

Журнал физической химии, 2019, T. 93, № 8, стр. 1206-1211
Неэмпирическое описание строения и энергии взаимодействия в димерах пергалогенметанов
Б. В. Руцкой a, *, Д. С. Безруков a, b
a Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
b Сколковский институт науки и технологий
Москва, Россия
* E-mail: bogdan.rutskoy@gmail.com
Поступила в редакцию 19.11.2018
После доработки 19.11.2018
Принята к публикации 11.12.2018
Аннотация
Неэмпирически исследованы межмолекулярные взаимодействия в димерах (CX4)2 (X = H, F, Cl, Br, I). Калибровочные значения энергий и длин связей димеров получены методом связанных кластеров с учетом одно- и двукратных возбуждений и неитерационной поправки на трехкратные возбуждения (CCSD(T)) с базисными наборами Даннинга, дополненными связевыми функциями, центрированными между атомами углерода, с последующей экстраполяцией к бесконечному базисному пределу. Анализ построенных сечений ППЭ димеров (CX4)2 позволил существенно уточнить энергии диссоциации для (СCl4)2, (CBr4)2 из предыдущих расчетов, а также впервые получить оценку этой величины для димера (CI4)2. Построенные корреляции между рассчитанными энергиями диссоциации димеров (CX4)2 и поляризуемостями CX4 продемонстрировали определяющую роль поляризуемости отдельных молекул в вариации физических свойств димеров CX4 при изменении X от фтора к иоду.
Понимание межмолекулярных сил, определяющих физико-химические особенности галогензамещенных алканов, важно для их применений во многих областях, от нефтехимии до фармакологии. Считается, что их особенность – проявление специфического взаимодействия “галоген–галоген”. Простейшие представители систем, природа взаимодействий которых полностью определяется этими силами, – димеры пергалогенметанов (CX4)2 (X = F, Cl, Br, I). В этот ряд можно добавить димер метана, который в каком-то смысле является “нулевым” соединением в ряду галогеналканов. Сложность проблемы корректного теоретического описания специфичных ван-дер-ваальсовых взаимодействий привела к включению метана в тестовый набор молекул для анализа качества функционалов плотности S22 [1], а использование высокоточных методов квантовой химии позволило оценить энергию межмолекулярного взаимодействия как 0.51 ккал/моль с погрешностью менее 1% [2], что позволяет применять данные методы для построения силовых полей с последующим использованием их при моделировании как газофазных характеристик (вириальные коэффициенты, термодинамические функции), так и состояний конденсированной фазы (фазовые переходы, подвижность, матричная изоляция).
Галогензамещенные системы изучены не так основательно, при этом точность описания падает при изменении заместителя от фтора к иоду. Так, наилучшей оценкой энергии межмолекулярного взаимодействия для димера (CF4)2 является значение, полученное методом связанных кластеров с учетом одно- и двукратных возбуждений и неитерационной поправки на трехкратные возбуждения (CCSD(T)) для базисного набора Сузуки, которое составило 0.74 ккал/моль [3]. Энергия взаимодействия между молекулами CCl4 была рассчитана на уровне метода теории возмущений Меллера–Плессета второго порядка (МП2) с экстраполяцией на бесконечный базисный набор и составила 3.523 ккал/моль [4]. Димер (CBr4)2 изучен еще хуже, для него энергия взаимодействия между мономерами определена методом МП2 с базисным набором aug(df)-6-311G* и составляет 3.702 ккал/моль [5]. Данные по димеру периодметана в литературе отсутствуют.
Некоторым аналогом этих ван-дер-ваальсовых систем могут быть димеры инертных газов, для которых показано [6–8], что наилучшее описание достигается в случае применения для расчета энергии межмолекулярного взаимодействия методов F12-CCSD(T) или CCSD(T) с базисными наборами, содержащими связевые функции. Это объясняется тем, что основной вклад в энергию взаимодействия вносят эффекты, обусловленные наличием межмолекулярной электронной корреляции, которые на сегодняшний день могут быть корректно учтены для больших систем только указанными методами [6]. Оба подхода, однако, не равнозначны. Явнокоррелированный метод F12-CCSD(T) наиболее точно описывает ориентационные и индукционные взаимодействия, возникающие между полярными молекулами, а дисперсионные взаимодействия, характерные для неполярных систем, включая димер метана, наиболее эффективно могут быть определены методом CCSD(T) с добавлением связевых функций [9].
Цель настоящей работы – проведение высокоточных расчетов структурных и энергетических свойств димеров (CX4)2 (X = H, F, Cl, Br, I) в рамках единой методологии, основанной на применении метода связанных кластеров CCSD(T) с дополнительным набором связевых функций; верификация результатов по доступным данным (X = H, F); сравнение полученных данных с известными расчетами более низкого уровня (X = = Cl, Br); проверка полученных данных на основе корреляционного анализа по известным экспериментальным данным.
ИССЛЕДУЕМЫЕ СИСТЕМЫ И ФИЗИЧЕСКИЕ ВЕЛИЧИНЫ
Основной интерес представляют потенциалы взаимодействия димеров (CX4)2 как функции расстояния r(C–C). Так как энергия взаимодействия ван-дер-ваальсовых систем определяется в первую очередь поляризуемостями отдельных молекул, то для установления корреляций в исследуемом ряду рассчитывались также поляризуемости атомов X и молекул CX4. Для оценки качества описания проверялись оценки равновесного расстояния C–X в индивидуальных молекулах CX4.
ДЕТАЛИ НЕЭМПИРИЧЕСКОГО МОДЕЛИРОВАНИЯ
Для описания электронной волновой функции использовались корреляционно-согласованные базисные наборы Даннинга cc-pVNZ и aug-cc-pVNZ (где N = D, T, Q). На предварительном этапе тестировались следующие варианты описания атомов галогенов. Для атомов хлора использовались как полноэлектронные (ПЭ) базисные наборы, так и комбинируемые с эффективными остовными потенциалами (ECP) 10SDF [10] и 10MWB [11]. Для описания атома брома использовались только эффективные остовные потенциалы 10MDF [12], 28MDF [13] и 28MWB [11]. В случае атома иода также применялись эффективные остовные потенциалы 28MDF [14] и 46MDF [13]; дополнительно были получены результаты с использованием ПЭ базисного набора aug-cc-pwCVTZ-DK3 [15], учитывающего скалярные релятивистские эффекты.
Поляризуемости были найдены методом конечного поля. Для атомов галогенов в состоянии 2Po рассчитывались обе компоненты анизотропной поляризуемости в нерелятивистском случае, поскольку, как было показано в работе [16], изотропная компонента слабо меняется при учете спин-орбитальных эффектов, а анизотропная составляющая мала. Симметрия компоненты волновой функции для атомов галогенов определялась заселенностью орбиталей; при расчете использовалась симметрия ${{C}_{{{\text{2}}v}}}$. Равновесную геометрию CX4 находили методом градиентной оптимизации с пороговым значением 1 × 10–6 ат. ед. Полученные значения энергии были экстраполированы к пределу бесконечного базисного набора при помощи двухпараметрической зависимости от обратного значения максимального орбитального момента базиса в кубической степени, предложенной Хельгакером [17].
Наиболее трудоемкой задачей было построение сечений ППЭ димеров (CX4)2. Предварительные тесты показали, что все исследуемые комплексы имеют равновесную геометрию симметрии D3d, ранее описанную в литературе (рис. 1). В методах МП2, CCSD, CCSD(T) с использованными в работе базисными наборами сечение строилось только вдоль расстояния C–С, при этом геометрия мономеров CX4 оставалась постоянной. Для описания систем X = Cl, Br, I использовался только один вариант эффективных остовных потенциалов, выбранный по результатам исследования индивидуальных молекул.

Для более полного описания межмолекулярной электронной корреляции применялся хорошо зарекомендовавший себя прием добавления дополнительных базисных функций [7, 18, 19] на середину расстояния C–C. Соответствующий базисный набор [3s3p2d2f1g] был взят из работы [7]. Полученные значения энергии взаимодействия экстраполировались к пределу бесконечного базисного набора способом, аналогичным использованному для отдельных молекул. Все расчеты выполнены в программном комплексе Molpro [20].
РЕЗУЛЬТАТЫ РАСЧЕТОВ
Расчет компонент поляризуемостей отдельных атомов X (X = H, F – I) показал, что уже на уровне метода Хартри–Фока во всех рассмотренных системах разброс значений не превышает 10% относительно результатов, полученных методом CCSD(T) с базисным набором aug-cc-pVQZ. Применение различных вариантов ECP практически не влияло на результаты. Переход к оценкам, полученным на уровне МП2 с использованием трижды валентно-расщепленных базисных наборов, дает оценки поляризуемости с погрешностью менее 3% во всех рассмотренных случаях. Поскольку для описания ван-дер-ваальсовых взаимодействий будет необходим более глубокий уровень учета эффектов электронной корреляции, то можно заключить, что погрешности описания отдельных молекул на данном этапе будут существенно меньше ошибки описания энергии межмолекулярного взаимодействия.
При исследовании молекул CX4 равновесные расстояния C–X были рассчитаны на уровне метода CCSD(T) в пределе бесконечного базисного набора и составили 1.30, 1.75, 1.94 и 2.10 Å для фтора, хлора, брома и иода соответственно. Средние величины поляризуемости, найденные методом CCSD(T) в базисном наборе aug-cc-pVTZ, для CF4, CCl4, CBr4, CI4 составили 18.96, 66.20, 94.06, 159.46 $a_{0}^{3}$, что в пределах 5% согласуется с экспериментальными данными для первых трех молекул: 19.31 [21], 70.92 [22], 101.29 $a_{0}^{3}$ [23]. Данные о поляризуемости периодметана в литературе отсутствуют.
На основании расчетов индивидуальных атомов и молекул CX4 был сделан вывод о том, что остовная корреляция не играет существенной роли в определении геометрии и значений поляризуемости исследованных систем. Так, в случае хлорсодержащих систем проводилось сравнение результатов, полученных как с использованием ECP для 10 остовных электронов (в модельный потенциал включены 1s-, 2s- и 2p-оболочки), так и полноэлектронных расчетов, причем в последнем случае расчеты методами МП2, CCSD и CCSD(T) сравнивались с результатами, полученными как с учетом возбуждений с остовных орбиталей, так и с учетом возбуждений только с валентных 3s- и 3p-оболочек. Было показано, что и геометрия, и поляризуемость наиболее чувствительны к выбору метода учета электронной корреляции, в то время как отклонения в оценках поляризуемости не превышают 2% при сравнении данных, полученных с использованием ECP и полноэлектронного расчета с учетом возбуждений со всех остовных орбиталей. Аналогичный вывод был сделан для брома, где сравнивались результаты, полученные с использованием ECP для описания 10 (1s, 2s, 2p) и 28 (1s, 2s, 2p, 3s, 3p, 3d) электронов; погрешность в данном случае составила 1.7% для значения поляризуемости. В случае атома иода обоснованность использования псевдопотенциала Дирака–Фока, учитывающего скалярные релятивистские эффекты, была подтверждена сравнением данных, полученных методом CCSD(T) для эффективных остовных потенциалов и для описания всех электронов в рамках формализма Дугласа–Кролла второго порядка в базисном наборе aug-cc-pwCVTZ-DK3. Различие в значениях поляризуемости составило 2.1%, относительное отклонение равновесного расстояния C–I не превысило 0.2%.
На основании полученных данных для дальнейшего исследования димеров выбраны эффективные остовные потенциалы, содержащие максимальное число остовных и валентных электронов в модельной части: 10SDF, 28MWB и 46MDF для хлор-, бром- и иодзамещенных метанов.
Наиболее важные результаты, найденные при расчете сечений ППЭ вдоль расстояния C–C, представлены в табл. 1 и 2. Во всех случаях подразумевается, что расчеты проведены с использованием дополнительных связевых функций, расположенных посередине расстояния C–C.
Таблица 1.
Энергия диссоциации (ккал/моль) в димерах пергалогенметанов
| Метод / базис | CH4 | CF4 | CCl4 | CBr4 | CI4 |
|---|---|---|---|---|---|
| МП2/CBS | 0.481 | 0.646 | 3.31 | 4.05 | 7.15 |
| CCSD/CBS | 0.393 | 0.578 | 1.61 | 2.05 | 2.94 |
| CCSD(T)/aug-cc-pVTZ | 0.525 | 0.810 | 2.18 | 2.90 | 4.11 |
| CCSD(T)/CBS | 0.519 | 0.805 | 2.24 | 2.94 | 4.14 |
Таблица 2.
Равновесное расстояние C–C (Å) в димерах пергалогенметанов
| Метод / базис | CH4 | CF4 | CCl4 | CBr4 | CI4 |
|---|---|---|---|---|---|
| МП2/CBS | 3.68 | 4.01 | 4.59 | 4.99 | 5.25 |
| CCSD/CBS | 3.74 | 4.04 | 4.86 | 5.20 | 5.61 |
| CCSD(T)/aug-cc-pVTZ | 3.65 | 3.94 | 4.76 | 5.12 | 5.49 |
| CCSD(T)/CBS | 3.65 | 3.94 | 4.73 | 5.10 | 5.43 |
Влияние уровня квантово-химического описания на примере димера метана проиллюстрировано на рис. 2; на рис. 3 приведены сечения ППЭ, полученные в выбранном референсном методе (CCSD(T)/aug-cc-pVTZ + bf[33221]) для всей исследованной серии димеров.
Рис. 2.
Сечения ППЭ димера перфторметана для разных базисных наборов со связевыми функциями и без них.
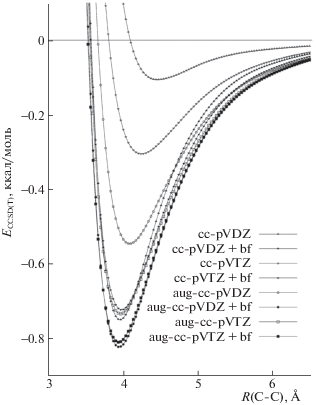
Рис. 3.
Сечения ППЭ димеров (CX4)2, X = H (1), F (2), Cl (3), Br (4), I (5), рассчитанные методом CCSD (T).

Использование двукратно валентно-расщепленных базисных наборов приводит к систематическому росту ошибки от фтора к иоду. При переходе от aug-cc-pVDZ к aug-cc-pVTZ различия между соседними сечениями ППЭ составляют ~1, 1.5, 5, 10 и 20% для X = H, F, Cl, Br и I соответственно.
Основным фактором, влияющим на межмолекулярное взаимодействие в системах (CX4)2, является межмолекулярная электронная корреляция. Таким образом, метод CCSD(T) с эффективным остовным потенциалом, включающим в себя все остовные и субвалентные электроны, с использованием базисного набора aug-cc-pVTZ, дополненного связевыми функциями [33221], дает калибровочные результаты. Значение энергии диссоциации димера перфторметана на 12% отличается от оценки, полученной тем же методом, но с использованием существенно более бедного базисного набора [3]. Полученные в настоящей работе энергии взаимодействия мономеров методом МП2, экстраполированные к бесконечному базисному набору, соответствуют значениям энергии диссоциации перхлорметана и пербромметана 3.523 и 3.702 ккал/моль, оцененным тем же методом [4, 5], но завышают в 1.6 и 1.3 раза соответственно, калибровочные оценки метода CCSD(T).
ПОЛЯРИЗАЦИОННЫЕ КОРРЕЛЯЦИИ
Энергия дисперсионного взаимодействия между молекулами A и B согласно формуле Слэтера–Кирквуда в интерпретации Мавроянниса [24] определяется поляризуемостями αA и αB молекул A и B, расстоянием Re между ними и числом электронов NA и NB, участвующих в ван-дер-ваальсовом взаимодействии:
(1)
${{D}_{{\text{e}}}} = {\text{3}}{{\alpha }_{{\text{A}}}}{{\alpha }_{{\text{B}}}}{{[{\text{2}}R_{{\text{e}}}^{6}({{({{\alpha }_{{\text{A}}}}{\text{/}}{{N}_{{\text{A}}}})}^{{{\text{1}}/{\text{2}}}}} + {{({{\alpha }_{{\text{B}}}}{\text{/}}{{N}_{{\text{B}}}})}^{{{\text{1}}/{\text{2}}}}})]}^{{ - {\text{1}}}}}.$В случае одинаковых молекул CX4 дисперсионная энергия пропорциональна поляризуемости CX4 в степени 1.5. Построенная зависимость ${{D}_{{\text{e}}}}R_{{\text{e}}}^{6}$ от поляризуемости CX4 (X = F, Cl, Br, I) в степени 1.5 представлена на рис. 4 и описывается линейной функцией с коэффициентом корреляции 0.9963. Из параметров линейной зависимости также можно получить эмпирическую оценку числа электронов, которая составила ~31. В соединении CX4 (за исключением метана) присутствует 28 валентных электронов от атомов галогенов, а также по одному электрону можно дополнительно отнести к каждому атому галогена за счет образования полярной ковалентной связи. Таким образом, формулу Слэтера–Кирквуда можно применять для описания сложных систем, рассматривая только валентные электроны атомов, участвующих в ван-дер-ваальсовом взаимодействии, с учетом поправки на дополнительную поляризацию молекулы. Корреляция поляризуемости CX4 с поляризуемостями X, X–, X2, показанная на диаграмме (рис. 5), указывает на то, что первая определяется поляризуемостью заместителей X, средней между поляризуемостью атома X и аниона X–.
Рис. 4.
Зависимость ${{D}_{{\text{e}}}}R_{{\text{e}}}^{6}$ для парного взаимодействия между молекулами CX4 от поляризуемости CX4 (X = F, Cl, Br, I) в степени 1.5.


Таким образом, реализован прецизионный подход для расчета энергии взаимодействия в димерах СX4 (X= H, F, Cl, Br, I). На основании построенных корреляций установлено, что значительный рост поляризуемости атомов в ряду фтор – иод является главным фактором резкого изменения свойств веществ в ряду перфторметан – периодметан. Димер периодметана – неустойчивое химическое соединение – в настоящей работе теоретически изучен впервые. Методом CCSD(T) в пределе бесконечного базисного набора с использованием остовного псевдопотенциала 46MDF определена энергия его диссоциации, которая составила 4.14 ккал/моль.
Список литературы
Molnar L.F., He X., Wang B., Merz K.M. Jr. // J. Chem. Phys. 2009. V. 131. P. 0651002.
Li A. H., Chao S. D. // J. Mol. Struct. THEOCHEM. V. 897. 2009. P. 90.
Biller M.J., Mecozzi S. // Mol. Phys. 2012. V. 110. P. 377.
Te-Li A.H., Huang S.-C., Chao S.D. // J. Chem. Phys. 2010. V. 132. P. 024506.
Mahlanen R., Jalkanen J.-P., Pakkanen T.A. // Ibid. 2005. V. 313. P. 271.
Cybulski S.M., Toczyłowski R.R. // Ibid. 1999. V. 111. P. 10520.
Tao Fu-Ming, Pan Yuh-Kang // Ibid. 1992. V. 97. P. 4989.
Patkowski K., Szalewicz K. // Ibid. 2010. V. 133. № 9. P. 094304.
Patkowski K. // Ibid. 2013. V. 138. P. 154101.
Igel-Mann G., Stoll H., Preuss H. // Mol. Phys. 1988. V. 65. P. 1321.
Bergner A., Dolg M., Küchle W. et al. // Ibid. 1993. V. 80. P. 1431.
Peterson K.A., Figgen D., Goll E. et al. // J. Chem. Phys. 2003. V. 119. P. 11113.
Stoll H., Metz B., Dolg M. // J. Comput. Chem. 2002. V. 23. P. 767.
Peterson K.A., Shepler B.C., Figgen D., Stoll H. // J. Phys. Chem. A. 2006. V. 110. P. 13877.
Bross D.H., Peterson K.A. // Theor. Chem. Acc. 2013. V. 133. P. 1434.
Fleig T, Sadlej A.J. // Phys. Rev. A. V. 2002. V. 65 P. 032506.
Helgaker T., Klopper W., Koch H., Noga J. // J. Chem. Phys. 1997. V. 106. P. 9639.
Buchachenko A.A., Stepanov. N.F. // Ibid. 1997. V. 106. P. 10134.
Buchachenko A.A., Roncero O., Stepanov N.F. // Rus. J. Phys. Chem. 2000. V. 74. P. 193.
Werner H.-J., Knowles P.J., Knizia G. et al. MOLPRO, Version 2010.1, A Package of ab initio Programs (2010).
Au J.W., Burton G.R., Brion C.E. // Chem. Phys. 1997. V. 221. P. 151.
Olney T.N., Cann N.M., Cooper G., Brion C.E. // Ibid. 1997. V. 223. P. 59.
Blanch E.W., Keir R.I., Ritchie G.L.D. // J. Phys. Chem. A. 2002. V. 106. P. 4257.
Mavroyannis C., Stephen M.J. // Mol. Phys. 1962. V. 5. P. 629.
Hättig C., Heß B.A. // J. Chem. Phys. 1998. V. 108. P. 3863.
Holka F., Urban M. // Ibid. 2014. V. 141. P. 214303.
Maroulis G. // Mol. Phys. 1992. V. 77. P. 1085.
Archibong E.F., Thakkar A.J. // Chem. Phys. Lett. 1993. V. 201 P. 485.
Maroulis G., Thakkar A.J. // Mol. Phys. 1991. V. 73 P. 1235.
FI-Kader M.S.A., Kalugina Y.N. // Chem. Phys. Lett. 2015. V. 639. P. 93.
Miller T.M., Bederson B. // Adv. At. Mol. Phys. 1978. V. 13. P. 1.
Gussoni M., Rui R., Zerbi G. // J. Mol. Struct. 1998. V. 447. P. 163.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии