никеля способствует разложению лигнина с выделением более тяжелых ароматических соединений, тогда как под влиянием железа активируются в большей мере алкильные фрагменты лигнина и следует ожидать в продуктах разложения более легких соединений. Повышение содержания металлов до 2.0% приводит к частичному разложению лигнина, причем в присутствии железа это происходит существенно эффективнее, чем в присутствии никеля.
Более наглядно это демонстрируют диаграммы, представленные на рис. 5 и в табл. 2. Диаграммы строились по данным нормированных по внутреннему стандарту ИК-спектров поглощения образцов лигнина с металлами до и после УР, в табл. 2 приведено процентное содержание, сохранившихся функциональных групп лигнина после УР, рассчитанное по относительным интенсивностям отдельных полос спектров.
Рис. 5.
Диаграммы изменения относительных интенсивностей полос валентных колебаний связей СН в алифатических группах лигнина (2930 см–1): 1 – до превращения, 2 – после превращения при МВИ; и скелетных колебаний ароматических колец лигнина (1604 см–1): 3 – до превращения, 4 – после превращения при МВИ. а – лигнин с добавками частиц Ni, б – лигнин с добавками частиц Fe.
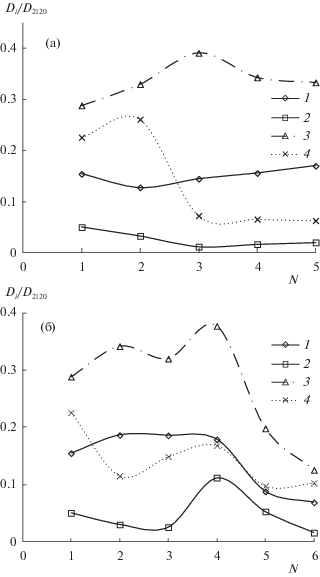
Таблица 2.
Содержание разных функциональных групп в составе сухого остатка после УК риформинга различных образцов лигнина
| № эксп. на диагр. рис. 5 | Образец | Состав функциональных групп в продукте УР лигнина (%) | ||||
|---|---|---|---|---|---|---|
| С=О (1713 см–1) |
С–О–Ph (1214 cм–1) |
С–О–Alk (1058 cм–1) |
Ph (1604 cм–1) |
–CH2–, –CH3 (2852 cм–1) |
||
| 1 | Лигнин | 25 | 77 | 45 | 78 | 31 |
| Лигнин + Fe (%) | ||||||
| 2 | 0.1 | 12 | 27 | 26 | 34 | 18 |
| 3 | 0.5 | 15 | 52 | 37 | 47 | 14 |
| 4 | 1.5 | 12 | 49 | 32 | 45 | 9 |
| 5 | 2.0 | 20 | 66 | 47 | 49 | 65 |
| 6 | 2.78 (МПС) | 32 | 98.5 | 66 | 82 | 29 |
| Лигнин + Ni (%) | ||||||
| 2 | 1.0 | 26 | 184 | 58 | ||
| 3 | 1.5 | 4.5 | 68 (32) | 23 | 18 | 10 |
| 4 | 2.0 | 1.0 | 66 | 28 | 19 | 12 |
| 5 | 1.5 (МПС) | 6 | 77 | 18 | 19 | 18 |
Анализ данных, представленных на диаграммах и в таблице, показывает, что превращение лигнина, содержащего частицы металла, под влиянием МВИ проходит достаточно глубоко по сравнению с лигнином, не содержащим металла. При этом природа металла оказывает существенное влияние на механизм протекания риформинга, поскольку состав остающихся в твердом остатке функциональных групп в образцах содержащих никель сильно отличается от такого же состава в образцах с железом. На диаграммах хорошо видно, как резко уменьшается содержание и ароматических и алифатических групп в лигнине при введении в него железа более 2 мас. %, введение соли никеля не приводит к такому эффекту. Причем это не связано с природой аниона при металле, поскольку образцы с нанесенными металлическими частицами из коллоидного раствора, полученного методом МПС, (№ 4 для никеля и № 6 для железа) не содержали солей металлов.
При этом диаграммы на рис. 5 отчетливо показывают, что введение 2.0% соли железа и особенно металлического железа из коллоидного органического раствора, полученного МПС (эксперимент 6), приводит к значительному уменьшению содержания как ароматических, так и алифатических функциональных групп лигнина, в то время как при внесении в лигнин никеля такого явления не наблюдается. Возможно, это связано с протеканием окислительных процессов в лигнине под влиянием железа, легко вступающего в окислительно-восстановительные процессы.
Высокая конверсия лигнина в ходе УР-образцов с малым (0.1–0.5%) содержанием железа по сравнению с аналогичными никельсодержащими образцами, можно объяснить анализом спектральных данных, представленных на рис. 6. В спектрах образцов с малым количеством соли металла (рис. 6), как и в образцах с более высоким содержанием лигнина (1.5%, рис. 4) прослеживается одинаковая тенденция увеличения интенсивности отдельных полос при внесении в лигнин соли металла. При этом отчетливо просматривается эффект поляризации ароматических связей (полосы 1604 см–1) под влиянием никеля и алифатических (полосы 2840–2970 см–1, 1360–1440 см–1, 1000–1100 см–1) – под влиянием железа. После риформинга в спектрах образцов с разными металлами также прослеживается существенное различие. В спектре образца с никелем полосы от ароматических колец и алкильных групп падают по интенсивности в среднем на 40–50%, но при этом возникают новые полосы с достаточно высокой интенсивностью в области поглощения связей Ph–O (1180–1300 cм–1). В спектре образца с железом все полосы падают по интенсивности более чем на 60%, и только в области 1040 см–1 возникает новая достаточно интенсивная полоса от простой эфирной связи Alk–O. Из этих данных следует, что конверсия лигнина в образцах с железом существенно выше, чем в образцах с никелем, при этом в образце с железом значительно утрачиваются как алкильные, так и ароматические группы лигнина и возникают новые простые алкильные эфиры. В образце с никелем алкильные группы тоже в значительной степени утрачиваются, а фенильные группы претерпевают перегруппировки с возникновением новых замещенных эфиров со связями Ph–O.

Исследование конвективного нагрева лигнина, содержащего микродисперсные частицы железа методом ИКДО
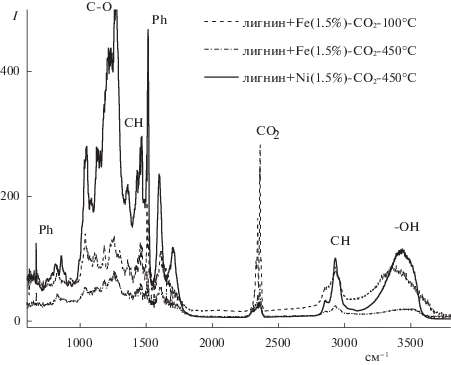
Исследование спектров ИКДО для образцов лигнина, содержащего никель, показало, что при конвективном нагреве этих образцов происходит разложение лигнина с образованием ванилиновых производных (кислота и спирт), которые прочно удерживаются на поверхности лигнина за счет водородного связывания [14]. Спектры ИКДО всех образцов с частицами железа были зарегистрированы в тех же условиях, что и никельсодержащие образцы, описанные в [14]. Ни в одном из образцов лигнина, пропитанных комплексом железа, спектры которых регистрировались в температурном диапазоне 20–450°C, в токе аргона и в токе углекислого газа, не было обнаружено спектральных признаков ванилиновых производных. На рис. 7 и 8 представлены спектры ИКДО образцов Fe-лигнин, зарегистрированные при различных температурах в токе СО2 и Ar в сравнении со спектром ИКДО Ni-лигнин при 450°C в токе СО2. В спектрах ИКДО Fe-содержащих образцов лигнина по мере повышения температуры происходит заметное падение всех полос спектра и при 450°C интенсивность спектра составляет не более 40% от интенсивности в спектре образца при комнатной температуре. Для сравнения на рисунке приведен спектр Ni-содержащего образца, зарегистрированного в тех же условиях при 450°C, в котором наблюдаются очень интенсивные полосы от смеси ванилиновой кислоты и ванилинового спирта, удерживаемых на поверхности лигнина за счет ассоциации с оставшимися в лигнине полярными функциональными группами. Эти данные также демонстрируют различные маршруты реакции разложения лигнина, содержащего железо или никель, при конвективном нагреве. Очевидно, что частицы железа в составе лигнина способствуют образованию легких углеводородов и/или оксигенатов, неспособных к прочной адсорбции на поверхности лигнина и легко уносимых газом среды (аргоном или углекислым газом) (рис. 7 и 8). С другой стороны, на никельсодержащем лигнине в этих условиях образуются ванилиновые производные, которые, как было показано в первом разделе настоящей статьи, способны формировать прочные ассоциаты между собой и с функциональными группами лигнина. Достаточно жесткие температурные условия и тем более условия МВИ способствуют переходу ванилиновых производных в мономерную форму, переходя при этом в газовую фазу они легко разлагаются с образованием синтез-газа.
Рис. 7.
Спектры ИКДО образцов Fe-лигнин, зарегистрированные при разных температурах в токе СО2 в сравнении со спектром ИКДО Ni-лигнин при 450°C в токе СО2.

Исследование фазового состава и зарядового состояния железа методами магнитных измерений, мессбауэровской спектроскопии и ПЭМ показали, что частицы железа при нанесении из ацетилацетонатного комплекса представляют собой, преимущественно, парамагнитные частицы оксида железа (Fe3+) с размером не более 1–2 нм, а из коллоидного органического раствора металлического железа – суперпарамагнитные частицы оксида железа несколько большего размера 3–4 нм [11]. В процессе риформинга, стимулированного МВ-излучением, частицы железа, нанесенные из коллоидного раствора, представляют частицы ~Fe2O3, гомогенно распределенные на поверхности органического остатка с узким распределением по размерам ~5–6 нм. Железосодержащие частицы, полученные путем нанесения Fe(acac)3, в процессе риформинга при микроволновом излучении представлены в виде двух модификаций: частицы с конфигурацией ядро–оболочка и наноразмерные частицы ферримагнитного гамма оксида железа. Первый тип железосодержащих частиц с размером 5–6 нм представляет собой частицы гамма оксида железа, покрытые тонкой пленкой нестехиометрического карбида Fe–Cn с размером, не превышающим 2 нм. При этом увеличение количества нанесенного железа от 0.1 до 2.0% приводит к увеличению относительной концентрации частиц γ-Fe2O3 и снижению концентрации частиц с конфигурацией ядро-оболочка. Увеличение глубины превращения органической массы лигнина в синтез-газ симбатно возрастает с повышением количества нанесенного железа, независимо от метода нанесения [11, 12].
Полученные в настоящей работе результаты позволяют предположить, что формирование наноразмерных частиц оксида железа как в среде Ar так и в среде СО2, связано со взаимодействием ионов железа с поверхностными атомами кислорода и углерода лигнина с формированием наноразмерных частиц оксида железа. На характер такого взаимодействия указывают два фактора: первый заключается в образовании суперпарамагнитных частиц оксида железа; второй – стабилизация частиц на поверхности, обладающих устойчивостью к агломеризации при протекании процесса углекислотного риформинга, при котором наблюдается достаточно глубокое превращение органической массы.
Из полученных в данной работе результатов можно однозначно утверждать, что образование синтез-газа из лигнина в условиях УР под влиянием МВИ протекает по различным маршрутам и с образованием различных интермедиатов в зависимости от природы металла, внесенного в лигнин.
Таким образом, из полученных в данной работе результатов можно однозначно утверждать, что образование синтез-газа из лигнина в условиях УР под влиянием МВИ протекает по различным маршрутам и с образованием различных интермедиатов в зависимости от природы металла, внесенного в лигнин. Наноразмерные частицы никеля, внесенные в лигнин, в большей мере активируют ароматические фрагменты структурных единиц лигнина, что приводит к разложению лигнина с образованием ванилиновых производных (кислота и спирт), которые прочно удерживаются на поверхности лигнина за счет водородного связывания. В условиях МВИ водородные связи, удерживающие ванилиновые производные на поверхности лигнина разрушаются, и происходит УР с образованием синтез-газа. В присутствии наноразмерных частиц железа в составе лигнина интермедиаты в виде ванилиновых производных не реализуются на поверхности лигнина, при этом происходит активация концевых алкильных и алкокси-групп в составе лигнина, что способствует образованию легких продуктов (углеводородов и оксигенатов), которые в условиях МВИ с высокой эффективностью превращаются в синтез-газ.
Список литературы
Rubio M.A., Rodrigez De R.J., Roque D.P. et al. // Appl. Energy. 2011. T. 88. № 3. C. 630.
Zhou C.-H., Xia X., Lin C.-X., Shen D. // Chem. Soc. Rev. 2011. T. 40. C. 5588.
Stoklosa R.J., Hodge D.B. // BioEnergy Res. 2015. № 8. C. 1224.
Toledano A., Serrano L., Garcia A. et al. // Chem. Eng. J. 2010. № 157. C. 93.
Mussatto S.I., Fernandes M., Roberto I.C. // Carbohydr. Polym. 2007. № 70. C. 218.
Patwardhan P.R., Brown R.C., Shanks B.H. // ChemSusChem. 2011. № 4. C. 1629.
Azadi P., Inderwildi O.R., Farnood R., King D.A. // Renew. Sustainable Energy Rev. 2013. V. 21. P. 506.
Zakzeski J., Bruijnincx P.C.A., Jongerius A.L., Weckhuysen B.M. // Chem. Rev., 2010. V. 110. P. 3552.
Gaspar A., Evtuguin D.V., Pascoal N.C. // Proceedings of 12th ISWPC. Wisconsin. 2003. V. 2. P. 83.
Арапова О.В., Цодиков М.В., Чистяков А.В. и др. // Докл. АН. 2017. Т. 475. № 4. С. 405.
Tsodikov M.V., Ellert O.G., Nikolaev S.A. et al. // J. Nanopart. Res. 2018. T. 20. № 3. P. 1.
Tsodikov M.V., Ellert O.G., Arapova O.V. et al. // Chem. Eng. Trans. 2018. V. 65. P. 367.
Tsodikov M.V., Ellert O.G., Nikolaev S.A. et al. // Chem. Eng. J. 2017. № 309. P. 628.
Арапова О.В., Бондаренко Г.Н., Чистяков А.В., Цодиков М.В. // Журн. физ. химии. 2017. Т. 91. № 9. С. 1520.
Vasil’kov A.Yu., Naumkin D.A., Belyakova O.A. et al. // Mendeleev Commun. 2016. № 26. C. 187.
Вест В. Применение спектроскопии в химии. М.: Изд-во иностр. лит., 1959.
Свердлов Л.М., Ковнер М.А., Крайнов Е.И. Колебательные спектры многоатомных молекул. М.: Наука, главная редакция физико-математической литературы, 1970.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии