

Журнал физической химии, 2020, T. 94, № 12, стр. 1752-1766
Химическое осаждение сульфидов металлов из водных растворов: от тонких пленок до коллоидных частиц
Н. С. Кожевникова a, b, *, В. Ф. Марков a, Л. Н. Маскаева a
a Уральский федеральный университет имени первого Президента России Б.Н. Ельцина
Екатеринбург, Россия
b Российская академия наук, Уральское отделение, Институт химии твердого тела
Екатеринбург, Россия
* E-mail: kozhevnikova@ihim.uran.ru
Поступила в редакцию 03.03.2020
После доработки 25.03.2020
Принята к публикации 14.04.2020
Аннотация
В обзоре представлены основные достижения последних лет по синтезу полупроводниковых сульфидов металлов методом химического осаждения из водных растворов одновременно с двух точек зрения: химической и коллоидно-конденсационной. Показано, что данный метод является наиболее универсальным и перспективным приемом синтеза различных форм сульфидов металлов, включая поликристаллические тонкие пленки, наноструктурированные порошки, изолированные частицы в коллоидных растворах. Установлено, что в зависимости от вида сульфидизатора, условий проведения процесса можно получать сульфиды металлов с различным размером частиц. Рассмотрены основные механизмы химических реакций, протекающих в водных растворах при образовании сульфида металла, и коллоидно-химические механизмы формирования различных поликристаллических структур на его основе.
Методу химического осаждения сульфидов металлов из водных растворов с применением тиоамидов уже более полутора веков. Первым полупроводниковым соединением, полученным этим методом, был сульфид свинца PbS [1], а первые публикации относятся к концу XIX в. и первым десятилетиям XX в. [1–4]. Именно в это время благодаря работам Дж. Рейнольдса [2], Г. Брюкмана [3], Ф. Кисински [4] и Х. Пика [5] химическое осаждение становится достаточно известным. В середине 40-х гг. XX в. этот метод был впервые использован для получения тонких пленок PbS, предназначенных для детекторов инфракрасного излучения [6, 7], и впоследствии нашел широкое применение для изготовления высокочувствительных ИК-датчиков [7].
В 30–50-е гг. ХХ в. на кафедре физической и коллоидной химии Уральского политехнического института (УПИ) имени С.М. Кирова проф. С.Г. Мокрушиным создается и активно развивается новое для тех лет направление “Экспериментальные и теоретические исследования ламинарных систем”, которое связано с исследованием процессов самопроизвольного образования тонких пленок на границе раздела жидкость–газ [8–11] и жидкость–твердое тело [12, 13]. Именно в те годы было установлено, что эти процессы имеют коллоидно-химический характер и аналогичны процессам получения пленок катодным распылением или испарением в вакууме [12, 14]. Проведенные исследования свидетельствовали об универсальности механизма образования пленок нерастворимых соединений и впервые показали, что его следует рассматривать именно с коллоидно-химической точки зрения [13]. Подобный взгляд на механизм образования пленок на межфазных границах, как будет показано ниже, остается актуальным и сегодня.
На 60–80-е гг. ХХ в. приходится первая волна работ по химическому осаждению из растворов. В лаборатории тонких пленок Индийского технологического института в Дели под руководством К.Л. Чопры и С.Р. Даса удалось разработать ряд перспективных технологий изготовления тонкопленочных солнечных элементов [15–23]. Развитие метода связано также с работами Д.И. Боуда с соавт., выполненными в научно-исследовательском центре г. Санта-Барбара США [7, 24], а также с работами основателя воронежской школы химиков-неоргаников проф. Я.А. Угая в ВГУ [25, 26]. Огромный вклад в формирование научных основ химического осаждения из растворов внесли исследования, проведенные в Уральском политехническом институте им. С.М. Кирова под руководством проф. Г.А. Китаева, ученика проф. С.Г. Мокрушина, и одного из основателей уральской школы тонкопленочного синтеза [9, 27–35].
В ответ на бытовавшее в середине 60-х годов прошлого века мнение о принадлежности химического способа получения сульфидных пленок скорее к искусству, чем к науке, Г.А. Китаев в [35] отмечал его связь с недостатком знаний о химических процессах в водных растворах. Он считал, что опыт, накопленный кафедрой физической и коллоидной химии УПИ, позволяет говорить о возможности эффективно управлять ими.
Именно изучение физико-химических закономерностей процессов образования осадков и получения пленок халькогенидов металлов химическим осаждением было главной целью работ, проводимых под руководством проф. Китаева Г.А. [35]. Еще одной, не менее важной задачей, было “привлечение внимания физиков и технологов, работающих в различных областях радиоэлектроники, к тонким пленкам халькогенидов элементов I–V групп периодической системы Д.И. Менделеева…” [35]. За три четверти века уральскими химиками были проведены исследования и выполнены разработки, послужившие основой как для фундаментального понимания процессов химического осаждения, так и для создания ряда технологий нанесения тонких пленок в промышленности. До сих пор данный метод остается одним из наиболее популярных при синтезе халькогенидов металлов. По этой причине представляется важным обобщить имеющиеся на сегодняшний день представления об этом методе и о физико-химических механизмах процессов, протекающих в растворе и на межфазной поверхности при образовании различных форм сульфидов металлов.
Для оценки интереса современных исследователей к данной тематике нами был проведен анализ научных публикаций, посвященных химическому осаждению за период 1975–2019 гг. с использованием библиографической базы данных Web of Science (WoS) на платформе Web of Knowledge. Анализ проведен по работам, в названии, аннотации и ключевых словах которых был указан термин “Chemical bath deposition”. Кроме того, проанализировано распределение работ по тематикам прикладного значения сульфидов металлов путем поиска по таким терминам, как “Thin solid films”, “Sulfide thin film solar cells”, “Sulfide photocatalysis” и “Sulfide nanoparticles and colloidal quantum dots”.
К концу ХХ в. метод химического осаждения активно применялся для получения как легированных, так и нелегированных тонкопленочных материалов для изготовления инфракрасных детекторов, химических сенсоров, светодиодов, фотопотенциометров, полупроводниковых лазеров. Однако в последние два десятилетия наблюдается “ренессанс” химического осаждения из растворов (рис. 1, кривая 2 (thin solid films)). С середины 90-х гг. изменения хода кривой 1 (сhemical bath deposition) практически повторяют изменения кривых 3, 4 и 5 (sulfide thin film solar cells, sulfide photocatalysis и sulfide nanoparticles and colloidal quantum dots).
Рис. 1.
Распределение публикаций, посвященных химическому осаждению, за период 1975–2019 гг. по данным библиографической базы данных научно-технической информации WoS на платформе Web of Knowledge; 1 – chemical bath deposition, 2 – thin solid films, 3 – sulfide thin film solar cells, 4 – sulfide photocatalysis, 5 – sulfide nanoparticles and colloidal quantum dots.
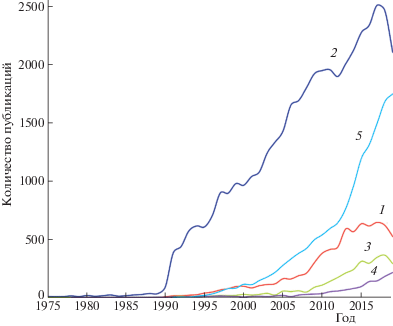
Возобновление интереса к методу связано, во-первых, с успехами в осаждении материалов для тонкопленочных преобразователей солнечного излучения четвертого поколения на основе соединений Сu–Zn–Sn(S,Se) с кестеритной структурой, имеющих по лабораторным данным КПД преобразования от 10 до 18% [36, 37] и теоретический предел до 32% [38, 39], что делает их лидерами среди аналогов по эффективности, доступности и стабильности работы (рис. 1, кривая 3). Во-вторых, после 1996 г. интерес к методу химического осаждения из растворов вышел далеко за пределы тематики тонких пленок в целом и тонкопленочных солнечных элементов в частности. Причина растущего интереса к методу заключалась в возможности получения сульфидных частиц из растворов в наноструктурированном состоянии, что может приводить к значительному изменению их физико-химических свойств [40]. Полупроводниковые нанокристаллы элементов II–VI и III–V групп Периодической системы и имеющие размеры меньше радиуса экситона Бора [41], представляют собой класс квазинульмерных объектов – “квантовых точек” (КТ). Вследствие пространственного ограничения носителей заряда или экситонов в них возникает эффект размерного квантования энергетического спектра [42], который обеспечивает проявление уникальных оптических и электронных свойств. Успехи химического синтеза коллоидных КТ заданного размера позволили их внедрять в органические и неорганические матрицы, открыли широкие возможности практического использования КТ для создания фотовольтаических систем, светоизлучающих диодов, полупроводниковых лазеров, хемо- и биосенсоров и биомедицинских приложений [42–49], что привело после 2000 г. к резкому росту публикационной активности в этой области (рис. 1, кривая 5).
В-третьих, гетерогенный фотокатализ с использованием полупроводниковых материалов на основе сульфидных частиц находит все большее применение в окислительно-восстановительных процессах [50–52], например, в реакциях фотокаталитического восстановления CO2 [52–56], фотоэлектрохимических реакциях образования циклических соединений из низкомолекулярных органических веществ [57] и в синтезе молекул пребиотиков [58–63].
Таким образом, рост исследований в области использования сульфидов металлов в различных сферах современной техники и их новые функциональные приложения спровоцировали возвращение интереса к методу химического осаждения из водных растворов как одному из перспективных. Большинство работ, в том числе и обзорных, посвященных методу химического осаждения касается его использования для получения тонких пленок на металлических, полупроводниковых и изолирующих поверхностях различной природы и конфигурации [64–72].
В настоящем обзоре авторы, опираясь на достижения последних лет в области синтеза, представляют метод химического осаждения как универсальный метод получения сульфидов металлов не только в виде тонкопленочных структур, но и в виде наночастиц и коллоидных КТ. При этом метод рассматривается одновременно с двух точек зрения: химической (непосредственно как химический процесс) и конденсационно-коллоидной (рассматривающей закономерности и особенности образования различных сульфидных форм). Такой подход позволяет глубже разобраться в сути процессов, протекающих при образовании сульфида металла и формировании различных структур на его основе. Первая часть обзора посвящена механизмам химических реакций, протекающих в водном растворе при образовании труднорастворимых сульфидов. Во второй части описаны физико-химические закономерности формирования отдельных сульфидных форм: поликристаллических тонких пленок, наноструктурированных частиц и коллоидных КТ.
ОБРАЗОВАНИЕ СУЛЬФИДА МЕТАЛЛА В ВОДНОМ РАСТВОРЕ: МЕХАНИЗМЫ ХИМИЧЕСКИХ РЕАКЦИЙ
Реакционная смесь при химическом осаждении халькогенидов металлов, в том числе и сульфидов, представляет собой водный раствор одной или нескольких солей металла Меn+, сульфидизатора (источника серы) и комплексообразующего агента. Упрощенно образование сульфида металла можно описать следующими четырьмя стадиями [71]:
1) образование равновесной гомогенной системы “комплексообразующий агент – вода”;
2) образование и диссоциация комплексных форм металла [MеLi]n+ik (где L – один или несколько лигандов, n – степень окисления металла Ме, i – число лигандов, k – заряд лиганда);
3) гидролитическое разложение сульфидизатора и/или взаимодействие его или продуктов разложения с комплексными формами металла;
4) реакция образования сульфида металла.
С целью регулирования скорости зарождения сульфидной фазы, а также для предотвращения процесса гидролиза водорастворимой соли металла, его предварительно связывают в комплексы за счет добавления различных комплексообразующих агентов, физико-химическая природа которых во многом определяет скорость и механизм процесса образования сульфида. В результате процесса гидролитического разложения сульфидизатора (стадия 3) происходит взаимодействие продуктов его разложения с реакционноспособными ионами металла; образования твердой фазы сульфида MеmSn на стадии 4 приводит к сдвигу равновесия процесса диссоциации комплексных ионов и снижению их концентрации. Подобный механизм реализуется при образовании труднорастворимых соединений, так как стадия 4 представляет собой практически необратимую реакцию.
В качестве сульфидизатора можно использовать любое соединение, которое в водном растворе может быть источником ионов S2–, например, тиоамиды, водорастворимые тиосульфаты, сульфиды и гидросульфиды щелочных и щелочно-земельных металлов или сероводород [64–79]. Универсальный сульфидизатор подобрать не удается, поскольку для каждого из них характерны определенные скорость и механизм разложения и формирования сульфида, а также оптимальные условия по щелочности среды, температуре, при которых происходит образование сульфида металла в виде пленки, порошка, коллоидного раствора. Как будет показано ниже, для получения сульфида металла в тонкопленочном состоянии одно из необходимых условий – низкая скорость поступления ионов S2– в зону реакции. Этому условию удовлетворяет группа органических серусодержащих соединений – тиоамидов, среди которых наиболее широкое использование получили тиокарбамид (диамид тиоугольной кислоты, тиомочевина) (NH2)2CS и его производные (тиосемикарбазид H2NCSNHNH2, тиокарбазид (NH2)2(NH)2CS, тиоацетамид CH3CSNH2, N-аллилтиомочевина C3H5NHCSNH2, ацетилтиомочевина C3H6N2OS) [72–79]. При этом изолированные наночастицы сульфидов можно получать с использованием более широкого круга сульфидизаторов, в том числе являющихся сильными электролитами. По типу сульфидизатора химическое осаждение можно условно разделить на гомогенное и гетерогенное. Принципиальное отличие первого от второго состоит в том, что осадитель (ион S2–) не приливают к раствору, содержащему соль металла, а он постепенно образуется за счет протекающих реакций разложения сульфидизатора в объеме раствора.
Существующие современные представления о химизме взаимодействия ионов металла с сульфидизатором и гидролитическом разложении сульфидизатора при гомогенном осаждении (стадию 3) можно описать следующими механизмами [72]:
– механизм комплексообразования, на первой стадии предполагается образование комплексов металла с сульфидизатором типа Ме[(NH2)2CS]$_{m}^{{n + }}$ либо [х(NH2)2CS · уМе(OH)2], которые в последующем диссоциируют с образованием сульфида металла;
– механизм диссоциации, на первом этапе идет гидролитический распад сульфидизатора, в результате чего образуются анионы S2– или HS–, которые взаимодействуют с комплексными ионами металлов, в результате чего образуются соответствующие сульфиды [Mе(L) ]n+ik + S2– → MеmSn.
Остановимся на этих точках зрения более подробно.
Механизм комплексообразования
Одним из первых исследователей механизма процесса образования сульфидов из водных растворов был Г. Брюкман [3], придававший большое значение образованию в водных щелочных растворах комплексных соединений тиомочевины с ионами свинца с координацией через серу: (NH2)2–C = S–Pb. Впоследствии точку зрения об образовании промежуточных комплексов типа (NH2)2CSs–Men+–OH$_{h}^{ - }$ (где s – количество растворенных молекул (NH2)2CS, h – количество гидратированных анионов ОН–, образующих комплекс) разделили и другие исследователи [4, 5, 80–82]. Сульфидизатор может также адсорбироваться на поверхности твердых частиц Cd(OH)2 либо на частицах состава [(CdOH)2(NH3)2], формируя поверхностные метастабильные комплексные соединения, которые затем разлагаются с образованием сульфида [83, 84].
С использованием квантово-химических расчетов было показано [85–88], что нуклеофильное присоединение к углероду гидроксид-иона активирует разложение (NH2)2CS. Образование комплекса [(NH2)2CS–OH]– приводит к появлению на молекуле (NH2)2CS адиабатической зарядовой флуктуации, вызывающей избирательное возбуждение связи S=C. Рассчитанная энергия связи сера–углерод уменьшается при этом с 10.7 до 8.9 эВ. Это приводит к повышению реакционной способности (NH2)2CS по отношению к халькофильным элементам за счет облегчения отщепления серы. Присутствие иона ОН– облегчает реакцию образования сульфида, при этом можно предположить следующий механизм реакции [72, 85] для случая двухвалентного металла. Сначала происходит атака гидроксид-иона на атом углерода в комплексе [(NH2)2CS–Ме]2+, в результате чего нарушается π-система центрального фрагмента (NH2)2CS и образуется sp3-гибридизованный атом углерода, характеризующийся тетраэдрической координацией
Далее происходит разрыв связи С–S, отщепляется сульфид металла и одновременно образуется цианамид или мочевина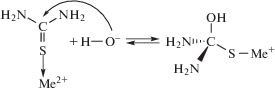
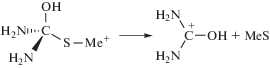
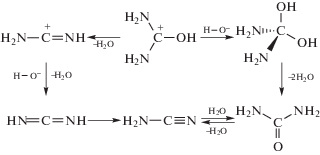
Таким образом, в механизме комплексообразования реакционные комплексы металла с тиоамидом рассматриваются как обязательные переходные (промежуточные) состояния в процессах синтеза конечного продукта. Лимитирующей стадией является трансформация молекулы (NH2)2CS, адсорбированной на атоме металла, в результате которой образуется прочная химическая связь Ме–S (вместо координационной (NH2)2CS–Ме) и разрывается связь S–C в молекуле (NH2)2CS.
Механизм диссоциации
Анализ публикаций, касающихся гидролитического разложения (NH2)2CS, показывает, что экспериментально установлено более двух десятков конечных продуктов и интермедиантов процесса. Среди них наиболее часто встречаются сероводород, цианамид, мочевина, гуанилмочевина, гуанидин, карбонат аммония [72, 89–92]. Однако, по мнению большинства исследователей, основными продуктами разложения (NH2)2CS при температурах вблизи комнатных все же являются сероводород H2S и цианамид H2CN2, а процесс может быть обратим [35, 90–92]
(1)
${{{\text{(N}}{{{\text{H}}}_{{\text{2}}}}{\text{)}}}_{{\text{2}}}}{\text{CS}} \Leftrightarrow {{{\text{H}}}_{{\text{2}}}}{\text{S}} + {{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{N}}}_{2}}.$(2)
$\begin{gathered} {{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{N}}}_{{\text{2}}}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \to {{({{{\text{H}}}_{{\text{2}}}}{\text{N}})}_{2}}{\text{CO}}{\kern 1pt} ; \\ {{({{{\text{H}}}_{{\text{2}}}}{\text{N}})}_{2}}{\text{CO}} + 2{\text{O}}{{{\text{H}}}^{ - }} \to 2{\text{N}}{{{\text{H}}}_{3}} + {\text{CO}}_{3}^{{2 - }}. \\ \end{gathered} $(3)
${{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{N}}}_{2}} + {\text{N}}{{{\text{H}}}_{{\text{4}}}}{\text{OH}} = {{({{{\text{H}}}_{{\text{2}}}}{\text{N}})}_{2}}{\text{C}}{\kern 1pt} = {\kern 1pt} {\text{NH}}_{2}^{ + } + {\text{O}}{{{\text{H}}}^{--}}.$(4)
${{{\text{(N}}{{{\text{H}}}_{{\text{2}}}}{\text{)}}}_{{\text{2}}}}{\text{CS}} \Leftrightarrow {\text{NH}}_{4}^{ + } + {\text{SC}}{{{\text{N}}}^{--}}.$(5)
$\begin{gathered} ({\text{C}}{{{\text{H}}}_{{\text{3}}}}){\text{NHC}}({\text{S}}){\text{NH}}({\text{C}}{{{\text{H}}}_{{\text{3}}}}) + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \Leftrightarrow \\ \Leftrightarrow ({\text{C}}{{{\text{H}}}_{{\text{3}}}}){\text{NHC}}({\text{O}}){\text{NH}}({\text{C}}{{{\text{H}}}_{{\text{3}}}}) + {{{\text{H}}}_{{\text{2}}}}{\text{S}}; \\ \end{gathered} $(6)
$\begin{gathered} ({{{\text{C}}}_{{\text{3}}}}{{{\text{H}}}_{{\text{5}}}}){\text{NHC}}({\text{S}}){\text{N}}{{{\text{H}}}_{{\text{2}}}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \Leftrightarrow \\ \Leftrightarrow ({{{\text{C}}}_{{\text{3}}}}{{{\text{H}}}_{{\text{5}}}}){\text{NHC}}({\text{О}}){\text{N}}{{{\text{H}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{S}}{\kern 1pt} . \\ \end{gathered} $Относительно недавно для синтеза сульфидов металлов в кислой среде при рН < 4 стали применять тиосульфат натрия Na2S2O3. Изучению устойчивости тиосерной кислоты в водных растворах посвящено много публикаций, обзор которых содержится в монографии [95]. Из многочисленных схем и гипотез о механизме процесса признание получили представления о двух первичных реакциях: распад тиосерной кислоты под действием сильных кислот на элементарную серу и сернистую кислоты и распад на сероводородную и серную кислоты в результате гидролиза [78, 79]:
(7)
${{{\text{H}}}_{{\text{2}}}}{{{\text{S}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{3}}}} \Leftrightarrow {\text{S}} + {{{\text{H}}}_{{\text{2}}}}{\text{S}}{{{\text{O}}}_{3}},$(8)
${{{\text{H}}}_{{\text{2}}}}{{{\text{S}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{3}}}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \Leftrightarrow {{{\text{H}}}_{{\text{2}}}}{\text{S}} + {{{\text{H}}}_{{\text{2}}}}{\text{S}}{{{\text{O}}}_{4}}.$Еще один сульфидизатор, о котором стоит упомянуть – это тиоацетамид CH3CSNH2, распространенный в аналитической практике реагент для осаждения сульфидов металлов. Это соединение в отличие от упомянутых выше сульфидизаторов, подвергается гидролизу во всей области рН. Однако взгляды на механизм гидролиза CH3CSNH2 у современных групп исследователей расходятся, часть из которых [76] придерживается классической реакции [96, 97]:
(9)
${\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CSN}}{{{\text{H}}}_{{\text{2}}}}\, + \,{\text{2}}{{{\text{H}}}_{{\text{2}}}}{\text{O}}\, \Leftrightarrow \,{\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{COOH}}\, + \,{{{\text{H}}}_{{\text{2}}}}{\text{S}}\, + \,{\text{N}}{{{\text{H}}}_{3}},$(10)
${\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CSN}}{{{\text{H}}}_{{\text{2}}}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \Leftrightarrow {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CON}}{{{\text{H}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{S}},$(11)
${\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CSN}}{{{\text{H}}}_{{\text{2}}}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \Leftrightarrow {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CSОH}} + {\text{N}}{{{\text{H}}}_{3}},$Другие авторы считают [98], что появление ионов S2– в растворах тиоацетамида обусловлено реакцией
(12)
${\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CSN}}{{{\text{H}}}_{{\text{2}}}} \Leftrightarrow {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CN}} + {{{\text{H}}}_{{\text{2}}}}{\text{S}}.$Количественной характеристикой реакции разложения любого сульфидизатора является термодинамическая константа равновесия, выраженная через равновесные концентрации участников реакции. Например, для реакции (1)
(13)
${{K}_{c}} = \frac{{{\text{[}}{{{\text{H}}}_{{\text{2}}}}{\text{S][}}{{{\text{H}}}_{{\text{2}}}}{\text{NCN]}}}}{{{\text{[(NH}}{{{\text{)}}}_{{\text{2}}}}{\text{CS]}}}}.$(14)
${\text{p}}{{K}_{c}}(\left( {{\text{N}}{{{\text{H}}}_{2}}{{)}_{2}}{\text{CS}}} \right) = \frac{{12\,554}}{T} - 14.255.$Механизм диссоциации сульфидизатора и экспериментальное определение величины Kс легли в основу метода термодинамического расчета условий осаждения сульфидов металлов с использованием тиоамидов, предложенного Г.А. Китаевым с сотр. [90]. Метод позволяет до начала эксперимента расчетным путем, используя при этом справочные термодинамические константы устойчивости комплексных форм и константы ионизации компонентов системы, величины произведений растворимости, определить условия образования сульфида металла, а также его труднорастворимых солей в выбранной реакционной смеси. Основные принципы термодинамического метода расчета граничных условий образования сульфидов металлов подробно представлены в [72, 101, 102]. Такой подход открыл широкие возможности для прогнозирования возможности осаждения сульфида металла как в виде осадка, так и в виде слоев/пленок на твердых подложках.
Вне зависимости от механизма взаимодействия катионов металла с сульфидизатором при гомогенном химическом осаждении сульфида металла молекулы сульфидизатора, взаимодействуя с водой, подвергаются гидролизу. Скорость этого процесса во многом определяется щелочностью среды. Так, результирующую химическую реакцию образования сульфида двухвалентного металла с использованием тиомочевины и нейтрального лиганда L можно представить следующим образом:
(15)
$\begin{gathered} {\text{МеL}}_{x}^{{2 + }} + {{({\text{N}}{{{\text{H}}}_{{\text{2}}}})}_{2}}{\text{CS}} + 2{\text{О}}{{{\text{Н}}}^{--}} = \\ = {\text{МеS}}{\kern 1pt} \downarrow + \;х{\text{L}} + {{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{N}}}_{{\text{2}}}} + 2{{{\text{H}}}_{{\text{2}}}}{\text{O}}. \\ \end{gathered} $Механизм образования твердой фазы
С учетом описанных выше представлений о взаимодействии ионов металла с сульфидизатором и его гидролизе возможные механизмы образования твердой фазы сульфида, например, с использованием (NH2)2CS, сводятся к следующим четырем [70, 103].
1) Ионный механизм
– диссоциация комплексного иона металла
(16)
${{[{\text{Ме}}{{{\text{L}}}_{i}}]}^{{2 + }}}^{{ik}} \Leftrightarrow {\text{М}}{{{\text{е}}}^{{2 + }}} + i{{{\text{L}}}^{k}},$– гидролиз сульфидизатора
(17)
${{({\text{N}}{{{\text{H}}}_{{\text{2}}}})}_{2}}{\text{CS}} + 2{\text{O}}{{{\text{H}}}^{ - }} \Leftrightarrow {{{\text{S}}}^{{2 - }}} + {{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{N}}}_{2}} + 2{{{\text{H}}}_{{\text{2}}}}{\text{O}},$– реакция ионного обмена
(18)
${\text{М}}{{{\text{е}}}^{{2 + }}} + {{{\text{S}}}^{{2--}}} = {\text{МеS}}{\kern 1pt} \downarrow {\kern 1pt} .$2) Кластерный (гидроксидный) механизм
– образование кластеров гидроксида металла
(19)
$n{\text{M}}{{{\text{e}}}^{{2 + }}} + 2n{\text{O}}{{{\text{H}}}^{--}} \Leftrightarrow {{[{\text{Me}}{{({\text{OH}})}_{{\text{2}}}}]}_{n}},$– гидролиз сульфидизатора по реакции (17),
– реакция ионного обмена
(20)
${{[{\text{Me}}{{({\text{OH}})}_{2}}]}_{n}} + {{{\text{S}}}^{{2--}}} = n{\text{MeS}}{\kern 1pt} \downarrow + \;2n{\text{O}}{{{\text{H}}}^{--}}.$3) Ионный механизм диссоциации промежуточного комплекса
– образование комплексного соединения металла с (NH2)2CS
(21)
${{({\text{N}}{{{\text{H}}}_{{\text{2}}}})}_{2}}{\text{CS}} + {\text{M}}{{{\text{e}}}^{{2 + }}} \Leftrightarrow {{[{{({\text{N}}{{{\text{H}}}_{{\text{2}}}})}_{2}}{\text{CS}}{\kern 1pt} --{\kern 1pt} {\text{Me}}]}^{{2 + }}},$– диссоциация комплексного соединения
(22)
$\begin{gathered} {{[{{({\text{N}}{{{\text{H}}}_{2}})}_{2}}{\text{CS}}{\kern 1pt} --{\kern 1pt} {\text{Me}}]}^{{2 + }}} + 2{\text{O}}{{{\text{H}}}^{ - }} = \\ = {\text{MeS}}{\kern 1pt} \downarrow + \;{{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{N}}}_{2}} + 2{{{\text{H}}}_{{\text{2}}}}{\text{O}}. \\ \end{gathered} $4) Кластерный механизм диссоциации промежуточного комплекса
– образование промежуточных форм гидроксида металла с (NH2)2CS
(23)
$\begin{gathered} {{[{\text{Me}}{{({\text{OH}})}_{2}}]}_{n}} + {{({\text{N}}{{{\text{H}}}_{{\text{2}}}})}_{2}}CS \Leftrightarrow \\ \Leftrightarrow {{[{\text{Me}}{{({\text{OH}})}_{2}}]}_{n}}_{{--1}}{{({\text{OH}})}_{2}}{\text{Me}}{\kern 1pt} --{\kern 1pt} {\text{S}}{\kern 1pt} --{\kern 1pt} {\text{C}}{{({\text{N}}{{{\text{H}}}_{{\text{2}}}})}_{2}}, \\ \end{gathered} $– диссоциация комплексного соединения
(24)
$\begin{gathered} {{[{\text{Me}}{{({\text{OH}})}_{2}}]}_{n}}_{{--1}}{{{\text{(OH}})}_{2}}{\text{Me}}{\kern 1pt} --{\kern 1pt} {\text{S}}{\kern 1pt} --{\kern 1pt} {\text{C}}{{({\text{N}}{{{\text{H}}}_{{\text{2}}}})}_{2}} = \\ = {{[{\text{Me}}{{({\text{OH}})}_{2}}]}_{n}}_{{--1}}{\text{MeS}}{\kern 1pt} \downarrow + \;{{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{N}}}_{2}} + 2{{{\text{H}}}_{{\text{2}}}}{\text{O}}, \\ \end{gathered} $Первые два механизма реакции образования сульфида основаны на представлении о гидролитическом распаде сульфидизатора, в котором лимитирующей стадией является гидролиз его молекул; третий и четвертый – на представлении об образовании промежуточных комплексов металла с сульфидизатором и разрыве связи С–S без образования свободных анионов S2–, а лимитирующей стадией является трансформация молекулы тиоамида, взаимодействующей с атомом металла. Можно предположить, что при химическом осаждении возможна реализация процесса по одному-двум или даже всем четырем механизмам. Однако вклад каждого из них в конечный результат процесса будет определяться условиями его проведения.
ФОРМИРОВАНИЕ ОТДЕЛЬНЫХ СУЛЬФИДНЫХ ФОРМ: КОЛЛОИДНО-КОНДЕНСАЦИОННЫЕ ОСОБЕННОСТИ И ЗАКОНОМЕРНОСТИ
Химическое осаждение наноструктурированных частиц сульфидов металлов
Для получения химическим осаждением наночастиц сульфидов металлов с заданной морфологией (размером, формой) решающее значение имеют стадии: первичного химического взаимодействия с образованием системы “осадок–раствор” и “старения” системы, обусловленные протеканием вторичных процессов взаимодействия осадка с раствором.
С коллоидно-химической точки зрения, можно считать установленным, что размеры частиц образующейся твердой фазы определяются такими условиями как температура, растворимость, концентрация и скорость перемешивания реагентов. Влияние этих факторов можно качественно объяснить в предположении, что размеры частиц связаны с относительным пересыщением по осаждаемому сульфиду: Δ = (Q – S)/S, где Q – концентрация растворенного вещества в какой-либо момент времени, S – его растворимость в момент достижения состояния равновесия [104]. В процессе образования сульфидной фазы добавление каждой порции сульфидизатора приводит к мгновенному пересыщению. В большинстве случаев при этом происходит образование порошкообразного осадка. Основные принципы термодинамического расчета величины пересыщения основаны на представлении о гидролитическом распаде сульфидизатора и подробно представлены в [72, 90, 101].
Согласно классическим представлениям, химическое взаимодействие с образованием системы “осадок–раствор” при гомогенном фазообразовании включает две стадии [104, 105]: образование центров кристаллизации (зародышей) и их рост. Соотношение их скоростей определяет в итоге размеры частиц осадка. По Веймарну [106], скорости образования и роста зародышей зависят от степени пересыщения Δ и, следовательно, от величины химического сродства реакции А. При прочих равных условиях скорость образования зародышей возрастает с увеличением Δ по экспоненциальному закону, в то время как скорость их роста связана с Δ линейно [104]. Согласно количественной теории образования кристаллических зародышей М. Фольмера [107, 108], с увеличением Δ раствора наиболее устойчивыми в нем становятся все более мелкие зародыши, что и обусловливает более высокую скорость их возникновения и меньший размер выросших частиц.
Метод химического осаждения позволяет получать частицы с размерами от нескольких нанометров до десятков микрон. Экспериментальное изучение влияния начальных условий процесса осаждения на коллоидно-химический механизм образования наночастиц PbS со структурой NaCl при гомогенном и гетерогенном химическом осаждении из водных растворов (NH2)2CS и Na2S показал следующее [109]. Размер первоначально образующихся частиц PbS при гетерогенном осаждении незначительно уменьшается с увеличением химического сродства и соответственно с увеличением пересыщения. В то же время при гомогенном осаждении размер наночастиц PbS находится в прямо пропорциональной зависимости от величины Δ, в свою очередь, пропорциональной значению химического сродства (рис. 2).
Рис. 2.
Зависимости между размером наночастиц 〈DPbS〉 и химическим сродством реакции A: 1 – при гомогенном (Pb(OH)Cit2– + (NH2)2CS + OH– = = PbS + Cit3– + H2NCN + H2O), 2 – при гетерогенном (Pb2+ + S2-= PbS) химическом осаждении. Размер 〈DPbS〉 определен непосредственно после синтеза [109].
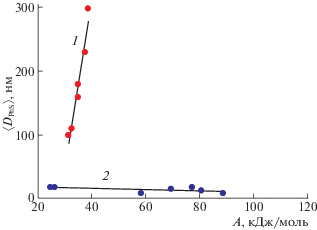
При гомогенном осаждении лимитирующей стадией процесса является реакция гидролиза сульфидизатора, например, (NH2)2CS [35, 72]. Эта реакция имеет достаточно большой индукционный период, и образование сульфида протекает с заметно меньшей скоростью, чем при использовании раствора Na2S, содержащего “готовые” ионы S2– и НS–. Вследствие этого степень пересыщения невелика, и первичных центров кристаллизации появляется сравнительно немного. В результате реакция гидролиза сульфидизатора приводит не столько к возникновению новых центров кристаллизации новой фазы, сколько к росту уже образовавшихся зародышей. При этом скорость роста зародышей прямо пропорциональна исходным концентрациям, т.е. величине пересыщения.
Процессы физического старения системы “сульфид–раствор”, связанные с самопроизвольным изменением размера наночастиц при контакте с раствором, представлены на рис. 3.
Рис. 3.
Старение системы “PbS–раствор” в условиях химического осаждения. Зависимость скорости роста w наночастиц PbS от химического сродства реакции A. На вставке: влияние времени нахождения в растворе наночастиц PbS на величину 〈DPbS〉 при значениях A = 26.28 (1), 24.56 (2), 77.06 (3), 88.47 кДж/моль (4) [109].
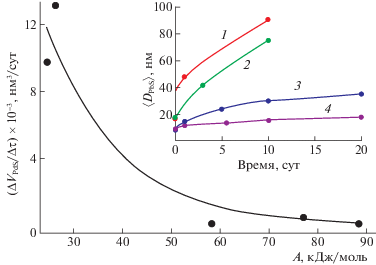
При больших значениях химического сродства размер 〈DPbS〉 практически не изменяется, тогда как при меньших его значениях 〈DPbS〉 возрастает в ∼4 раза. Скорость роста $w = \Delta {{V}_{{{\text{PbS}}}}}{\text{/}}\Delta \tau $ наночастиц обратно пропорциональна А ($\Delta {{V}_{{{\text{PbS}}}}}$ − изменение объема наночастицы PbS за время $\Delta \tau $). Таким образом, при малых пересыщениях старение системы “сульфид–раствор” соответствует механизму оствальдовского созревания [108], при котором рост более крупных частиц происходит за счет растворения более мелких. Рост сульфидных частиц при старении системы “наночастицы–раствор” при малых Δ и А, с коллоидной точки зрения, легко объясним. Известно, что каждая частица в растворе вследствие адсорбции на своей поверхности ионов несет на себе положительный или отрицательный заряд [105]. Например, частица PbS отрицательно заряжена в растворе, содержащем даже небольшой избыток ионов S2–, благодаря их предпочтительной адсорбции. Степень адсорбции возрастает с увеличением концентрации ионов и соответственно величины Δ. В результате поверхность каждой частицы насыщается. При химическом осаждении сульфида с небольшим избытком S2–-ионов на поверхности частиц они формируют первичный отрицательно заряженный адсорбционный слой. Далее под действием электростатических сил образуется слой противоинов, в результате чего формируется двойной электрический слой, благодаря которому частицы отталкиваются, и рост частиц прекращается.
Таким образом, тип сульфидизатора, а, следовательно, и тип химического осаждения позволяют регулировать количество центров кристаллизации и скорость их роста. При введении в исходный водный раствор “готовых” ионов S2– и НS– процесс образования частиц сульфида подчиняется классической теории образования осадков и правилу осаждения Веймарна–Габера [110]: размер наночастиц увеличивается с уменьшением Δ. Использование молекулярных сульфидизаторов, подвергающихся гидролизу, способствуют росту образовавшихся зародышей и приводит к получению более крупных частиц, размер которых возрастает с увеличением Δ.
Химическое осаждение тонкопленочных сульфидов металлов
С момента выхода первых публикаций по химическому осаждению пленок полупроводниковых соединений, в которых слои PbS, Cu‑S и Sb‑S были получены из водных тиосульфатных растворов ацетата свинца, сульфата меди и виннокислой сурьмы [1, 111], этот метод широко используется для синтеза тонкопленочных материалов. Для синтеза тонких пленок существует большое количество методов, среди которых термическое, магнетронное, атомно-лучевое, электронно-лучевое испарение, спрей-пиролиз, электроосаждение и др. Однако в них, как правило, применяются дорогие прекурсоры, высокая температура, сложное оборудование, а в некоторых − при синтезе образуются опасные побочные продукты. Существенным недостатком методов испарения является также относительно низкая воспроизводимость свойств. По сравнению с ними для растворных методов, в том числе и химического осаждения, свойственны комнатные температуры проведения процесса, возможность нанесения пленок на подложки любого размера и формы, экономичность.
Согласно имеющимся представлениям [69–72, 83, 84, 112], химическое осаждение пленок из растворов может идти по следующим механизмам: ионному обмену, кластерному (гидроксидному, коллоидному) и ионно-кластерному (смешанному) [70, 113]. Механизм ионного обмена схематически представлен на рис. 4 на примере образования пленки CdS. Образование частиц сульфидной пленки идет непосредственно на подложке, а их адгезия к подложке в общем случае обеспечивается силами Ван-дер-Ваальса. В отдельных случаях возможно взаимодействие за счет сил химической и электрической природы [70, 113]. Кластерный (коллоидный) механизм предполагает образование зародышей твердой фазы и последующее формирование частиц этой фазы непосредственно в растворе, затем их рост, коагуляцию и адсорбцию на подложке.
Рис. 4.
Схематическое изображение ионного механизма образования пленки CdS на подложке: диффузия ионов Cd2+ и S2– к подложке (1), их взаимодействие с образованием зародыша твердой фазы CdS (2). Рост зародыша за счет адсорбции ионов Cd2+ и S2– (3), коагуляция частиц CdS вследствие ван-дер-ваальсовых взаимодействий (4) [70, 113].
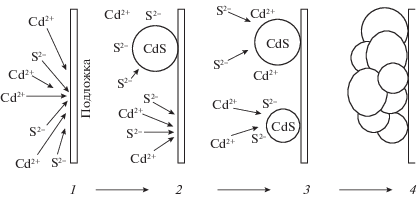
С коллоидно-химической точки зрения, этот механизм образования пленок на межфазной границе, как было впервые показано в работах С.Г. Мокрушина [13, 114], состоит из следующих стадий: образование коллоидно-дисперсного вещества, адсорбция коллоидных частиц на поверхности раздела фаз, укрупнение их в результате поверхностной коагуляции.
Химический состав образующихся коллоидных частиц (кластеров) определяется условиями синтеза. При химическом осаждении из растворов при рН < 5 с использованием в качестве сульфидизаторов CH3CSNH2 и тиосульфат-иона S2O$_{3}^{{2 - }}$ это – кластеры сульфида металла. В щелочных растворах это может быть гидроксид металла, образования которого экспериментально трудно избежать, несмотря на использование комплексообразующих агентов. В [28, 30, 33] Г.А. Китаевым с соавторами установлено, что условием зарождения сульфидной пленки является самопроизвольное возникновение на поверхности гидрофильной подложки зародышей гидроксида металла, которые выступают центрами кристаллизации и последующего роста фазы сульфида (рис. 5). Особенно это характерно для металлов, образующих прочные гидроксокомплексы (свинец, кадмий, олово, цинк, ртуть). Такой механизм получил название “гидроокисная схема” зарождения пленки сульфида металла. При формировании пленки образование сульфидных частиц идет не только на поверхности подложки, но и в объеме раствора. Важную роль здесь играет скорость их накопления в растворе: чем она выше, тем меньше толщина слоя.
Рис. 5.
Схематическое изображение кластерного (гидроксидного, коллоидного) механизма образования пленки CdS на подложке: диффузия коллоидных гидроксидных частиц за счет броуновского движения к подложке (1), на которой происходят их адсорбция (2) и взаимодействие с ионами S2– (3). На поверхности подложки и в объеме раствора идет реакция обмена гидроксид-ионов на сульфид-ионы (4) и формирование пленки CdS (5) [70, 113, 114].
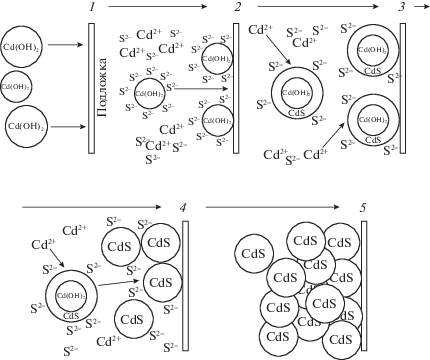
Ионный и коллоидный механизмы предполагают гомогенную нуклеацию. Однако зарождение пленки сульфида металла может происходить одновременно как по гомогенному, так и гетерогенному механизму, т.е. по смешанному механизму. Принципиальное отличие смешанного механизма от простого гидроксидного заключается в том, что синтез сульфида металла идет не только за счет образующихся в растворе ионов S2–. В этом случае сульфидизатор, в частности (NH2)2CS, может адсорбироваться на поверхности частиц гидроксида металла, формируя поверхностные метастабильные комплексы типа [Me(OH)2]n–1(OH)2 Me–S–C(NH2)2, которые затем разлагаются с образованием сульфида. Наличие неподеленных пар электронов у атомов серы и азота в (NH2)2CS обусловливает высокую адсорбционную активность (NH2)2CS по отношению к гидроксидной фазе и последующую ее сульфидизацию, учитывая значительные различия в произведениях растворимости гидроксида и сульфида металла. В экспериментальных исследованиях это проявляется в наличии значительного индукционного периода процесса формирования сульфидной фазы на подложке [35, 68, 72, 115]. Стадия сульфидизации гидроксида является лимитирующей, и скорость ее протекания фактически определяется постоянством содержания сульфидной серы в системе, связанной с константой разложения тиоамида в заданных условиях.
Схема смешанного механизма химического осаждения пленок сульфидов металлов представлена на рис. 6 [115]. В ходе реакции разложения промежуточного комплекса [(NH2)2CS–Me]n+ в объеме раствора образуются кластерные частицы сульфида металла фрактального строения (1). При помещении подложки в щелочную реакционную смесь на ней происходит образование подслоя гидроксида металла (2). Фрактальные частицы в объеме принимают участие в броуновском движении, коагулируют, образуя более крупные частицы (5), и при столкновении с поверхностью подложки адсорбируются на ней (4).
Рис. 6.
Схема образования пленок сульфидов металлов при химическом осаждении по смешанному механизму: i – реакция в объеме; ii – реакция на поверхности; iii – седиментация [115].
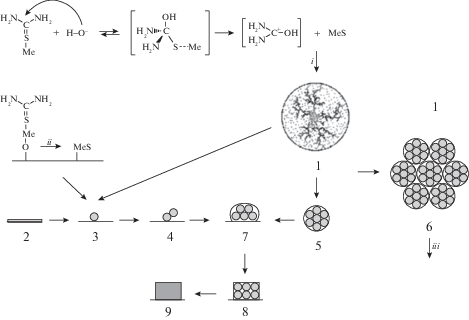
Вместе с тем возможно образование сульфида металла и на поверхности подложки в результате сульфидизации слоя гидроксида металла с образованием первичных поверхностных частиц сульфидной фазы (3). В результате броуновского движения частиц вида (1) и (5) происходит дальнейшее агрегирование частиц сульфида металла на поверхности (7). Одновременно коагуляция полимолекулярных частиц (5) в объеме раствора приводит к образованию достаточно крупных агрегатов (6), которые седиментируют. Частицы, закрепившиеся на поверхности подложки (7), постепенно кристаллизуются (8) и (9) [115].
Синтез квантовых точек сульфидов металлов коллоидно-химическим осаждением
В основе синтеза квантовых точек сульфидов металлов лежит получение коллоидных растворов с монодисперсными частицами путем химической конденсации твердой фазы из пересыщенного раствора. В [116] впервые был опубликован метод синтеза монодисперсных коллоидных наночастиц халькогенидов, основанный на высокотемпературном пиролизе металлорганических прекурсоров в координирующей среде алкилфосфинов. Несмотря на то, что указанная технология показала свою эффективность, ее главными недостатками являются токсичность и пирофорность используемых прекурсоров, а также их высокая стоимость. В связи с этим переход к недорогим и более экологически безопасным компонентам позволит сделать технологию производства КТ более доступной. Отсюда большой интерес представляет метод химического осаждения, в котором стабильные водные коллоидные растворы КТ могут быть получены по схеме one-pot. Кроме того, химическое осаждение дает возможность синтезировать наночастицы непосредственно в водной среде, т.е. с гидрофильной поверхностью без необходимости проведения операций по замене лигандов или других способов перевода частиц из неполярной среды в водную.
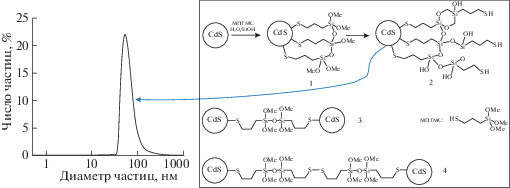
Основная сложность синтеза агрегативно и седиментационно устойчивых коллоидных растворов сульфидных КТ – их гидрофобность, поэтому синтез КТ непосредственно в водном растворе сопровождается интенсивными процессами коагуляции и агломерации частиц. Для предотвращения этих процессов в раствор вводят стабилизирующие агенты, которые служат лигандами для ионов Меn+. Среди них фосфаты, тиолы, производные аминокислот, гидрофильные полимеры, а в качестве сульфидизаторов используют водорастворимые сульфиды щелочных металлов [113, 117–122]. Самый распространенный тип стабилизирующих лигандов в таких методиках – водорастворимые короткоцепочечные меркаптокислоты [123–125]. В работе [126] экспериментально продемонстрирована возможность получения стабильного водного золя люминесцентных КТ CdS при использовании 3-меркаптопропилтриметоксисилана (МПТМС). Известно, что меркаптогруппа связывается с поверхностью наночастиц за счет образования связи Me–S–R (где Me – Cd, Zn, R–алкоксисилановый фрагмент). В результате образуется покрытие с алкоксисилановыми группами на поверхности, которые в дальнейшем гидролизуются в присутствии воды, образуя полисилоксановую оболочку 1 (рис. 7). При этом может протекать и конкурирующая реакции поликонденсации с другими молекулами МПТМС, находящимися в растворе. Таким образом, будут образовываться наночастицы CdS, содержащие на поверхности меркаптогруппы 2 (рис. 7). Процесс коагуляции можно объяснить агрегацией за счет образования полисилоксановых связей между несколькими наночастицами 3. По причине высокой склонности меркаптогрупп к окислению кислородом воздуха наночастицы CdS могут агрегировать за счет взаимодействия поверхностных меркаптогрупп с образованием дисульфидных производных 4 (рис. 7) [126]. Такой коллоидный раствор остается агрегативно устойчивым в течение первых 24 ч, при этом седиментационная стабильность и флуоресцентные свойства золя сохраняются около двух недель.
Рис. 7.
Схема поверхностной модификации наночастиц CdS при использовании МПТМС и возможные пути агрегации золя CdS. Динамический размер коллоидных частиц 66 ± 30 нм [126].
Существенно более длительную стабильность придают полидентантные соединения, такие как этилендиаминтетрауксусная кислота (ЭДТА), которая была также успешно применена для синтеза коллоидных растворов CdS [122, 127], стабильных в течение нескольких месяцев. Для получения мицелл CdS размером ∼15 нм использовались сульфид натрия Na2S, хлорид кадмия CdCl2 и ЭДТА C10H16N2O8 (H4edta). При этом под мицеллой понимают частицы дисперсной фазы, которые состоят из нерастворимого в данной среде ядра очень малого размера (агрегата мицеллы), окруженного стабилизирующей оболочкой адсорбированных ионов и молекул растворителя [105]. При смешении растворов в получившемся растворе протекают две реакции:
(25)
$\begin{gathered} {\text{C}}{{{\text{d}}}^{{2 + }}}{\text{(aq)}} + {{{\text{H}}}_{{\text{2}}}}{\text{edt}}{{{\text{a}}}^{{2--}}}{\text{(aq)}} = \\ = {\text{Cdedt}}{{{\text{a}}}^{{2--}}}{\text{(aq)}} + 2{{{\text{H}}}^{ + }}{\text{(aq)}}, \\ \end{gathered} $(26)
${\text{Cdedt}}{{{\text{a}}}^{{2--}}}{\text{(aq)}} + {{{\text{S}}}^{{2--}}}{\text{(aq)}} = {\text{CdS}}{\kern 1pt} \downarrow + \;{\text{edt}}{{{\text{a}}}^{{4--}}}{\text{(aq)}}.$Долговременная стабильность раствора обусловлена тем, что ионы Na+, H+, Cl– не могут прочно присоединиться к поверхности кристалла, тогда как ионы еdta4–, находящиеся в растворе, образуют с поверхностными ионами Cd2+, принадлежащими кристаллу, прочные моноядерные и протонированные комплексонаты. Ионы CdHedta– и Cdеdta2– ориентируют вокруг себя полярные молекулы воды, создавая дополнительную гидратную оболочку. При этом формируются гидрофильные оболочки, и возникает адсорбционно-сольватный фактор устойчивости коллоидного раствора. Однако образование прочных комплексонатов приводит к интенсификации процессов распада (растворения) дисперсной сульфидной фазы. В результате образование и рост агрегата мицеллы, т.е. наночастиц CdS, по реакции (26) затормаживается, и равновесие этой реакции сдвигается влево. Следовательно, присутствие ионов H2еdta2– и H3еdta– обеспечивает протекание двух параллельных реакций: комплексообразования и растворения дисперсной фазы. Формула мицеллы, состоящей из агрегата CdS, слоя потенциалопределяющих ионов, а также адсорбционного и диффузного слоев гидратированных противоионов может быть записана следующим образом:
(27)
$\begin{gathered} \{ {{[{\text{CdS}}]}_{m}} \cdot 2n{\text{Cdedt}}{{{\text{a}}}^{{2--}}} \cdot (2n--x){\text{N}}{{{\text{a}}}^{ + }}{\text{(aq)}} \cdot \\ \cdot \;\left( {2n--x} \right){{{\text{H}}}^{ + }}{\text{(aq)}}{{\} }^{{2x--}}} \cdot x{\text{N}}{{{\text{a}}}^{ + }}{\text{(aq)}} \cdot x{{{\text{H}}}^{ + }}({\text{aq}}). \\ \end{gathered} $Таким образом, метод химического осаждения позволяет получать стабильные водные коллоидные растворы гидрофобных сульфидных наночастиц. Их водорастворимость достигается введением стабилизирующих агентов (лигандов). Благодаря молекулярным лигандам возникает адсорбционно-сольватный, а лигандам-электролитам – еще и электростатический факторы устойчивости коллоидного раствора.
ЗАКЛЮЧЕНИЕ
Сравнительно давно появившийся метод химического осаждения сульфидов металлов из водных растворов с применением тиоамидов, остается актуальным и сегодня. Анализ современных научных публикаций, посвященных химическому осаждению, показывает возвращение интереса к химическому осаждению сульфидов металлов из водных растворов как перспективному и развивающемуся методу синтеза новых функциональных неорганических материалов для создания солнечных элементов, фотокатализаторов, фотовольтаических и сенсорных устройств на основе тонких пленок, наноструктурированных частиц и коллоидных КТ.
В настоящем обзоре впервые предпринята попытка представить метод химического осаждения одновременно с двух точек зрения: химической и коллоидно-конденсационной. С химической точки зрения, осаждение можно условно разделить на два типа: гомогенное и гетерогенное. Принципиальное отличие состоит лишь в том, что осадитель (ион S2–) либо приливают к раствору, содержащему осаждаемые комплексные ионы металла, либо он постепенно образуется за счет протекающей реакции гидролиза сульфидизатора во всем объеме раствора. Гидролизующиеся сульфидизаторы используют для синтеза пленок, а сульфидизаторы-электролиты – для синтеза коллоидных растворов. Метод химического осаждения позволяет регулировать как скорость образования, так и скорость роста сульфидных частиц, благодаря чему удается получать частицы размерами от нескольких нанометров до десятков микрон, а также синтезировать различные поликристаллические структуры на основе сульфидов металлов: от тонких пленок до коллоидных КТ.
Несмотря на то, что химическое осаждение давно привлекает внимание химиков, многие вопросы, касающиеся закономерностей формирования той или иной формы сульфида, все еще не вполне ясны. Дальнейшее развитие метода обеспечит увеличение контроля за ходом химического процесса, позволит повысить воспроизводимость результатов, а также расширит области практического использования получаемых материалов.
Обзор посвящается создателям уральской школы тонкопленочного синтеза халькогенидов металлов из растворов проф. С.Г. Мокрушину и проф. Г.А. Китаеву.
Список литературы
Puscher C. // Polytech. J. 1868. V. 190. № 5. 421.
Reynolds J.E. // J. Chem. Soc., Trans. 1884. V. 45. P. 162.
Brükmann G. // Kolloid Zs. 1933. Bd. 61. № 1. S. 1.
Kisinski F. // Chemistry and Industry. 1948. V. 67. № 4. P. 54.
Pick H. // Zs. Phys. 1949. Bd. 126. № 1. S. 12.
Cashman R.J. // J. Opt. Soc. Am. 1946. V. 36. P. 356.
Боде Д.Е. Детекторы на основе солей свинца // Физика тонких пленок. Пер. с англ. М., 1968. Т. 3. С. 299.
Мокрушин С.Г., Коняев П.С. // Журн. физ. химии. 1935. Т. 6. С. 95.
Мокрушин С.Г., Китаев Г.А. // Коллоид. журн. 1957. Т. 19. № 1. С. 93.
Мокрушин С.Г., Ткачев Ю.Д., Шабалина О.К. // Там же. 1957. Т. 19. № 5. С. 597.
Мокрушин С.Г., Китаев Г.А. // Там же. 1959. Т. 21. № 1. С. 80.
Казаков Е.М., Китаев Г.А., Мокрушин С.Г. // Там же. 1960. Т. 22. № 1. С. 23.
Казаков Е.М., Китаев Г.А., Мокрушин С.Г. // Изв. вузов. Химия и хим. технол. 1961. Т. 4. № 3. С. 411.
Мокрушин С.Г., Китаев Г.А., Шабалина О.К. // Докл. АН СССР. 1954. Т. 94. № 6. С. 1109.
Sharma N.C., Pandya D.K., Sehgal H.K., Chopra K.L. // Mater. Res. Bull. 1976. V. 11. № 9. P. 1109.
Sharma N.C., Pandya D.K., Mukerjee A.K. et al. // Appl. Optics. 1977. V. 16. № 11. P. 2945.
Sharma N.C., Pandya D.K., Sehgal H.K., Chopra K.L. // Thin Solid Films. 1977. V. 42. № 3. P. 383.
Sharma N.C., Pandya D.K., Sehgal H.K., Chopra K.L. // Ibid. 1979. V. 59. № 2. P. 157.
Sharma N.C., Kainthla R.C., Pandya D.K., Chopra K.L. // Ibid. 1979. V. 60. № 1. P. 55.
Sharma N.C., Pandya D.K., Sehgal H.K., Chopra K.L. // Ibid. 1979. V. 62. № 1. P. 97.
Sharma N.C., Chopra K.L. // Solid State Electronics. 1980. V. 23. № 8. P. 869.
Чопра К., Дас С. Тонкопленочные солнечные элементы. М.: Мир, 1986. 440 с.
Chopra K. Thin Film Device Applications. New York: Springer US, Plenum Press, 1983. 300 p.
Bode D.E., Johnson T.H., Maclian B.N. // J. Appl. Opt. 1965. V. 4. P. 327.
Угай А.Я., Яценко О.Б., Авербах Е.М. // Твердотельная радиоэлектроника. 1973. С. 77.
Угай А.Я., Яценко О.Б., Максименков Н.В., Авербах Е.М. // Изв. АН СССР. Неорган. материалы. 1975. Т. 11. № 5. С. 947.
Мокрушин С.Г., Китаев Г.А. // Коллоид. журн. 1961. Т. 23. № 4. С. 438.
Китаев Г.А., Мокрушин С.Г., Урицкая А.А. // Там же. 1965. Т. 27. № 1. С. 51.
Урицкая А.А., Китаев Г.А. Мокрушин С.Г. // Там же. 1965. Т. 27. № 5. С. 767.
Урицкая А.А., Китаев Г.А. Мокрушин С.Г. // Журн. физ. химии. 1965. Т. 39. № 8. С. 2065.
Китаев Г.А., Лундин А.Б., Мокрушин С.Г. // Изв. вузов. Химия и хим. технол. 1966. Т. 9. № 4. С. 411.
Китаев Г.А., Больщикова Т.П. // Неорган. материалы. 1966. Т. 2. № 1. С. 65.
Китаев Г.А., Фофанов Г.М., Лундин А.Б. // Там же. 1967. Т. 3. № 3. С. 473.
Щербакова В.Я., Двойнин В.И., Китаев Г.А., Урицкая А.А. // Там же. 1977. Т. 13. № 9. С. 1719.
Китаев Г.А. Дис. … д-ра хим. наук. Свердловск: УПИ им. С.М. Кирова, 1971. С. 9–10.
Barkhouse D.A.R., Gunawan O., Gokmen T. et al. // Progress in Photovoltaics: Research and Application. 2012. V. 20. № 1. P. 6.
Adewoyin A.D., Olopade M.A., Chendo M. // Optik. 2017. V. 133. P. 122.
Scragg J.J., Dale P.J., Peter L.M. // Thin Solid Films. 2009. V. 517. P. 2481.
Sarswat P.K., Snure M., Free M.L., Tiwari A. // Ibid. 2012. V. 520. P. 1694.
Хайрутдинов Р.Ф. // Успехи химии. 1998. Т. 67. № 2. С. 125.
Олейников В.А., Суханова А.В., Набиев И.Р. // Российские нанотехнологии. 2007. № 2. С. 160.
Хребтов А.И., Талалаев В.Г., Werner P. и др. // Физика и техника полупроводников. 2013. Т. 47. № 10. С. 1356.
Целиков Г.И., Тимошенко В.Ю., Пленге Ю. и др. // Там же. 2013. Т. 47. № 5. С. 633.
Бричкин С.Б., Разумов В.Ф. // Успехи химии. 2016. Т. 85. № 12. С. 1297.
Олейников В.А. // Биоорганическая химия. 2011. Т. 37. № 2. С. 171.
Александрова О.А., Галиева Д.М., Дробинцева А.О. и др. Наночастицы, наносистемы и их применение. Ч. 1. Коллоидные квантовые точки. Уфа: Аэтерна, 2015. 236 с.
Коржевский Д.Э., Кирик О.В., Сухорукова Е.Г., Гиляров А.В. // Морфология. 2010. Т. 137. № 3. С. 71.
Мусихин С.Ф., Александрова О.А., Лучинин В.В., Максимов А.И., Мошников В.А. // Биотехносфера. 2012. № 3–5 (23–24). С. 40.
Самохвалов П.С., Артемьев М.В., Набиев И.Р. // Российские нанотехнологии. 2013. Т. 8. № 5–6. С. 119.
Henglein A., Gutierrez M., Fischer C.H. // Ber. Bunsenges. Phys. Chem. 1984. V. 88. P. 170.
Zhang F., Wang H., Liu H. et al. // Appl. Sci. 2019. V. 9. P. 2489.
Kanemoto M., Shiragami T., Pac C. et al. // J. Phys. Chem. 1992. V. 96. P. 3521.
Zhou R., Guzman M.I. // J. Phys. Chem. C. 2014. V. 118. № 22. P. 11649.
Eggins B.R., Robertson P.K.J., Stewart J.H., Woods E. // J. Chem. Soc. Chem. Commun. 1993. V. 24. P. 349.
Eggins B.R., Robertson P.K.J., Murphy E.P. et al. // J. Photochem. Photobiol. A. 1998. V. 118. P. 31.
Johne P., Kisch H. // Ibid. 1997. V. 111. P. 223.
Zhou R., Basu K., Hartman H. et al. // Sci. Reports. 2017. V. 7. № 1. P. 533.
Zhang X.V., Ellery S.P., Friend C.M. et al. // J. Photochem. Photobiol. A. 2007. V. 185. P. 301.
Guzman M.I., Martin S.T. // Int. J. Astrobiol. 2008. V. 7. P. 271.
Guzman M.I. // Astrobiology. 2009. V. 9. P. 833.
Guzman M.I., Martin S.T. // Chem. Commun. 2010. V. 46. № 13. P. 2265.
Zhou R., Guzman M.I. // J. Phys. Chem. C. 2016. V. 120. № 13. P. 7349.
Zhang X.V., Martin S.T. // J. Am. Chem. Soc. 2006. V. 128. № 50. P. 16032.
Lokhande C.D. // Mater. Chem. Phys. 1991. V. 27. P. 1.
Savadogo O. // Solar Energy Mater. Cells. 1998. V. 52. P. 361.
Lincot D., Froment M., Cachet H. In: Adv. Electrochem. Sci. Eng. V. 6. (Eds. R.C. Alkire, D.M. Kolb). Wiley-VCH, Verlag GmbH, 1999. P. 165.
Mane R.S., Lokhande C.D. // Mater. Chem. Phys. 2000. V. 65. P. 1.
Herrero J., Gutierrez M.T., Guillen C. et al. // Thin Solid Films. 2000. V. 361–362. P. 28–33.
Hodes G. // Phys. Chem. Chem. Phys. 2007. V. 9. P. 2181.
Hodes G. Chemical Solution Deposition of Semiconductor Films. New York: Marcel Dekker Inc., 2002.
Pawar S.M., Pawar B.S., Kim J.H. et al. // Current App. Phys. 2011. V. 11. P. 117.
Марков В.Ф., Маскаева Л.Н., Иванов П.Н. Гидрохимическое осаждение пленок сульфидов металлов: моделирование и эксперимент. Екатеринбург: Изд-во УрО РАН, 2006. 217 с.
Китаев Г.А., Урицкая А.А., Ятлова Л.Е., Миролюбов В.Р. // Журн. прикл. химии. 1994. Т. 67. С. 1612.
Bhaskar P.U., Babu G.S., Kumar Y.B. K. et al. // Mater. Chem. Phys. 2012. V. 134. P. 1106.
Преображенская Т.Ю., Юсупов Р.А. // Журн. физ. химии. 1974. Т. 48. № 3. С. 724.
Bhaskar P.U., Babu G.S., Kumar Y.B. K. et al. // Mater. Chem. and Phys. 2012. V. 134. P. 1106.
Ятлова Л.Е., Китаев Г.А. // Изв. вузов. Химия и хим. технология. 1969. Т. 12. № 6. С. 709.
Китаев Г.А., Урицкая А.А. // Журн. прикл. химии. 1999. Т. 72. С. 569.
Китаев Г.А., Урицкая А.А., Белова Н.С. // Там же. 2000. Т. 73. С. 1433.
Norr M.K. // J. Phys. Chem. 1961. V. 65. P. 1278.
Bauer R., Wehling I. // Fresen. Z. Anal. Chem. 1964. V. 199. P. 171.
Семенов В.Н. // Журн. прикл. химии. 1992. Т. 65. № 7. С. 1489.
Dona J.M., Herrero J. // J. Electrochem. Soc. 1997. V. 144. P. 4081.
Ortega-Borges R., Lincot D. // Ibid. 1993. V. 140. P. 3464.
Промежуточный комплекс в химических реакциях / Ю.Н. Макурин, Р.Н. Плетнев, Д.Г. Клещев, Н.А. Желонкин // Свердловск: УрО АН СССР, 1990. 197 с.
Китаев Г.А., Макурин Ю.Н., Двойнин В.И., Миролюбов В.Р. // Журн. физ. химии. 1975. Т. 49. № 4. С. 1011.
Китаев Г.А., Макурин Ю.Н., Двойнин В.И. // Там же. 1976. Т. 50. № 12. С. 3074.
Макурин Ю.Н., Двойнин В.И., Китаев Г.А. // Теорет. и эксперим. химия. 1976. Т. 12. № 2. С. 267.
Mertschenk B., Beck F., Bauer W. Thiourea and Thiourea Derivatives. In: Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2005.
Китаев Г.А., Больщикова Т.П., Фофанов Г.М. и др. // Кинетика и механизм образования твердой фазы. Сб. тр. УПИ № 170. Свердловск, 1968. С. 113.
Романов И.Т., Китаев Г.А. // Физико-химия процессов на межфазных границах. Свердловск: Изд-во УПИ, 1976. С. 6.
Hariskosa D., Powalla M., Chevaldonnet N. et al. // Thin Solid Films. 2001. V. 387. P. 179.
Shaw W.H.R., Walker D.G. // J. Am. Chem. Soc. 1956. V. 78. P. 5769.
Лундин А.Б., Булатов Н.К., Дариенко Н.И., Якимова Л.Б. // Изв. вузов. Химия и хим. технол. 1976. Т. 19. № 7. С. 1059.
Волынский Н.П. Тиосерная кислота. Политионаты: реакция Вакенродера. М.: Наука, 1971.
Swift E.H., Butler E.A. // Anal. Chem. 1956. V. 28. P. 146.
Butler E.A., Peters D.G., Swift E.H. // Ibid. 1958. V. 30. P. 1379.
Sugimoto T., Dirige G.E., Muramatsu A. // J. Colloid & Interface Sci. 1995. V. 176. P. 442.
Марков В.Ф., Маскаева Л.Н., Дивинская Г.Г., Морозова И.М. // Вестн. УГТУ-УПИ. Сер. хим. 2003. С. 120.
Виноградова Т.В., Марков В.Ф., Маскаева Л.Н. // Журн. общ. химии. 2010. Т. 80. № 11. С. 1878.
Марков В.Ф., Маскаева Л.Н. // Журн. физ. химии. 2010. Т. 84. № 8. С. 1421–1426.
Федорова Е.А., Маскаева Л.Н., Марков В.Ф. и др. // Там же. 2019. Т. 93. № 1. С. 142.
García-Valenzuela J.A. // Comments on Inorganic Chemistry. 2017. V. 37. № 2. P. 99.
Skoog D.A., West D.M., Holler F.J., Crouch S.R. // Fundamentals of Analytical Chemistry. Belmont, CA: Brooks/Cole, Cengage Learning, 9th ed., 2014. 1072 p.
Фролов Ю.Г. Курс коллоидной химии. Поверхностные явления и дисперсные системы, М.: Химия, 1989, 464 с.
von Weimarn P.P. // Chem. Rev. 1925. V. 2. № 2. P. 217.
Фольмер М. Кинетика образования новой среды. М.: Наука, 1986. 205 с.
Leite E.R., Ribeiro C. // Crystallization and Growth of Colloidal Nanocrystals. New York; Dordrecht; Heidelberg; London: Springer, 2012. P. 36−39.
Кожевникова Н.С., Урицкая А.А., Ремпель А.А. // Докл. АН. Серия Физическая химия. 2013. Т. 453. № 2. С. 167.
Barlow D.A., Baird J.K., Su C.-H. // J. Crystal Growth. 2004. V. 264. P. 417.
Puscher C. // Dingl. J. 1869. V. 190. P. 421.
O’Brien P., McAleese J. // J. Mater. Chem. 1998. V. 8. P. 2309.
Кожевникова Н.С., Ворох А.С., Урицкая А.А. // Успехи химии. 2015. Т. 84. № 3. С. 225.
Мокрушин С.Г., Ткачев Ю.Д. // Коллоид. журн. 1961. Т. 23. № 5. С. 438.
Марков В.Ф., Маскаева Л.Н. // Изв. АН. Сер. хим. 2014. № 7. С. 1523.
Murray C.B., Norris D.J., Bawendi M.G. // J. Am. Chem. Soc. 1993. V. 115. P. 8706.
Spoerke E.D., Voigt J.A. // Adv. Funct. Mater. 2007. V. 17. P. 2031.
Zou L., Fang Z., Gu Z., Zhong X. // J. Luminescence. 2009. V. 129. P. 536.
Zhuang Z., Lu X., Peng Q., Li Y. // J. Am. Chem. Soc. 2010. V. 132. P. 1891.
Nelwamondo S.M.M., Moloto M.J., Krause R.W., Moloto N. // Mater. Res. Bull. 2012. V. 47. P. 4392.
Huang P., Jiang Q., Yu P., Yang L., Mao L. // ACS Appl. Mater. & Interfaces. 2013. V. 5. P. 5239.
Кожевникова Н.С., Ворох А.С., Ремпель А.А. // Журн. общ. химии. 2010. Т. 80. С. 365.
Jana N.R., Earhart C., Ying J.Y. // Chem. Mater. 2007. V. 19. P. 5074.
Torimoto T., Reyes J.P., Iwasaki K. et al. // J. Am. Chem. Soc. 2003. V. 125. P. 316.
Hirai T., Okubo H., Komasawa I. // J. Mater. Chem. 2000. V. 10. P. 2592.
Кожевникова Н.С., Демин А.М., Краснов В.П., Ремпель А.А. // Докл. АН. 2013. Т. 452. № 1. С. 47.
Ремпель А.А., Кожевникова Н.С., Ремпель С.В. // Изв. АН. Сер. хим. 2013. Т. 62. С. 400.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии