

Журнал физической химии, 2020, T. 94, № 1, стр. 143-152
Мехнизм лазерного флешфотолиза ридимерных e-таутомеров 4-N,N-диэтиламиноазобензола в среде пропан-2-ола
Ю. А. Михеев a, *, Ю. А. Ершов b
a Российская академия наук, Институт биохимической физики им. Н.М. Эмануэля
Москва, Россия
b Московский государственный технический университет им. Н.Э. Баумана
Москва, Россия
* E-mail: mik@sky.chph.ras.ru
Поступила в редакцию 12.03.2019
После доработки 12.03.2019
Принята к публикации 09.04.2019
Аннотация
Проведен анализ разностных транзитных абсорбционных спектров (TAS), полученных при флешфотолизе изопропанольных растворов 4-N,N-диэтиламиноазобензола (DEAB) пучками лазерного излучения длительностью 15 нс с длинами волн λex = 265 и 353 нм. Показана несостоятельность построения механизма транс (t) → цис (c)-фотоизомеризации DEAB на основе идеи о мономерном строении этого красителя. На основе ридимерной концепции аминоазобензольных красителей, к каковым относятся ридимеры DEAB2, дано адекватное объяснение природы сигналов TAS, структура которых кардинально изменяется при изменении длины волны λex. При этом использован метод сопоставления TAS-сигналов DEAB2 с ультравысокоскоростными (субпико- и пикосекундными) сигналами TAS ридимеров референтного красителя аминоазобензола (AAB2). Установлено, что кардинальное различие структуры TAS-сигналов DEAB2, генерируемых импульсами с указанными длинами волн λex, связано с возбуждением разных ридимерных e-таутомеров. При этом малоустойчивый в основном состоянии ридимерный e-таутомер приобретает повышенную устойчивость в пропаноле и, поглощая радиацию с λex = 353 нм, изменяет строение одного из своих мономеров на хиноидное, оставаясь ридимером. Показано, что более устойчивый в основном состоянии ридимерный e-таутомер диссоциирует при возбуждении импульсами радиации с λex = 265 нм на индивидуальные мономеры, один из которых изомеризуется с образованием молекул цис-DEAB.
В работах [1–9] показано, что простые красители на основе аминоазобензола в своем основном состоянии являются ридберговскими димерами (ридимерами). Межмономерная ковалентная связь нового типа в ридимерах образуется в результате спаривания электронов, промотированных с sp2-орбиталей атомов N азогрупп на ридберговские 3s-орбитали азогрупп. При этом хромогенами, определяющими цветность аминоазобензольных красителей, служат принадлежащие ридимерам катионы фениламинильного типа (CPhAT). Эти катионы и поглощающие УФ-излучение фенильные группы ридимеров, способны в результате фотовозбуждения индуцировать фотохимические превращения. Например, на основе анализа спектров резонансной рамановской спектроскопии установлен факт значительного усиления кислотности у дипротонированных ридимеров AABH$_{2}^{{ + + }}$ при фотовозбуждении их катионов CPhAT и фенильных групп [6]. В работах [7–9] впервые объяснена природа спектров транзитных состояний AAB2 и метилоранжа (MOD2), полученных методами ультравысокоскоростной лазерной транзитной абсорбционной спектроскопии (TAS) и ультравысокоскоростных транзитных линз (UTL). При этом были раскрыты собенности t → c-фотоизомеризации AAB2 [7, 8] и причины ее отсутствия у растворенного в воде MOD2 [9].
Новые сведения о ридимерном строении [1–6] аминоазобензольных красителей и их фотовозбужденных состояний [7–9], установленные путем анализа результатов ультравысокоскоростных методик исследования, позволяют улучшить понимание также и результатов менее скоростных (наносекундных) методов. В настоящей работе с целью детализации фотохимических свойств ридимеров и в развитие ридимерной концепции аминоазобензольных красителей используются опубликованные в литературе данные по лазерному флешфотолизу 4-N,N-диэтиламиноазобензола. UV–VIS-спектры продуктов фотолиза этого красителя наблюдали в работе [10], но не получили адекватного объяснения в силу сложившегося представления о мономерном строении фотовозбуждаемых аминоазобензольных красителей. В [10] основному состоянию этого красителя традиционно (и неверно) приписывали формулу мономера DEAB
где R = CH3CH2, тогда как в действительности это – ридимер DEAB2 [5].
Феноменология спектральных трансформаций диэтиламиноазобензола DEAB2в интерпретации [10]
Авторы [10] проводили флешфотолиз изопропанольных растворов DEAB2 с применением неодимового лазера в режиме генерации импульсов излучения с длинами волн λex = 265 и λex = 353 нм длительностью 15 нс и с энергией ≈100 мДж (соответственно 1.33 × 1016 и 1.78 × 1016 фотон/импульс). В работе [10] даны разностные транзитные спектры, зафиксированные в конце импульсов возбуждения. Эти спектры, полученные с раствором DEAB2 в насыщенном азотом изопропаноле и приведенные в [10] на разных рисунках, сопоставлены на рис. 1. Авторы [10] отмечают интенсивную полосу отбеливания в разностном спектре 1 в интервале 380–480 нм и две полосы поглощения при 360 и 510 нм. Этот спектр практически не изменяется в течение 10 мкс после импульса, и замена азота на воздух в растворе практически не отражается на интенсивности и времени жизни наблюдаемого спектра. Природа положительных полос 360 и 510 нм на разностном спектре не обсуждается (ниже показано, что они принадлежат с-DEAB). Полоса отбеливания (рис. 1, λex = 265 нм) приписывается расходованию молекул t-DEAB в результате фотохимического разрыва связей С–NEt2. При этом не исключается возможность образования незначительного количества с-DEAB.
Рис. 1.
Разностные спектры диэтиламиноазобензола (раствор в изопропаноле), полученные в конце импульсов лазерного излучения с длинами волн λex = = 265 (1) и 353 нм (2). Данные [10].

Кроме отмеченных результатов, авторы [10] сообщают о наличии двух видов флуоресценции растворов DEAB2, насыщенных азотом. В частности, излучение с λex = 270 нм приводит к эмиссии с λem = 320 нм, которую связывают с флуоресценцией неизвестной примеси. Другой вид флуоресценции наблюдается при коротком времени задержки после импульса с λex = 265. В этом случае флуоресцентная эмиссия охватывает область λem = 360–440 нм и имеет время полураспада t0.5 = = 20 нс (время жизни τ = 3 × 10–8 с). Характерно, что эта эмиссия вновь появляется после затухания при быстром повторном облучении с λex = = 265, хотя и не достигает начальной интенсивности. Этот вид флуоресценции авторы [10] приписывают неизвестному продукту фотоизомеризации молекул DEAB.
В итоге авторы [10] ограничиваются предположением, что разностный спектр 1 (рис. 1) является суммарным спектром, по крайней мере, двух компонентов, из которых один, возможно, соответствует с-DEAB, а другой неизвестному соединению. При этом подчеркивается, что в соответствии со спектром 1, реакция фотоизомеризации под действием импульса с λex = 265 нм значительно менее важна, чем под действием импульса с λex = 353 нм. (Ниже показано, что имеет место обратная ситуация.)
Авторы [10] отмечают, что транзитная полоса поглощения с λm = 410 нм в разностном спектре 2 (рис. 1, λex = 353 нм) по своему положению практически соответствует полосе исходного красителя. Однако, опираясь на неверно трактуемые данные работ [11, 12], эту полосу неадекватно относят к цис-форме DEAB, образующейся путем фотоизомеризации мнимых молекул t-DEAB. О наличии флуоресценции от излучения с λex = 353 нм не сообщается. Следует отметить, что работы [11, 12], на которые ссылаются авторы [10], посвящены оптическим свойствам транс- и цис-форм азобензола (AB), которые не имеют аминогрупп в качестве заместителей и поэтому не являются референтными при обсуждении фотоиндуцированных реакций аминоазобензольных ридимеров, включая DEAB2 и AAB2. В действительности спектры молекул c-DEAB и c-AAB имеют при λ = = 400–410 нм минимумы поглощения (см. ниже). Это исключает связь полосы поглощения на разностном спектре 2 (рис. 1) с молекулами c-DEAB. Таким образом, трактовки спектров 1 и 2 (рис. 1) нельзя признать обоснованными.
Цель настоящего анализа – обсуждение представленных на рис. 1 данных [10] на основе ридимерного строения DEAB2. Основанием для этого служит механизм фотоизомеризации ридимерного “родительского” красителя – аминоазобензола (AAB2) [7, 8], рассматриваемого нами в качестве референтного соединения.
ФОТОНИКА DEAB2 И AAB2
Сопоставление спектров растворов DEAB2 и AAB2
Авторы [10] приводят спектр DEAB2, растворенного в изопропаноле (рис. 2, кривая 1), вместе с разностными спектрами (рис. 1), не сопровождая их спектрами фотостационарных состояний фотохимических превращений. Учитывая общий характер электронных (e) таутомерных равновесий аминоазобензольных красителей [1–9], мы используем приведенные в [13] спектры референтного красителя AAB2. К последним относятся спектры 2, 3 и 4 (рис. 2) растворов AAB2 в диэтиловом эфире (DEE), метаноле и этиленгликоле (EG) соответственно. Авторы [13] накапливали c-AAB, реализуя фотостационарное состояние (за время ∼ 10 с) с помощью излучения непрерывного лазера мощностью ∼200 мДж/с, λex = 405 нм. На рис. 2 дан спектр 5, характеризующий фотостационарное состояние для AAB2 в DEE, и разностный спектр 6, полученный вычитанием спектра 2 из спектра 5.

Из сопоставления спектров DEAB2 (рис. 2, кривая 1, раствор в изопропаноле) и AAB2 (рис. 2, кривая 2, раствор в DEE) следует, что максимум более широкой абсорбционной полосы DEAB2 (λm = 410 нм) батохромно смещен относительно максимума полосы AAB2 (λm = 380 нм) и, что немаловажно, интенсивность на длинноволновом спаде полосы поглощения у DEAB2 в изопропаноле значительно выше чем у AAB2 в среде DEE.
Авторы [13], отмечая наличие относительно продолжительного спада (“хвоста”) в спектре AAB2 в среде DEE (до λ > 500 нм) (рис. 2, кривая 2), дают для него традиционное объяснение как проявление слабой полосы n → π*-перехода. То же объяснение сохраняется и в отношении более выраженных по интенсивности “хвостов” в спектрах AAB2 в среде метанола (рис. 2, кривая 3) и EG (рис. 2, кривая 4). Между тем, показано, что реально полосы, обусловленные n → π*-переходами в азобензоле (AB) и диметиламиноазобензольных ридимерах, находятся в UV-области спектра при 300–330 нм [4] (что естественно относится и к DEAB2 в изопропаноле). В этой связи наличие указанных “хвостов” и увеличение их оптических плотностей в спиртах нельзя связывать с влиянием полярности среды на интенсивность n → π*-полос, которых нет в данной области спектра. Объяснение данного эффекта следует искать с учетом влияния среды на равновесие в основном состоянии ридимеров между их электронными (e-) таутомерными формами.
e-Таутомерные формы основного состояния ридимеров
Согласно [7–9], для фотопревращений аминоазокрасителей наиболее значимы равновесные e-конфигурации:

В схеме 1: R = H в AAB2 и R = C2H5 в DEAB2, точки под атомами N – электроны на sp2-орбиталях, точки над N – электроны на pz-орбиталях. Между азогруппами мономеров показаны ридберговские ковалентные связи из двух электронов. В каждом мономере sp2-орбиталь, потерявшая электрон, наделяет свой атом N положительным зарядом. На pz-орбиталь этого атома N переходит электрон с pz-орбитали соседнего атома N. В свою очередь, возникшие на pz-орбиталях соседних атомов N положительные заряды поляризуют электронные pz-системы в сопряженных кольцах с образованием катионов фениламинильного типа (CPhAT).
Ридимеры (1б) в схеме 1 образуются вследствие того, что атомы азота аминных групп в (1a) отдают по электрону в соседние фениленовые кольца, которые становятся донорами электронов для CPhAT (Ph+N+) в противолежащих мономерах. В итоге, связь между мономерами в ридимере усиливается, и возникают наиболее устойчивые e-таутомеры (1б), в которых одноэлектронные связи между противолежащими катионами мономеров фиксируют четыре катиона фениламинильного типа [3–9] с положительными зарядами, делокализованными в орто- и пара-положениях фенильных и фениленовых колец. Катионы подобного типа имеют не только связывающие и разрыхляющие молекулярные π-орбитали (МО), но и вакантные несвязывающие МО (НСМО) с нулевой энергией. В таких катионах переход электронов с высших занятых π-орбиталей (ВЗМО) происходит на НСМО под действием VIS-света (π → π*-переход). В результате этого в спектрах красителей AAB2 и DEAB2 появляются полосы поглощения, распространяющиеся в VIS-область. В AAB2 такая полоса имеет λm = = 380 нм (рис. 2, спектр 2), в DEAB2 – λm = 410 нм (рис. 2, спектр 1).
Появление в указанных полосах длинноволновых “хвостов”, интенсивность которых несколько увеличивается в спиртовой среде, можно связать со смещением e-таутомерного равновесия в сторону ридимерной формы (1а), обладающей в случае AAB2 UV-полосой при 320 нм и VIS-полосой при 500 нм [7–9]. Не исключено, что форма (1а) становится более устойчивой в полярной спиртовой среде вследствие изменения в ней характера межмономерных связей и появления у аминогрупп R2N$^{{ \bullet \bullet }}$ определенной степени свободы для образования водородных связей с молекулами спиртов.
Смещение равновесия (1б) в сторону (1а) важно тем, что оно не только приводит к наблюдаемому уширению спектров AAB2 и DEAB2 в спиртовой среде, но и создает запас e-таутомеров (1а), обладающих крайне слабым поглощением света в VIS-полосе e-таутомеров (1б). Повышение в спиртовой среде запаса таутомеров (1а), не абсорбирующих излучения, которое поглощается e-таутомерами (1б), следует учитывать при объяснении результатов флешфотолиза DEAB2. Дело в том, что каждый акт фотопревращения e-таутомера (1б) вызывает акт перехода (1а) → (1б) из наличного запаса e-таутомеров (1а). Сходным образом, расходование таутомера (1а) может стимулировать акт перехода (1б) → (1а). Такое пополнение фотолизующихся ридимеров за счет ридимеров, поглощающих радиацию в другой области спектра, может отразиться на форме разностных транзитных спектров.
Фотодиссоциация и транс → цис-изомеризация ридимеров
Механизм фотопревращения референтного ридимерного красителя AAB2–химической основы простых аминоазобензольных красителей – был установлен в работах [7–9]. В [7–9] показано, что импульсное ультравысокоскоростное лазерное излучение с λex = 400 нм индуцирует изомеризацию AAB2 в цис-аминоазобензол (с-AAB) в результате диссоциации ридимера AAB2 на мономеры. Это излучение абсорбируется преимущественно e-таутомером (1б), диссоциация которого создает две неодинаковые франк-кондоновские (FK) пары мономеров примерно в равных количествах:
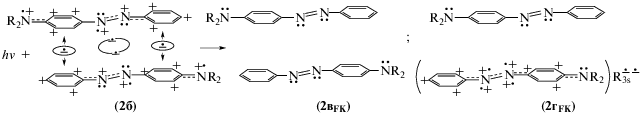
Здесь R = H в AAB2 и R = C2H5 в DEAB2. При этом гомогенная пара (схема 2, (2вFK)) образуется в результате разрыва R3s–R3s-связи фотовозбужденного ридимера (2б), что приводит к возвращению освободившихся электронов на sp2-орбитали атомов N обоих мономеров. Эта пара – промежуточное FK-состояние.
Образование гетерогенной пары (2гFK) связано с тем, что только одна ${\text{R}}_{{3{\text{s}}}}^{ \bullet }$ -орбиталь из разорванной при фотолизе R3s–R3s-связи возвращает электрон на sp2-орбиталь атома N. Вторая одноэлектронная ${\text{R}}_{{3{\text{s}}}}^{ \bullet }$-орбиталь захватывает электрон из одноэлектронной связи ридимера (2б). В результате возникает пара (2гFK), состоящая из мономера t-AAB и поляризованного мономера с двумя катионами фениламинильного типа и с сильно ослабленной связью ${\text{N}}_{{ \bullet + }}^{{ + \bullet }}$ - - ${\text{N}}_{{ \bullet + }}^{{ \bullet + }}$ в азогруппе. Заряды поляризованного мономера уравновешиваются двумя отрицательными зарядами электронов, находящимися на ридберговской орбитали ${\text{R}}_{{3{\text{s}}}}^{{\underline { \bullet \bullet } }}$.
Поляризованные мономеры из пары (2гFK) имеют преимущество вступать в t–c-изомеризацию по сравнению с электронейтральными мономерами t-AAB (с группой N=N в парах (2вFK) и (2гFK)), так как их поляризованные группы ${\text{N}}_{{ \bullet + }}^{{ + \bullet }}$ - -${\text{N}}_{{ \bullet + }}^{{ \bullet + }}$ имеют ослабленную азосвязь в сочетании с сильным кулоновским отталкиванием положительно заряженных атомов азота. Именно их изомеризация с последующей рекомбинацией зарядов объясняет наличие субпикосекундных сигналов TAS (с τ1 = 0.2 пс [7]) и сигналов ультрабыстрых транзитных линз (с τ1 = 0.3 пс [8]), а также около 25% образующихся молекул c-AAB от числа всех мономеров, входящих в AAB2 (остальные 75% мономеров рекомбинируют в AAB2) [7, 8].
Рекомбинация FK гомогенных пар
FK-пары из неполяризованных мономеров (2вFK) превращаются в FK-ридимеры с транзитной полосой при λmax = 525 нм по схеме 3 [7–9].

В схеме 3 каждый мономер в паре (3вFK) отдает на создание межмономерной связи по электрону, промотированному на ридберговскую орбиталь. Образующиеся при этом FK-ридимеры несут на себе бинарные резонаторы из катионов Pћ+N$_{{ \bullet \bullet }}^{ + }$ (3av) и R2N$^{{ \bullet \bullet }}$Pћ+N$_{{ \bullet \bullet }}^{ + }$ ($3{\text{а}}_{2}^{{\mathbf{v}}}$), а также бинарные резонаторы из не имеющих заряда фениленовых и фенильных колец.
В каждой паре резонаторов происходит расщепление π*-уровней одиночных резонаторов (механизм Симпсона [1, 4–8]). По этой причине пары из незаряженных колец обусловливают суммарную широкую полосу при λmax ∼ 320 нм, а пары из заряженных колец – суммарную широкую полосу VIS-поглощения в области 500–750 нм [7–9]. В момент своего образования ридимеры (3av) ↔ ↔ ($3{\text{а}}_{2}^{{\mathbf{v}}}$) получают некоторый избыток колебательной (v) энергии. Избыток v-энергии релаксирует одновременно с релаксацией TAS-сигнала при λmax ∼ 525 нм (за ∼2 пс) в основное электронное, но термически неравновесное состояние (1а) за счет последовательных актов ($3{\text{а}}_{2}^{{\mathbf{v}}}$) → (3av) → → ($1{\text{а}}_{{\text{T}}}^{{\mathbf{v}}}$) → (1аT) [7–9]. После этого происходит релаксация структур типа (1аТ) в состояние равновесия (1аT) → (1а) ↔ (1б) (схема 1).
Что касается одиночных неполярных мономеров t-AAB в парах (2гFK), то их превращение в исходные ридимеры (1б), (2б) требует реализации более или менее длительной стадии самодиффузии для встречи и образования пар, соответствуюших (2вFK), (3вFK). В то же время мономеры t‑AAB обладают слабым UV–VIS-поглощением в спектральной области 375–480 нм (как и у молекул t-AB) по сравнению с исходными ридимерами (1б), (2б). Поэтому их вклад в транзитные и разностные спектры поглощения в области 375–480 нм невелик по сравнению с поглощением ридимеров (1б) и молекул цис-изомеров, образующиеся из мономеров (2гFK).
МЕХАНИЗМЫ ФЛЕШФОТОЛИЗА DEAB2
Изложенные выше основные моменты фотоники ридимеров, характеризуемые схемами 2 и 3, необходимы и достаточны для адекватного обсуждения представленных в [10] разностных спектров флешфотолиза DEAB2.
Механизм фотолиза импульсами с λex = 265 нм
Результат импульсного фотолиза DEAB2, зарегистрированного при действии вспышек с λex = = 265 нм на раствор красителя в изопропаноле представлен в [10] в виде разностного спектра 1 (рис. 1). Этот спектр качественно схож с разностным спектром 6 (рис. 2), который был получен в работе [13], вычитанием исходного спектра 2 (рис. 2) из фотостационарного спектра 5 (рис. 2). (Последний был зарегистрирован непосредственно под действием непрерывного излучения лазера с λex = 405 нм на раствор референтного красителя AAB2 в диэтиловом эфире (DEE)). Для спектров 1 (рис. 1) и 6 (рис. 2) характерно наличие интенсивных полос отбеливания, характеризующих расходование красителей. Исходные спектры обоих красителей представлены на рис. 2 (кривая 1, DEAB2) и на рис. 2 (кривая 2, AAB2). Согласно [13], содержание c-AAB под непрерывным излучением лазера (91.6%) многократно превышает фотостационарную долю исходного ридимера AAB2. Наличие у обоих красителей одинаково высокоинтенсивных полос отбеливания позволяет заключить, что излучение с λex = 265 нм тоже создает молекулы c-DEAB.
Следует отметить, что краситель DEAB2 обладает, как и диметиламинобензол и другие алкилированные по аминному азоту производные AAB2, характерными для диалкиланилиновых хромофоров полосами UV-поглощения при λm = 260 и 307 нм (из них вторая полоса менее интенсивна) [5]. В этой связи можно принять, что действие излучения с λex = 265 нм создает S3 (π,π*)-состояние диалкиланилинового хромофора DEAB2, энергия возбуждения которого конвертируется на S1 (π,π*)-уровень ридимера (1б) по правилу М. Каши, индуцируя тем самым процессы, представленные в схемах 2 и 3.
К сходству сопоставляемых разностных спектров 1 (рис. 1) и 6 (рис. 2) следует также отнести наличие в них положительных полос поглощения по обе стороны от отрицательных полос отбеливания: в случае молекул c-DEAB они находятся при λ = 370 и 500 нм, а в случае молекул c-AAB при λ = 325 и 475 нм. Единственное различие в данном случае заключается в батохромном смещении тех и других полос DEAB2 относительно таких полос AAB2. Сходное батохромное смещение полос имеет место и в спектрах исходных красителей, представленных на рис. 2: кривая 1 (DEAB2) и кривая 2 (AAB2). Оно обусловлено наличием в ридимерах DEAB2 диалкиламинных заместителей Et2N-, влияющих на абсорбционные полосы так же, как и заместители Me2N – в диметиламиноазобензоле [1, 5].
В этой связи следует обратить внимание на то, что у c-AAB положение максимумов полос поглощения (λmax = 330 и 455 нм на рис. 1, спектр 3) [13] соответствуют точкам пересечения спектров 5 и 2 (рис. 2), а также точкам пересечения разностных спектральных полос с осью абсцисс (при нулевой оптической плотности). С учетом этого из разностного спектра 1 (рис. 1) следует, что максимумы поглощения с-DEAB находятся при λmax = 380 и 480 нм. Следует отметить, что у не имеющих аминогрупп молекул c-азобензола (с-AB – референтное соединение для молекул c-AAB и с-DEAB) максимумы расположены в более коротковолновой области: 275 и 435 нм [14] (в [11] – 281 и 433 нм). Следует подчеркнуть, что длинноволновые полосы всех указанных цис-форм, наблюдаемые в VIS-области спектра, обусловлены π → π*-электронными переходами, но не традиционно постулируемыми n → π*-переходами [4, 15]. Поэтому указанный выше факт одновременного батохромного смещения длинноволновых и коротковолновых полос под влиянием электронодонорных аминогрупп отражает именно то, что длинноволновые полосы цис-производных AB соответствуют π → π*-полосам.
Отметим, что высокое содержание молекул c‑AAB (91.6%) в опытах [13] (рис. 2, спектры 5, 6) было получено вследствие их крайне низкого светопоглощения в полосе возбуждающего излучения с λex = 405 нм [13]. Это обеспечивало крайне низкую скорость обратной фотохимической цис → транс-изомеризации c-AAB в фотостационарном состоянии в среде DEE. Осуществленный выше анализ данных рис. 1 (спектр 1) и рис. 2 (спектры 2, 5 и 6), свидетельствует в пользу столь же высокого количества с-DEAB, получаемого в условиях флешфотолиза, и крайне низкой интенсивности поглощения молекул c-DEAB в полосе излучения с λex = 265 нм (в силу ее удаленности от UV-полосы поглощения с-DEAB с λmax = 380 нм). Таким образом, наличие в разностном спектре 1 (рис. 1) интенсивной полосы отбеливания и прилегающих к ней полос поглощения обусловлено, вопреки представлению авторов [10], именно образованием значительного количества с-DEAB.
Механизмы флуоресценции DEAB2 при действии излучения с λex = 270 и 265 нм
В [10] обнаружена флуоресценция на длине волны λem = 320 нм, сопровождающая действие излучения с λex = 270 нм на раствор DEAB2 в изопропаноле, но не детектируемая после окончания облучения. Авторы [10] эту флуоресценцию соотнесли с неизвестной флуоресцирующей примесью. Флуресценция другого вида, в области 360–440 нм, наблюдалась по окончании импульса с λex = 265 нм. Она имела характеристическое время затухания τ ≈ 3 × 10–8 c (период полураспада 20 нс) и вновь появлялась при повторном воздействии излучения с λex = 265 нм, однако не достигала исходной интенсивности. Этот вид флуоресценции приписан [10] продуктам фотоизомеризации DEAB неизвестного строения.
Между тем, на основании схемы 2 можно предположить, что оба вида флуоресценции принадлежат мономерам t-DEAB, образующимся одновременно с поляризованными мономерными предшественниками молекул с-DEAB (пара (2гFK)). В соответствии с этим, скорость образования мономеров t-DEAB должна быть равна скорости образования изомеризующихся поляризованных мономеров и соответственно такой же высокой, как скорость образования с-DEAB. Относительно оптического спектра t-DEAB следует отметить, что он не должен иметь полосы UV-поглощения с λmax = 380 нм, характерной для с-DEAB, в силу специфических хромофорных свойств фенильного и фениленового колец в t-DEAB. Так, хромофорная группа с фенильным кольцом PhN=N– в t-DEAB имеет UV-полосу λm = 259 нм [16], а диэтиланилиновый фрагмент Et2NPh– должен обеспечить полосы UV-поглощения, характерные для молекул бесцветных лейкокарбинолов трифенилметановых красителей и родамина B. Эти лейкокарбинолы тоже несут на себе фрагменты Et2NPh- и Me2NPh- и имеют интенсивную UV-полосу с λm = 263–264 нм вместе с более слабой полосой λm = 300–307 нм [17, 18]. По этой причине следует ожидать, что энергия излучения с λex = 265 нм (270 нм), поглощенная фенильными и фениленовыми группами, может индуцировать изомеризацию мономеров t-DEAB, увеличивая выход c-DEAB в дополнение к изомеризации поляризованных мономеров (2гFK). Кроме того, эта энергия может передаваться путем внутренней конверсии на хромофоры t-DEAB с λm = 300–307 нм, которые и будут эмиттерами флуоресценции с λem = 320 нм (правило М. Каши).
Флуоресценция в области 360–440 нм [10] с временем жизни τ ≈ 3 × 10–8 c соответствует излучательному переходу между π*-уровнем возбужденного состояния S1 и π-уровнем основного состояния S0 [19]. Этот вид флуоресценции тоже можно приписать молекулам t-DEAB, если учесть, что в них азогруппы N=N функционируют так же, как азогруппы N=N в t-AB.
Действительно, азогруппы N=N в t-DEAB обладают, как и в t-AB [15], ридберговскими 3s-орбиталями, которые с небольшой вероятностью генерируют процесс чрезвычайно высокоскоростной e-таутомеризации с образованием катионов фениламинильного типа (CPhAT). Как отмечалось выше, для CPhAT характерна НСМО, уровень которой расположен посередине между уровнями ВЗМО и нижней разрыхляющей МО. Функционирование этих е-таутомерных МО обеспечивает появление абсорбционных полос в интервале 360–450 нм ([1–5, 14, 15]). Следует подчеркнуть, что реализация процесса e-таутомеризации с появлением указанной системы MО способна обеспечить еще один путь передачи энергии e-возбуждения от фенильных и фениленовых абсорберов, возбужденных излучением с λex = 265–270 нм. Такие акты e-таутомеризации с образованием CPhAT реализуются с высокой эффективностью, например, в молекулах t-AB, возбужденных на уровень S2 [15]. Они же способны конкурировать с реализующимися по правилу М. Каши актами конверсии энергии возбуждения на уровни диэтиланилинового фрагмента Et2NPh- (хромофоров t-DEAB с λm = 300–307 нм, эмиттеров флуоресценции с λem = 320 нм). Именно уровни возбужденных состояний CPhAT, как наиболее низкие акцепторы энергии возбуждения, могут быть источником флуоресценции в области 360–440 нм.
Механизм фотолиза импульсами с λex= 353 нм
Результат флешфотолиза DEAB2 излучением с λex = 353 нм представлен в [10] в виде разностного спектра 2 (рис. 1). Он принципиально отличается от разностного спектра 1 (рис. 1), который, согласно изложенному выше, свидетельствует о количественном превращении ридимеров красителя DEAB2 в молекулы с-DEAB. Действительно, в разностном спектре 2 (рис. 1) присутствует интенсивная полоса поглощения при λm = 410 нм в той области спектра, в которой молекулы с-DEAB отличаются крайне незначительным поглощением. Кроме того, в спектре 2 (рис. 1) отсутствуют явные признаки полос поглощения с-DEAB в областях спектра 320–400 и 490–560 нм. Эти факты, вопреки утверждениям авторов [10], фактически аннулируют утверждение о принадлежности данного спектра молекулам с-DEAB.
Очевидно также, что наличие противоположно направленных главных полос в разностных спектрах 1, 2 (рис. 1) отражает принципиально неодинаковые результаты воздействия излучений с λex= 353 и 265 нм на раствор DEAB2 в изопропаноле при равновесии ридимерных форм (1а) и (1б). Выше отмечалось, что содержание формы (1а) ридимеров AAB2 и DEAB2 увеличивается в спиртовой среде, и этот факт следует учитывать при обсуждении спектральных изменений, связанных с фотолизом DEAB2. Дело в том, что ридимеры (1а) могут расходоваться двумя путями. Один из них – e-таутомеризация по схеме 1, направленная на восполнение убыли e-таутомеров (1б), диссоциирующих по схеме 2, другой – расходование за счет прямого их фотовозбуждения, что, в свою очередь, индуцирует также расходование и ридимеров (1б) за счет e-таутомеризации по схеме 1.
Любой из указанных выше способов воздействия на ридимеры (1а) приводит к снижению интенсивности их поглощения, отражаясь на разностных спектрах флешфотолиза в виде полос отбеливания (bleaching) в отрицательных областях соответствующих графиков. Например, в спектре 1 (рис. 1) наблюдается слабая полоса отбеливания в спектральной области 290–320 нм, которую можно связать с трансформацией ридимеров (1а) в ридимеры (1б) при фотолизе излучением с λex = = 265 нм. Эта полоса отбеливания имеет слабую интенсивность вследствие наложения на нее интенсивной полосы поглощения с λm = 380 нм от накапливающихся в большом количестве молекул c-DEAB.
В то же время разностный спектр 2 (рис. 1), полученный при воздействии излучения с λex = 353 нм, содержит интенсивную полосу отбеливания в интервале 300–370 нм и слабую полосу отбеливания в области 490–570 нм. Этот факт четко проявляется по той причине, что в разностном спектре 2 (рис. 1) не наблюдается явного присутствия сигналов от молекул c-DEAB.
Для наглядности на рис. 3, представлены в одной и той же шкале длин волн спектр 2 (из рис. 1) и спектр 1 (из рис. 2) исходной равновесной смеси ридимеров (1а) и (1б). Из рис. 3 следует, что действие импульсов с λex = 353 нм создает положительную по знаку разностную транзитную полосу 2, ставшую значительно уже полосы 1, характерной для равновесной смеси e-таутомеров DEAB2. В данном случае появление разностных полос отбеливания в интервалах 300–370 и 490–570 нм свидетельствует о значительном снижении содержания ридимеров (1а) после импульса с λex = 353 нм. Следует отметить, что длина волны радиации с λex = 353 нм приходится на область максимума UV-полосы ридимеров (1а), тогда как интенсивность поглощения этой радиации ридимерами (1б), должна быть значительно ниже, чем у ридимеров (1а). Об этом свидетельствует фактор значительного сужения интенсивной транзитной полосы с максимумом λm = 410 нм в спектре 2 (рис. 1 и рис. 3). Эта полоса наблюдается в отсутствие явных спектральных признаков молекул c-DEAB, и положение ее максимума соответствует максимуму полосы катионов CPhAT, которая характерна также для ридимеров (1б) (спектр 1 на рис. 2, рис. 3).
Рис. 3.
Спектры красителя DEAB2, растворенного в изопропаноле: 1 – исходный, 2 – разностный, полученный в конце импульса лазерного излучения с длиной волны λex = 353 нм. Данные [10].

Отмеченный факт, а также наличие разностных отрицательных полос отбеливания в интервалах 300–370 и 490–570 нм позволяет предположить, что положительная полоса катионов CPhAT с λm = 410 нм появляется в результате фотопревращения ридимеров (1а), ведущего к значительному снижению их концентрации одновременно с концентрацией ридимеров (1б). В пользу этого свидетельствует совпадение положений максимумов (λm = 410 нм) на положительной полосе спектра 2 (рис. 1, рис. 3) и на отрицательной полосе спектра 1 (рис. 1) ридимеров (1б), исчезнувших под действием импульсов с λex = = 265 нм. Это имеет место при том, что в области при λm = 410 нм ни молекулы c-DEAB, ни ридимеры (1а) практически не обладают поглощением.
Фотопревращение ридимеров (1а) можно представить схемой:

Согласно схеме 4, акт поглощения радиации с λex = 353 нм происходит в системе бинарных фениленовых резонаторов R2N$^{{ \bullet \bullet }}$PћN$_{{ \bullet + }}^{{ \bullet \bullet }}$ ридимеров (1а) за счет π → π*-перехода (S0 → S1). Он ведет к образованию транзитного ридимера (4atr) путем последовательных актов переноса двух электронов с pz-орбитали азота аминогруппы ридимера (1a) на заряженную pz-орбиталь азота азогруппы. При этом сначала происходит акт переноса электрона между атомами азота в азогруппе N$_{{ \bullet + }}^{{ \bullet \bullet }}$–N$_{{ \bullet \bullet }}^{ + }$ → N$_{{ \bullet + }}^{{ \bullet + }}$–N$_{{ \bullet \bullet }}^{ \bullet }$ (в схеме не показано), и на азоте рядом с фениленовой группой появляется заряженная pz-орбиталь. Эта заряженная pz-орбиталь акцептирует электрон с pz-орбитали азота аминогруппы и в результате появляется промежуточное возбужденное состояние (4а*). Далее таким же образом происходит перенос второго электрона с pz-орбитали азота аминогруппы ридимера (1a). В конечном итоге возникает состояние (4аtr) с положительно заряженной хиноидной структурой и катионом CPhAT, единственным абсорбером радиации с λm = 410 нм. Другие фрагменты ридимера (4аtr), а именно, фенильная и фениленовая группы, а также хиноидная структура, не имеют поглощения в области спектра с λm = 410 нм [1–3, 5]. Таким образом, образование ридимера (4аtr) с одним катионом CPhAT объясняет одновременное появление интенсивной положительной полосы и двух отрицательных полос на транзитном спектре 2 (рис. 1, рис. 3). Другими словами, радиация с λex = 353 нм вызывает смещение равновесия (1б) → (1a) + hv353 → (4a*) → (4аtr). Затем термически неустойчивый ридимер (аtr) релаксирует в состояние равновесия (схема 1) по мере охлаждения раствора, нагретого импульсом поглощенного излучения.
Таким образом, адекватно объяснены причины спектроскопических различий, наблюдавшихся при лазерном флешфотолизе диэтиламиноазобензола излучением с длинами волн λex = = 265 и 353 нм, благодаря, во-первых, учету ридимерного строения данного красителя и, во-вторых, учету аналогии между его транзитными электронными таутомерами и транзитными e-таутомерами референтного красителя аминоазобензола. Представленный механизм флешфотолиза ридимерного DEAB2 раскрывает несостоятельность сделанной в [10] попытки описать механизм на основе традиционной идеи о мономерной природе основного состояния аминоазобензольных красителей. Следует также отметить, что экспериментальные данные метода лазерного наносекундного флешфотолиза позволяют дополнить и детализировать стадии фотопревращения DEAB2 благодаря их комплексному анализу вместе с данными методов ультравысокоскоростной спектроскопии.
Список литературы
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Журн. физ. химии. 2017. Т. 91. № 4. С. 672.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Там же. 2017. Т. 91. № 10. С. 1683.
Михеев Ю.А., Ершов Ю.А. // Там же. 2018. Т. 92. № 2. С. 267.
Михеев Ю.А., Ершов Ю.А. // Там же. 2018. Т. 92. № 8. С. 1251.
Михеев Ю.А., Ершов Ю.А. // Там же. 2018. Т. 92. № 10. С. 1552.
Михеев Ю.А., Ершов Ю.А. // Там же. 2019. Т. 93. № 2. С. 313.
Михеев Ю.А., Ершов Ю.А. // Там же. 2019. Т. 93. № 6. С. 946.
Михеев Ю.А., Ершов Ю.А. // Там же. 2019. Т. 93. № 7. С. 1111.
Михеев Ю.А., Ершов Ю.А. // Там же. 2019. Т. 93. № 11. С. 1746.
Allen N.S., Binkley J.P., Parsons B.J. et al. // J. Photochemistry. 1984. V. 26. № 2–3. P. 213.
Gerson F., Heilbronner E., van Veen A., Wepster B.M. // Helvetica Chim. Acta. 1960. V. 43. № 7. P. 1889.
Schulze J., Gerson F., Murrel J.N., Heilbronner E. // Ibid. 1961. V. 44. № 2. P. 428.
Joshi N.K., Fuyuki M., Wada A. // J. Phys. Chem. B. 2014. V. 118. № 7. P. 1891.
Nägele T., Hohe R., Zinth W., Wachtveitl J. // Chem. Phys. Lett. 1997. V. 272. № 5, 6. P. 489.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Журн. физ. химии. 2015. Т. 89. № 11. С. 1773.
Robin M.B., Simpson W.T. // J. Chem. Phys. 1962. V. 36. № 3. P. 580.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Журн. физ. химии. 2008. Т. 82. № 9. С. 1770.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Там же. 2010. Т. 84. № 10. С. 1949.
Турро Н. Молекулярная фотохимия. М.: Мир, 1967.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии