

Журнал физической химии, 2019, T. 93, № 8, стр. 1199-1205
Молекулярно-динамическое моделирование сольватного окружения эфира 18-краун-6 в смешанных растворителях этанол–диметилсульфоксид
М. А. Волкова a, *, И. А. Кузьмина a, Е. Г. Одинцова b, В. А. Шарнин a
a Ивановский государственный химико-технологический университет
Иваново, Россия
b Российская академия наук, Институт химии растворов им. Г.А. Крестова
Иваново, Россия
* E-mail: mariia.a.volkova@gmail.com
Поступила в редакцию 14.11.2018
После доработки 14.11.2018
Принята к публикации 11.12.2018
Аннотация
С использованием метода молекулярной динамики определено число молекул этанола (EtOH) в сольватном окружении эфира 18-краун-6 (18C6) в зависимости от концентрации диметилсульфоксида (DMSO) в бинарной смеси EtOH–DMSO (χDMSO = 0.0–0.8 мол. доли); рассчитаны вероятности образования водородных связей (ВС), средние числа ВС и продолжительные времена жизни ВС между молекулами 18C6–EtOH, DMSO–EtOH и EtOH–EtOH. Предположено, что пересольватация макроцикла практически завершается при χDMSO ~ 0.6 мол. доли. Установлено, что способность молекул EtOH к образованию ВС с 18C6, DMSO и друг с другом уменьшается с ростом концентрации DMSO в бинарной смеси. Показано, что продолжительные времена жизни ВС EtOH–DMSO выше, чем для 18C6–EtOH и EtOH–EtOH.
Краун-эфиры аналогичны некоторым биолигандам, способным избирательно взаимодействовать с ионами металлов и нейтральными молекулами путем включения их в полость своей кольцевидной молекулы [1–4]. Данное свойство макроциклических соединений открывает широкие перспективы их использования в научных исследованиях в области химии координационных соединений [5–7], а также дает возможность применять их в хроматографии, экстракции, при разделении ионов и изотопов, в химической сенсорике, моделировании биохимических процессов и т.д. [8–11].
Процессы, протекающие в жидкой фазе, сопровождаются взаимодействиями “растворитель–растворенное вещество” и “растворитель–растворитель”. В соответствии с представлениями о растворах, в окружении сольватированного иона, молекулы, ассоциата, комплекса или другой подобной частицы структура растворителя меняется по мере удаления от центра сольватируемой частицы (ядра) [12]. Отличительная особенность сольватокомплексов, образующихся в смешанных растворителях, – соотношение молекул разных растворителей в сольватном окружении частицы и в массе раствора не одинаково. Поэтому определение сольватного окружения частиц – одна из важных задач при изучении сольватации в смешанных растворителях.
Интенсивное развитие новых расчетных методов, таких как молекулярно-динамическое моделирование (МД) [13–15], дает возможность детально изучить процессы, происходящие на молекулярном уровне, а именно: получить данные о сольватном окружении частиц в среде растворителей, исследовать динамику водородных связей (ВС), в которых участвует каждая молекула ансамбля в любой, произвольно выбранный момент времени и др.
Цель настоящего исследования – молекулярно-динамическое моделирование сольватного окружения молекулы эфира 18-краун-6 (18C6) в среде смешанных неводных растворителей этанол–диметилсульфоксид (EtOH–DMSO) при различных концентрациях второго компонента в бинарной смеси (χDMSO = 0.0 – 0.8 мол. доли).
EtOH и DMSO отличаются донорно-акцепторными свойствами [16] и способностью к образованию ВС. Известно, что в спиртах сольватация эфира 18C6 осуществляется, преимущественно благодаря образованию ВС между O-донорными атомами макроцикла и атомами водорода гидроксильных групп растворителя [17]. Вопрос об участии атомов водорода метильных групп DMSO в образовании ВС остается дискуссионным [18–21]. На практике данные растворители применяются как вспомогательные вещества для улучшения растворимости медицинских препаратов, поскольку обеспечивают полярность среды, близкую к таковой в биологических объектах [22, 23].
ДЕТАЛИ МОДЕЛИРОВАНИЯ
Классическое молекулярно-динамическое моделирование проведено в NVT-ансамбле с использованием программного пакета GROMACS-4.5.4 [24] в полноатомном силовом поле OPLSAA [25–27].
На первом этапе моделирования создавались кубические ячейки с периодическими граничными условиями, содержащие 512 молекул индивидуального или бинарного растворителя. Количество молекул DMSO в ячейке составляло 102, 205, 307 или 410, в зависимости от состава бинарной смеси (χDMSO = 0.2, 0.4, 0.6 и 0.8 мол. доли соответственно). В каждом случае размеры ячейки подбирались таким образом, чтобы соответствовать экспериментальным плотностям индивидуальных или смешанных растворителей при 298 К и 1 атм [28]. После минимизации энергии и уравновешивания системы в течение 0.5 нс к молекулам растворителя определенного состава добавлялась одна молекула краун-эфира. При этом длина ребра ячеек трехкомпонентной системы корректировалась таким образом, чтобы сохранялась плотность среды, после чего проводилась минимизация энергии и выполнялось уравновешивание системы в течение 0.5 нс. Общая продолжительность моделирования систем с требуемыми параметрами температуры и плотности составила 1 нс. Для интегрирования уравнений Ньютона использовался алгоритм Верле [29] с шагом по времени 1 фс, для поддержания постоянной температуры – термостат Нозе–Хувера [30, 31], для учета дальнодействующих электростатических взаимодействий – модифицированный метод суммирования Эвальда [32, 33], для ограничения по всем длинам связей – алгоритм LINCS [34].
Исходная геометрия молекул эфира 18-краун-6 (I), этанола (II) и диметилсульфоксида (III) получена путем оптимизации в программном пакете GAUSSIAN 03 [35] с использованием теории функционала электронной плотности в варианте B3LYP [36–38] с применением корреляционно-согласованного трехэкспонентного базисного набора cc-pVTZ [39]. Зарядовые характеристики молекул получены из распределения электронной плотности в рамках программы NBO 3.1 [40], входящей в состав программного комплекса GAUSSIAN 03.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Информацию об относительной вероятности нахождения пары атомов (или молекул) на определенном расстоянии один от другого в зависимости от межъядерного расстояния можно получить из функций радиального распределения (ФРР). В усредненном виде в них заложена вся информация о траекториях движения каждой отдельной частицы, а также обо всех возможных молекулярных конфигурациях.
В численном моделировании существование ВС между парой молекул определяется в соответствии с заранее сформулированными критериями их образования. Одним из наиболее широко используемых критериев, который позволяет оценить взаимное расположение частиц, является геометрический критерий [41, 42]. Пороговые значения критерия $R_{{{\text{OO}}}}^{c}{\text{ и }}R_{{{\text{OH}}}}^{c}$ оцениваются либо экспериментально, либо по положению первых минимумов на соответствующих ФРР, и поэтому считается, что они индивидуальны для каждого вещества. Для спиртов (метанола и этанола) приняты следующие значения: $R_{{{\text{OO}}}}^{c}$ = 3.5 Å, $R_{{{\text{OH}}}}^{c}$ = = 2.6 Å [43, 44].
На рис. 1 приведены усредненные ФРР по всем атомам кислорода молекулы эфира 18-краун-6 и атомам водорода и кислорода молекул EtOH (gO(I)–H(II)(r) и gO(I)–O(II)(r) на рис. 1а и 1б соответственно), на рис. 2 – ФРР центров масс 18C6 и EtOH, gI–II(r). Сравнение положения пиков на ФРР O(I)H(II) и О(I)O(II) (рис. 1) с пороговыми значениями, определяемыми геометрическим критерием ВС [41, 42] и высот пиков при изменении состава растворителя (рис. 1), анализ областей экстремального поведения gI–II(r) на ближних расстояниях и областей пологого спада на удаленных расстояниях (рис. 2) позволили в первом приближении оценить возможность образования ВС между молекулами 18C6 и EtOH при смене состава растворителя EtOH → (EtOH–DMSO).
Рис 1.
Атом-атомные ФРР: а) gO(I)–H(II)(r) и б) gO(I)–O(II)(r) при различных значениях χDMSO: 1 – 0, 2 – 0.2, 3 – 0.4, 4 – 0.6, 5 – 0.8 мол. доли.


Как следует из рис. 1, высокая степень локализации молекул EtOH вокруг молекулы макроцикла наблюдается лишь в индивидуальном растворителе. При переходе от чистого EtOH к его смесям с DMSO уменьшаются высоты соответствующих пиков на ФРР, что связано с уменьшением количества молекул EtOH в ближнем окружении молекулы 18C6 в результате процесса пересольватации.
Отсутствие пиков на ФРР O(I)H(II) и О(I)O(II) (рис. 1), а также отсутствие четко выделенных областей экстремального поведения на ближних расстояниях и областей пологого спада на удаленных расстояниях на gI–II(r) (рис. 2) при χDMSO > 0.4 мол. доли может свидетельствовать о том, что при данном составе смешанного растворителя возможность образования ВС 18C6–EtOH стремится к нулю. На основании данных рис. 1 и 2 можно предположить, что пересольватация эфира 18C6 практически завершается при χDMSO ~ 0.6 мол. доли.
Сделанные предположения подтверждаются расчетом вероятности образования ВС между молекулами 18C6 и EtOH в бинарной этанольно-диметилсульфоксидной смеси (fi на рис. 3, где i – это число ВС). Значения fi определялись как отношение суммарного количества шагов, на которых было зафиксировано существование молекулы растворенного вещества в состоянии i ВС с EtOH, к общему числу шагов наблюдения.
Рис. 3.
Вероятности образования ВС (fi, где i = 0–6) между молекулами 18C6 и EtOH в смешанных растворителях EtOH–DMSO; 1–4 – см. рис. 1.

Состав сольватного окружения 18C6 можно оценить через координационное число (КЧ), под которым понимают статистически усредненное количество частиц сорта β в сольватном окружении частицы сорта α. Средние КЧ (ncoord(I–II)), характеризующие число молекул EtOH в сольватном окружении 18C6 в зависимости от состава смешанного растворителя, получены интегрированием ФРР gI–II(r) (рис. 2) и приведены в табл. 1. Расчет ncoord(I–II) производился по формуле [45]:
Таблица 1.
Средние координационные числа (ncoord), и средние числа ВС (nHB) при значениях χDMSO = 0.2–0.8 мол. доли
| ni | 0.0 | 0.2 | 0.4 | 0.6 | 0.8 |
|---|---|---|---|---|---|
| ncoord(I–II) | 28.79 | 19.14 | 8.13 | 2.00 | – |
| nHB(I–II) | 4.58 | 1.73 | 0.56 | – | – |
| nHB(III–II) | – | 2.18 | 1.23 | 0.64 | 0.25 |
| nHB(II–II) | 1.78 | 0.82 | 0.32 | 0.08 | 0.02 |
С использованием геометрического критерия [41, 42] рассчитаны средние числа ВС, образуемых молекулой 18-краун-6 с молекулами EtOH, входящими в сольватное окружение макроцикла (nHB(I–II) в табл. 1).
Как следует из табл. 1, значения ncoord(I–II) изменяются от 28.79 в EtOH до 2.00 при χDMSO = = 0.6 мол. доли. Количество ВС 18C6-EtOH снижается от 4.58 в EtOH до 0.56 при χDMSO = 0.4 мол. доли (табл. 1). При χDMSO > 0.4 мол. доли ВС не образуются. Сравнение значений ncoord(I–II) и nHB(I–II) показывает, что в образовании ВС с молекулой 18C6 участвуют не все молекулы EtOH, находящиеся в ближнем окружении макроцикла. Это может быть обусловлено тем, что сольватация 18C6 осуществляется не только отдельными молекулами EtOH, но и молекулами спирта, связанными между собой.
Согласно [20], ассоциация молекул DMSO осуществляется, преимущественно, за счет диполь-дипольного взаимодействия по атомам серы и кислорода с образованием цепочечной структуры. В работе [21] отмечено, что атомы кислорода молекулы DMSO способны к образованию ВС с молекулами воды. Наличие в молекуле спирта, как и в молекуле воды, полярной химической связи O–H, позволило предположить, что между молекулами DMSO и EtOH возможно образование ВС O···H–O.
Сделанные предположения подтверждаются расчетами вероятности образования ВС между молекулами DMSO и EtOH (рис. 4) и между молекулами спирта (рис. 5) в системе 18С6–EtOH–DMSO. Как видно из рис. 4 и 5, вероятность образования ВС DMSO–EtOH и EtOH–EtOH варьируется от 2 до 3 и от 1 до 3 соответственно в зависимости от состава бинарной смеси.
Рис. 4.
Вероятности образования водородных связей (fi, где i = 0–3) между молекулами DMS = 0.2 (1), 0.4 (2), 0.6 (3) и 0.8 мол. доли (4).
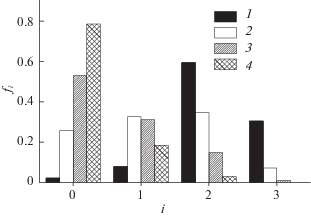
Рис. 5.
Вероятности образования водородных связей (fi, где i = 0–3) между молекулами этанола в системе 18C6–EtOH–DMSO при χDMSO = 0.0 (1), 0.2 (2), 0.4 (3), 0.6 (4) и 0.8 мол. доли (5).

Расчет средних чисел ВС между молекулами 18C6 и EtOH (nHB(I–II) в табл. 1), молекулами DMSO и EtOH (nHB(III–II) в табл. 1) и молекулами спирта (nHB(II–II) в табл. 1) показывает, что способность молекул EtOH образовывать ВС с 18C6, DMSO и друг с другом, уменьшается с ростом концентрации DMSO в трехкомпонентной системе. Значения nHB(III–II) при концентрациях DMSO от 0.2 до 0.8 мол. доли выше, чем значения nHB(II–II) и nHB(I–II). Это может быть связано с более высокой плотностью отрицательного заряда на атоме кислорода в молекуле DMSO по сравнению с таковыми в молекулах 18C6 и EtOH. Величины точечных зарядов на атомах кислорода составляют –0.961, –0.683 и –0.537 |e| в DMSO, EtOH и 18C6 соответственно.
В разное время были предложены различные подходы к расчету времени жизни ВС из данных численного эксперимента [46–48]. Широко распространены методы вычисления времен жизни ВС непосредственно из автокорреляционной функции параметра существования ВС [49, 50]. Время жизни ВС $(\tau _{{{\text{HB}}}}^{{\text{I}}})$ [49] длится до того момента, пока какая-то из молекул-партнеров не просто разорвет ее (т.е. в данной паре молекул нарушится геометрический критерий существования связи [41, 42]), а образует при этом ВС с другой молекулой; при этом промежуточные разрывы, не сопровождающиеся образованием новой связи, не принимаются во внимание, сколько бы они не длились.
Величины $\tau _{{{\text{HB}}}}^{{\text{I}}}$, рассчитанные для связей I–II, II–II и III–II в стандартном программном пакете GROMACS 4.5.4 [24] с использованием автокорреляционной функции параметра существования ВС, представлены в табл. 2. Из данных табл. 2 следует, что ВС 18C6–EtOH и EtOH–EtOH характеризуются значительно меньшей продолжительностью существования, чем ВС DMSO–EtOH.
Таблица 2.
Продолжительные времена жизни ВС ($\tau _{{{\text{HB}}}}^{{\text{I}}}$, пс) при значениях χDMSO = 0.2–0.8 мол. доли
| $\tau _{{{\text{HB}}}}^{{\text{I}}}$ | 0.0 | 0.2 | 0.4 | 0.6 | 0.8 |
|---|---|---|---|---|---|
| $\tau _{{{\text{HB}}}}^{{\text{I}}}$(I–II) | 98.82 | 96.75 | 54.25 | – | – |
| $\tau _{{{\text{HB}}}}^{{\text{I}}}$(II–II) | 41.74 | 42.54 | 50.14 | 52.33 | 56.21 |
| $\tau _{{{\text{HB}}}}^{{\text{I}}}$(III–II) | – | 134.25 | 142.49 | 147.04 | 160.68 |
Таким образом, повышение содержания DMSO в бинарной смеси приводит к снижению количества молекул EtOH в сольватном окружении макроцикла от 28.79 (в чистом EtOH) до 2.00 (при χDMSO = 0.6 мол. доли) и уменьшению числа ВС, образующихся между молекулами 18C6 и спирта от 4.58 (в чистом EtOH) до их полного отсутствия (при χDMSO > 0.4 мол. доли).
Предположено, что пересольватация макроцикла практически завершается при χDMSO ~ 0.6 мол. доли. Установлено, что способность молекул EtOH к образованию ВС с 18C6, DMSO и друг с другом, уменьшается с ростом концентрации DMSO в бинарной смеси. Показано, что продолжительные времена жизни ВС изменяются в ряду: $\tau _{{{\text{HB}}}}^{{\text{I}}}$(EtOH–EtOH) < $\tau _{{{\text{HB}}}}^{{\text{I}}}$(18C6–EtOH) < < $\tau _{{{\text{HB}}}}^{{\text{I}}}$(DMSO–EtOH). Сделан вывод, что изменение сольватного окружения молекулы эфира 18-краун-6 определяется не только взаимодействиями “растворитель–растворенное вещество”, но и взаимодействиями “растворитель–растворитель”, причем не только молекул одного сорта.
Список литературы
Pedersen C.J. // J. Am. Chem. Soc. 1967. V. 89. № 26. P. 7017. https://doi.org/10.1021/ja01002a035
Хираока М. Краун-соединения. Свойства и применения. М.: Мир, 1986. 363 с.
Стид Дж.В., Этвуд Дж.Л. Супрамолекулярная химия. Т. 1. М.: ИКЦ “Академкнига”, 2007. 480 с.
Davis F., Higson S. Macrocycles: Construction, Chemistry and Nanotechnology Applications. Chichester: John Wiley & Sons Ltd, 2011. 596 p.
Usacheva T.R., Sharnin V.A., Matteoli E. // Ch. 1 in Monograph “Glycine. Biosynthesis, Physiological Functions and Commercial Uses” / Ed. by W. Vojak. New York: Nova Science Pub. Inc. 2013. P. 1–33.
Usacheva T.R., Sharnin V.A., Matteoli E. // Ch. 5 in Monograph “Advances in Chemistry Research”. V. 22 / Ed. by J.C. Taylor. New York: Nova Science Pub. Inc. 2014. P. 127–157.
Kuz’mina I.A., Usacheva T.R., Volkova M.A. et al. // Ch. 7 in Monograph “Advances in Chemistry Research”. V. 33. Ed. by J.C. Taylor. New York: Nova Science Pub. Inc. 2016. P. 205–223.
Цивадзе А.Ю., Жилов В.И., Демин С.В. // Коорд. химия. 1996. Т. 22. № 4. С. 243.
Richens D.A., Simpson D., Peterson S. et al. // J. Chromatogr. A. 2003. V. 1016. P. 155. https://doi.org/10.1016/S0021-9673(03)01330-X
Gokel G.W., Leevy W.M., Weber M.E. // Chem. Rev. 2004. V. 104. P. 2723. https://doi.org/10.1021/cr020080k
Kakhki R.M.Z., Rounaghi G. // J. Chem. Eng. Data. 2011. V. 56. N 7. P. 3169. https://doi.org/10.1021/je200220d
Крестов Г.А. // Журн. ВХО им. Д.И. Менделеева. 1983. Т. 28. № 6. С. 70.
Товбин Ю.К. Метод молекулярной динамики в физической химии. М.: Наука, 1996. 334 с.
Virk A.S., Stait-Gardner T., Willis S.A. et al. // Front. Phys. 2015. V. 3. https://doi.org/10.3389/fphy.2015.00001
Horowitz S., Trievel R.C. // J. Biol. Chem. 2012. V. 287. № 50. P. 41576. https://doi.org/10.1074/jbc.r112.418574
Фиалков Ю.А. Не только в воде. Л.: Химия, 1989. 88 с.
Баранников В.П., Гусейнов С.С., Вьюгин А.И. // Коорд. химия. 2002. Т. 28. № 3. С. 163.
Fuchs R., McCravy G.E., Bloomfield J.J. // J. Am. Chem. Soc. 1961. V. 83. № 20. P. 4281. https://doi.org/10.1021/ja01481a043
Safford G.J., Schaffer P.C., Leung P.S. et al. // J. Chem. Phys. 1969. V. 50. № 5. P. 2140. https://doi.org/10.1063/1.1671344
Amey R.L. // J. Phys. Chem. 1968. V. 72. № 9. P. 3358. https://doi.org/10.1021/j100855a061
Верстаков Е.С., Ястремский П.С., Кесслер Ю.М. и др. // Журн. структур. химии. 1980. Т. 21. № 5. С. 91.
Гулякин И.Д., Оборотова Н.А., Печенников В.М. // Химико-фармацевтич. журн. 2014. Т. 48. № 3. С. 46.
Цицуашвили М.Д., Павлова С.И., Албегова Д.З., Козлов И.Г. // Международный журнал по иммунореабилитации (Int. J. Immunorehabilitation). 2010. Т. 12. № 2. С. 105a.
Apol E., Apostolov R., Berendsen H.J.C. GROMACS-4.5.4 // Sweden. 2001–2010. http://www.gromacs.org
Jorgensen W.L., Maxwell D.S., Tirado-Rives J. // J. Amer. Chem. Soc. 1996. V. 118. № 45. P. 11225.
Zwier M.C., Kaus J.W., Chong L.T. // J. Chem. Theory Comput. 2011. V. 7. P. 1189. https://doi.org/10.1021/ct100626x
Borin I.A., Skaf M.S. // J. Chem. Phys. 1999. V. 110. P. 6412. https://doi.org/10.1063/1.478544
Крестов Г.А., Афанасьев В.Н., Ефремова Л.С. Физико-химические свойства бинарных растворителей: Справ. изд. Л.: Химия, 1988. 688 с. ISBN 5-7245-0039-6
Allen M.P., Tildesley D.J. Computer Simulation of Liquids. L.: Clarendon Press, 1987.
Nose S. // Mol. Phys. 1984. V. 52. № 2. P. 255.
Hoover W.G. // Phys. Rev. A. 1985. V. 31. № 3. P. 1695.
Darden T., York D., Pedersen L. // J. Chem. Phys. 1993. V. 98. P. 10089.
Essmann U., Perera L., Berkowitz M.L. et al. // Idid. 1995. V. 103. P. 8577.
Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. // J. Comput. Chem. 1997. V. 18. P. 1463.
Frisch M.J., Trucks G.W., Schlegel H.B. et al. Gaussian 03, Revision B.03 // Gaussian, Inc., Pittsburgh PA, 2003.
Hertwig R.H., Koch W. // J. Chem. Phys. Lett. 1997. V. 268. № 5. P. 345.
Hehre W.J., Ditchfield K., Pople J.A. // J. Chem. Phys. 1972. V. 56. № 5. P. 2257.
Dill J.D., Pople J.A. // J. Chem. Phys. 1975. V. 62. № 7. P. 2921.
Dunning T.H. // J. Chem. Phys. 1989. V. 90. № 2. P. 1007.
Glendening E.D., Reed A.E., Carpenter J.E. et al. // QCPE Bull. V. 10. 1990. P. 58.
Arunan E., Desiraju G.R., Klein R.A., Sadlej J. et al. // Pure Appl. Chem. 2011. V. 83. № 8. P. 1619. https://doi.org/10.1351/PAC-REP-10-01-01
Arunan E., Desiraju G.R., Klein R.A., Sadlej J. et al. // Pure Appl. Chem. 2011. V. 83. № 8. P. 1637. https://doi.org/10.1351/PAC-REC-10-01-02
Guardia E., Marti J., Padro J.A. et al. // J. Mol. Liq. 2002. V. 96–97. P. 3.
Haughney M., Ferrario M., McDonald I.R. // J. Phys. Chem. 1987. V. 91. № 19. P. 4934.
Скрышевский А.Ф. Структурный анализ жидкостей и аморфных тел. М.: Высшая школа, 1980. 328 с.
Маленков Г.Г., Тытик Д.Л. Метод молекулярной динамики в физической химии / Под ред. Ю.К. Товбина. М.: Наука, 1996. 204 с.
Маленков Г.Г., Франк-Каменецкий М.М., Гривцов А.Г. // Журн. структур. химии. 1987. Т. 28. № 2. С. 81.
Волошин В.В., Маленков Г.Г., Наберухин Ю.И. // Там же. 2007. Т. 48. № 6. С. 1133.
Rapaport D.C. // Mol. Phys. 1983. V. 50. № 5. P. 1151.
Антипова М.Л., Петренко В.Е. // Журн. физ. химии. 2013. Т. 87. № 7. С. 1196.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии