

Журнал физической химии, 2020, T. 94, № 2, стр. 230-237
Кинетика и механизм реакций гидролиза бензилэфирных связей в водно-органических средах
Т. С. Селиверстова a, *, М. А. Кушнер a, Л. Г. Матусевич a
a Белорусский государственный технологический университет
Минск, Республика Беларусь
* E-mail: tamara.tsel@yandex.ru
Поступила в редакцию 05.04.2019
После доработки 05.04.2019
Принята к публикации 09.04.2019
Аннотация
Определены кинетические параметры (константа скорости, энергия и энтропия активации) реакций кислотно-каталитического гидролиза простых бензиловых эфиров (3,4-диметоксифенил)-(2-метоксифенокси)метана и (4-гидрокси-3-метоксифенил)метоксиметана в широком диапазоне составов смесей воды с органическими растворителями − диоксаном, ДМСО и уксусной кислотой. Показано, что кислотно-каталитический гидролиз указанных бензиловых эфиров в смесях воды с апротонными растворителями протекает как бимолекулярная реакция нуклеофильного замещения. В водных растворах уксусной кислоты механизм гидролиза может быть бимолекулярным либо мономолекулярным в зависимости от структуры эфира и содержания органического растворителя. Обcуждено влияние растворителей на скорость и механизм изученной реакции на основе сольватационных представлений.
Изучение реакций гидролиза простых бензил-эфирных связей обусловлено тем, что такая связь, наряду с другими типами простой эфирной связи и С–С-связями, является основой формирования и существования природного полимера лигнина, характеризующегося нерегулярным трехмерным сетчатым строением. Разрыв таких связей во многом определяет успех процессов делигнификации древесины и других растительных материалов с целью получения целлюлозных волокнистых материалов, например, для производства бумаги. Среди множества исследований альтернативных способов получения целлюлозы не теряют своей актуальности технологические процессы, относящиеся к так называемым органосольвентным способам, которые удовлетворяют возросшим требованиям к защите окружающей среды; снижению энергозатрат, имеют предпосылки к более полной рециклизации реагентов и увеличению выхода целлюлозы с приемлемыми потребительскими показателями [1–3]. Исследование превращений лигнина на соединениях, моделирующих основные типы химической связи его структурных фрагментов, позволяет вникнуть в суть процессов, протекающих при варке древесины с целью формирования оптимальных условий и прогнозирования свойств продуктов делигнификации, поскольку ранее обнаружена хорошая корреляция результатов, полученных при изучении реакций модельных соединений с результатами органосольвентных варок древесины [4].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В настоящей работе изложены результаты проведенных нами исследований по влиянию органических растворителей диоксана, ДМСО и уксусной кислоты на скорость и механизм кислотного гидролиза 4-гидрокси- и 4-алкоксибензиловых эфиров – (3,4-диметоксифенил)-(2-метоксифенокси)метана (1) и (4-гидрокси-3-метоксифенил)метоксиметана (2), моделирующих бензилэфирные структурные фрагменты лигнина:

Гидролиз эфиров осуществляли в смесях органический растворитель–вода переменного состава в диапазоне температур 50–90°C, в качестве катализатора использовалась HCl или H2SO4.
(3,4-Диметоксифенил)-(2-метоксифенокси)-метан (1) синтезирован из ванилина: метилированием диметилсульфатом получали вератровый альдегид, затем последний по реакции Канниццаро превращали в вератровый спирт, после чего замещали спиртовый гидроксил тионилхлоридом и полученный вератрилхлорид обрабатывали гваяколятом натрия по Вильямсону. Тпл = 72°C, что соответствует литературным данным [5].
4-Гидрокси-3-метоксифенилметоксиметан (2) синтезирован согласно [6] из ванилина восстановлением натрия боргидридом с последующим метилированием ванилинового спирта. Тпл = 34–35°C, что соответствует литературным данным.
Диоксан очищали согласно [7], ДМСО перегоняли под вакуумом, ледяную уксусную кислоту использовали без дополнительной очистки.
Кинетические исследования проводили ампульным методом. Пробы с реакционной смесью выдерживали в запаянных ампулах в термостате. Через определенные промежутки времени ампулы вскрывали и определяли концентрацию необходимых веществ. Кинетику гидролиза эфира 1 определяли по концентрации гваякола, эфира 2 – по концентрации ванилинового спирта. Концентрацию фенольных соединений определяли спектрофотометрическим методом при длине волны 612 нм [8], в основу которого положена способность фенолов с незамещенным пара-положением и гваяцилкарбинолов образовывать с хинонмонохлоримидом в щелочной среде окрашенные соединения.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Механизм гидролиза
Гидролиз бензилэфирных связей лигнина может быть отнесен к одному из наиболее распространенных типов органических реакций – нуклеофильному замещению (SN). Для нуклеофильного замещения у насыщенного атома углерода различают два крайних случая с точки зрения механизма: мономолекулярное замещение (SN1) и бимолекулярное замещение (SN2). Оба механизма для кислотного гидролиза бензилэфирной связи можно представить следующей схемой:
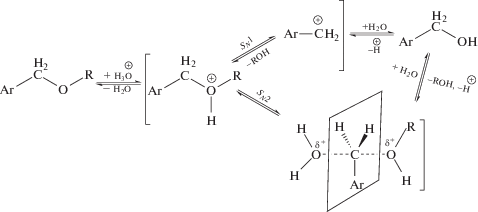
Однако в отличие от других SN-реакций изучению механизма гидролиза бензиловых эфиров не уделялось достаточно внимания, поскольку обычные диалкиловые и алкилариловые эфиры мало активны. Среди органических растворителей, используемых для органосольвентных способов делигнификации, особое внимание заслуживают апротонные растворители – диоксан, диметилсульфоксид (ДМСО), и протонный растворитель уксусная кислота.
На рис. 1 приведены зависимости скорости гидролиза бензиловых эфиров 1 и 2 от состава растворителя. Добавка к воде апротонных растворителей приводит к существенному уменьшению (почти на два порядка) скорости расщепления эфирной связи. Причем характер зависимости kэксп от мольной доли растворителя (Xs) практически одинаков для обоих эфиров как в среде водного диоксана, так и в среде водного ДМСО. Следовательно, в данных растворителях гидролиз 4-гидрокси- и 4-алкоксибензиловых эфиров протекает по одному и тому же механизму.
Рис. 1.
Зависимости константы скорости гидролиза простых бензиловых эфиров от состава смешанного растворителя: 1 – эфир 1, вода–диоксан и вода–ДМСО; 2 – эфир 2, вода–диоксан и вода–ДМСО; 3 – эфир 1, вода–уксусная кислота; 4 – эфир 2, вода–уксусная кислота.

Эффект влияния уксусной кислоты не тождественен у обоих эфиров. Для соединения 1 в водных растворах уксусной кислоты наблюдается постоянное увеличение скорости реакции с возрастанием концентрации уксусной кислоты (рис. 1). Скорость реакции гидролиза эфира 2 при добавке к воде уксусной кислоты первоначально снижается, достигая минимального значения в области Хs = 0.2 мольных долей, что соответствует 30–40% содержанию уксусной кислоты, а затем резко возрастает. Это свидетельствует в пользу того, что механизм гидролиза соединения 2 изменяется с возрастанием концентрации уксусной кислоты.
Математическая обработка экспериментальных результатов показала наличие линейной корреляции между логарифмом константы скорости кислотного гидролиза эфиров 1 (k1) и 2 (k2) в смесях вода–диоксан и вода–ДМСО при различном содержании органического растворителя, описываемой уравнением lgk1 = a + blgk2.
Соблюдение такой зависимости (рис. 2) указывает на единый механизм гидролиза этих эфиров в средах вода–апротонный растворитель. Отсутствие такой корреляции для смесей вода−уксусная кислота свидетельствует о различии механизмов гидролиза эфиров 1 и 2 в этой системе растворителей.

Термодинамические параметры реакции
На основании кинетических измерений для различных составов смешанных растворителей и различных температур рассчитаны термодинамические параметры гидролиза изученных эфиров – свободная энергия активация, энтальпия и энтропия активации (табл. 1).
Таблица 1.
Термодинамические параметры реакций кислотно-каталитического гидролиза простых бензиловых эфиров
| Органический растворитель | XS, мольная доля | ΔG≠, кДж/моль | ΔH≠, кДж/моль | ΔS≠, кДж/(моль K) |
|---|---|---|---|---|
| 1 | ||||
| Диоксан (0.01 М HCl) | 0 | 102.7 | 95.8 | –16.7 |
| 0.02 | 103.5 | 90.8 | –30.1 | |
| 0.12 | 107.3 | 87.0 | –50.2 | |
| 0.32 | 111.0 | 76.2 | –104.7 | |
| ДМСО (0.01 М HCl) | 0.03 | 103.7 | 91.3 | –37.8 |
| 0.15 | 108.5 | 91.3 | –49.0 | |
| 0.37 | 114.4 | 73.2 | –123.5 | |
| Уксусная кислота (0.01 М H2SO4) | 0 | 101.5 | 74.6 | –83.5 |
| 0.12 | 102.6 | 76.6 | –78.7 | |
| 0.22 | 102.4 | 78.4 | –72.8 | |
| 0.31 | 102.2 | 94.1 | –24.7 | |
| 0.44 | 100.5 | 100.9 | 2.84 | |
| 2 | ||||
| Диоксан (0.01 М HCl) | 0 | 108.0 | 108.9 | 3.7 |
| 0.02 | 109.7 | 104.9 | –10.5 | |
| 0.12 | 113.1 | 99.1 | –35.5 | |
| 0.32 | 115.6 | 94.7 | –55.9 | |
| ДМСО (0.01 М HCl) | 0.03 | 109.8 | 106.3 | –7.0 |
| 0.15 | 113.1 | 100.1 | –32.8 | |
| 0.37 | 114.4 | 95.2 | –57.0 | |
| Уксусная кислота (0.01 М H2SO4) | 0 | 102.6 | 111.1 | 35.2 |
| 0.03 | 101.5 | 109.5 | 33.7 | |
| 0.12 | 99.7 | 107.4 | 32.9 | |
| 0.22 | 98.0 | 105.3 | 31.5 | |
| 0.44 | 96.0 | 103.1 | 30.9 | |
Как свидетельствуют полученные данные, увеличение доли апротонного растворителя в бинарной смеси приводит к возрастанию свободной энергии активации гидролиза простых бензиловых эфиров. Энтальпия активации при этом снижается, а энтропия изменяется в сторону более высоких отрицательных значений. В средах вода–протонодонорный растворитель в случае гидролиза эфира 2 увеличение доли уксусной кислоты приводит к снижению свободной энергии активации, уменьшению энтальпии активации. Энтропия активации гидролиза данного эфира имеет положительные значения во всем исследованном диапазоне состава смешанного растворителя. В случае гидролиза эфира 1 с увеличением доли уксусной кислоты в бинарном растворителе свободная энергия активации сначала возрастает, а затем с Хs = 0.22 мол. доли снижается. Энтальпия активации при этом монотонно возрастает, а энтропия активации изменяется от высоких отрицательных значений до положительных.
Полученные значения термодинамических параметров реакции кислотного гидролиза простых бензиловых эфиров позволяют сделать вывод о механизме исследуемой реакции в смешанных растворителях. В смесях вода–апротонный растворитель для обоих эфиров более вероятен бимолекулярный механизм нуклеофильного замещения, о чем свидетельствуют более высокие значения ΔH≠. В средах вода–уксусная кислота для эфира 2 предпочтителен мономолекулярный механизм, на что указывают высокие значения ΔH≠ и положительные значения ΔS≠. В случае гидролиза эфира 1 добавка протонодонорного растворителя приводит к изменению механизма реакции от бимолекулярного к мономолекулярному, о чем свидетельствует характер зависимости ΔG≠ = f(Хs), а также изменение значений ΔS≠ от отрицательных до положительных.
Обнаружено также наличие линейной корреляции между величинами ΔH≠ и ΔS≠ описываемой уравнением ΔH≠ = a + bΔS≠ (рис. 3) для гидролиза простых бензиловых эфиров 1 и 2.
Рис. 3.
Зависимости ΔH≠ = f(ΔS≠) для кислотного гидролиза бензиловых эфиров в водно-органических средах переменного состава: 1 – эфир 1, вода–диоксан и вода–ДМСО; 2 – эфир 1, вода уксусная кислота; 3 – эфир 2, вода–диоксан и вода–ДМСО; 4 – эфир 2, вода уксусная кислота.

Влияние апротонных растворителей и диоксана и ДМСО для каждого из эфиров укладывается в одну линейную зависимость. Такое совпадение зависимостей для смесей вода–апротонный растворитель является признаком единого механизма гидролиза этих эфиров. В то же время в смесях вода–уксусная кислота такая линейная корреляция имеет место для каждого эфира в отдельности.
Роль полярности среды
Скорость многих гидролитических реакций, катализируемых кислотами, хорошо описывается электростатической теорией, учитывающей взаимосвязь константы скорости реакции с диэлектрической проницаемостью среды. Основным положением этой теории является тот факт, что в отсутствие специфической сольватации в растворе наблюдается прямолинейная зависимость lg k = f(1/ε). Отклонение от линейности или отсутствие таковой указывают на существенный вклад специфической сольватации. Данная зависимость была применена для описания электростатических взаимодействий в реакциях кислотно-каталитического гидролиза сложных эфиров и амидов [9]. Наблюдавшийся отрицательный наклон полученной зависимости был интерпретирован в качестве доказательства высокой полярности переходного состояния.
Нами при кислотном гидролизе простых бензиловых эфиров в присутствии апротонных растворителей обнаружена аналогичная зависимость константы скорости от полярности среды (рис. 4). Поэтому можно полагать, что в случае кислотного гидролиза исследуемых эфиров переходное состояние сильно полярно.
Рис. 4.
Зависимости lgk от (1/ε) для гидролиза простых бензиловых эфиров: 1 – эфир 1, вода–диоксан; 2 – эфир 1, вода–ДМСО; 3 – эфир 2, вода–диоксан; 4 – эфир 2, вода–ДМСО; 5 – эфир 1, вода–уксусная кислота; 6 – эфир 2, вода–уксусная кислота.

В области больших концентраций воды эта зависимость практически линейна, что указывает на преобладание влияния электростатических взаимодействий в растворе, без существенного вклада специфической сольватации.
Для учета электростатических взаимодействий в растворе используются эмпирические зависимости константы скорости и диэлектрической проницаемости среды [10]. Для ион-дипольного взаимодействия, к которому может быть отнесен гидролиз бензиловых эфиров, эта зависимость описывается уравнением lgkε = lgkε = ∞ + + [Z]eμ/(KTr≠ε), где kε = ∞ – константа скорости при бесконечно большом значении ε, Z – число единиц заряда иона, е – заряд электрона, μ – дипольный момент воды, K – постоянная Больцмана, Т – температура, r≠ – расстояние максимального сближения реагирующих частиц в лимитирующем акте реакции (радиус активированного комплекса).
Данное уравнение дает возможность определить расстояние наибольшего сближения между реагирующими частицами в лимитирующей стадии реакции r≠, значение которого также можно использовать для оценки наличия специфических эффектов среды. Для соединений 1 и 2 значения r≠ в водно-диоксановой среде 0.93 нм и 0.216 нм соответственно. Порядок этих величин говорит о том, что диэлектрическая проницаемость среды оказывает существенное влияние на кислотный гидролиз простых бензиловых эфиров, во всяком случае, роль ее сравнима с ролью специфической сольватации. В системе ДМСО–вода получены неразумно малые значения параметра r≠ (0.034 нм и 0.036 нм соответственно), что указывает на преобладающее влияние специфических эффектов среды.
В случае системы ДМСО–вода специфические эффекты значительно перекрывают влияние диэлектрической проницаемости. Для ДМСО характерно сильное взаимодействие с водой. Можно предположить, что чем сильнее взаимодействие апротонного растворителя с водой, тем больше будет ослабляться сольватация активированного комплекса. Данные физико-химических исследований смешанных растворителей [11] указывают на уменьшение силы взаимодействия между апротонным растворителем и водой в последовательности ДМСО > диоксан.
Для водно-уксуснокислых сред зависимость lgk = f(1/ε) (рис. 4) в случае эфира 1 практически прямолинейна в области небольшого содержания уксусной кислоты и имеет положительный наклон, что свидетельствует в пользу участия в лимитирующем акте положительного иона и указывает на карбокатионный механизм превращения (SN1). Рассчитанная величина r≠ (0.117 нм) показывает, что наряду со специфической сольватацией имеют место электростатические взаимодействия, которые оказывают значительное влияние на скорость реакции. Увеличение содержания уксусной кислоты вызывает перегиб зависимости lgk = f(1/ε), что может быть обусловлено специфической сольватацией реагирующих частиц и переходного состояния.
Для гидролиза эфира 2 на зависимости lgk от 1/ε можно выделить два практически прямолинейных отрезка. В смешанном растворителе с большим содержанием уксусной кислоты (Хs > > 0.32 мол. доли) получен положительный наклон этой зависимости, подтверждающий участие в реакции карбокатиона (механизм SN1). В области больших концентраций воды наклон зависимости отрицательный, что свидетельствует в пользу преобразования механизма реакции.
Интерпретация влияния уксусной кислоты на скорость гидролиза бензиларильной связи может быть дана на основе сольватационных представлений. Если один из компонентов бинарной смеси способен к специфической сольватации, в системе протекают параллельно и независимо друг от друга два процесса: с участием специфической сольватации M…Si и несольватированной M частиц. Суммарная скорость процесса определяется как V = k°[М] + ki[M…Si]. Следовательно, в растворителях с низким содержанием специфически сольватирущего компонента общая скорость реакции будет определяться произведением k°[M]. При переходе к смесям с большим содержанием компонента Si за счет сдвига сольватационных равновесий вклад концентрации M…Si в общую скорость процесса возрастает и может оказывать доминирующее влияние, если k° и ki сравнимы. В соответствии с приведенным уравнением в растворителе вода–уксусная кислота с низким содержанием специфически сольватирущего компонента – уксусной кислоты – можно предположить протекание реакции в основном с участием специфически несольватированной частицы по бимолекулярному механизму (SN2). В этом случае на скорость реакции сильное влияние оказывает полярность среды. С увеличением мольной доли уксусной кислоты в смешанном растворителе происходит понижение полярности среды, что и обуславливает уменьшение скорости реакции. В концентрированных растворах уксусной кислоты возрастает роль частиц специфически сольватированных уксусной кислотой. С увеличением доли уксусной кислоты в смешанном растворителе их концентрация возрастает за счет сдвига сольватационных равновесий, что и обусловливает увеличение скорости гидролиза.
Роль воды в реакции кислотного гидролиза бензиловых эфиров
Роль воды особенно отчетливо прослеживается при исследовании гидролиза в средах с апротонными растворителями. Для гидролитических реакций в водно-органических средах часто обнаруживают линейную логарифмическую зависимость между константой скорости и концентрацией воды либо ее активностью (аw), которая выражается уравнением lgk = А + nlgаw, где А и n – постоянные величины. Изучение данной зависимости дает возможность получить дополнительную информацию о механизме превращения. Данная зависимость имеет линейный характер, если коэффициенты активности исходного и переходного состояний изменяются синхронно при варьировании состава растворителя [12].
В случае гидролиза исследуемых эфиров 1 и 2 зависимости lgk от lgаw оказалась линейной лишь в области низких концентраций апротонного растворителя (~ до Хs = 0.2 мол. доли). При более высоком содержании диоксана и ДМСО наблюдается сильное отклонение от линейности.
Из сравнения численных значений постоянных А (табл. 2) можно сделать вывод, что изменение сольватации исходного и переходного состояний обоих эфиров в двух различных по полярности растворителях – диоксане и ДМСО – мало различается, наблюдается лишь незначительное уменьшение А при переходе от ДМСО к диоксану. Полученный результат подтверждает, что в данных растворителях кислотно-каталитический гидролиз простых бензиловых эфиров протекает по одному и тому же механизму с участием высоко полярного переходного состояния по свойствам подобно исходному состоянию [12].
Таблица 2.
Значения постоянных А и n в уравнении (3) для кислотно-каталитического гидролиза бензиловых эфиров в водно-органических средах
| Эфир | Растворитель | n | –A |
|---|---|---|---|
| 1 | диоксан–вода | 3.5 | 9.3 |
| ДМСО–вода | 3.2 | 8.9 | |
| 2 | диоксан–вода | 3.0 | 8.2 |
| ДМСО–вода | 2.1 | 6.7 |
Константа n интерпретируется как число молекул воды, принимающих участие в сольватации при переходе от исходного состояния к переходному. Для обоих эфиров наблюдается некоторая тенденция к уменьшению n при использовании ДМСО–вода, что указывает на более сильное взаимодействие ДМСО с водой.
Для кислотного гидролиза бензиловых эфиров оказалась также линейной и логарифмическая зависимость константы скорости от соотношения количества воды и апротонного растворителя в диапазоне концентраций апротонного растворителя Хs = 0.05–0.3 мол. доли (рис. 5).
Рис. 5.
Зависимости lgk = f(lg(Xs/(1 – Xs)) для гидролиза бензиловых эфиров в смесях воды с апротонными растворителями: 1 – эфир 1, 2 – эфир 2.

Тангенс угла наклона данной зависимости (m) интерпретируется как “истинное” количество молекул воды, непосредственно связанных с субстратом в активированном комплексе [12]. Для обоих изученных эфиров эта величина, рассчитанная по углу наклона прямолинейных участков этой зависимости, m = 1 и не зависит от природы растворителя. Следовательно, в смесях вода–апротонный растворитель с бензиловым эфиром в активированном комплексе прочно связана одна молекула воды, непосредственно принимающая участие в реорганизации электронных конфигураций, связанных с протеканием гидролиза.
Итак, на основании вышеизложенных экспериментальных фактов можно сделать вывод относительно влияния растворителей на скорость и механизм исследуемой реакции. Так кислотно-каталитический гидролиз бензиловых эфиров в смесях воды с апротонными растворителями диоксаном и ДМСО протекает как бимолекулярная реакция нуклеофильного замещения (SN2). Однако, естественно, это не означает, что образование новой и разрыв старой связи должен протекать строго синхронно. Переходное состояние в данном процессе обладает высокой полярностью –связь бензильного атома с уходящей группой более рыхлая, чем вновь образующаяся связь. В этом процессе первостепенную роль играет влияние апротонного растворителя на изменение сольватации исходных веществ и переходного состояния. Снижение скорости гидролиза происходит вследствие уменьшения сольватации переходного состояния за счет связывания молекул воды молекулами ДМСО и диоксана. Поскольку вода отличается от апротонных растворителей в основном способностью быть донором протонов, можно предположить, что при сольватации переходного состояния наряду с электростатическими силами важную роль играют водородные связи.
В водно-уксуснокислых средах механизм гидролиза бензиловых эфиров зависит от структуры эфира и состава среды. В высококонцентрированных растворах ионизирующего растворителя уксусной кислоты реакция протекает через образование карбокатиона в лимитирующей стадии (SN1). Этот же механизм характерен и для гидролиза бензиловой алкильной связи в разбавленных растворах уксусной кислоты, где гидролиз бензиларильной простой эфирной связи протекает по бимолекулярному механизму.
Список литературы
Шахгелдиев Ф.Х., Адилова Л.И., Сафарова Г.М., Махмудова Р.А. // Современные исследования. 2018. № 2. С. 6. http://www.nauka.org.ru/wp-content/uploads.
Барбаш В.А. // Энерготехнологии и ресурсосбережение. 2012. № 1. С. 27.
Гермер Э.И. // Лесной журнал. 2003. № 4. С. 99.
Дёмин В.А. Теоретические основы отбелки целлюлозы. СПб.: СПбГЛТУ, 2013. 100 с.
Larsson P., Lindberg B. // Acta chem. Scand. 1962. V. 16. № 7. P. 1757.
Mikawa N. // Bull. Chem. Soc. Jap. 1954. V. 27. № 1. P. 53.
Денеш И. Тирование в неводных средах. М.: Мир, 1971. 413 с.
Резников В.М., Якубовский С.Ф. // Химия древесины. 1972. Вып. 11. С. 61.
Laidler K. // Suomen kem. 1960. V. A33. № 2. P. 44.
Амис Э.А. Влияние растворителя на скорость и механизм химических реакций. М.: Мир, 1968. 328 с.
Наберухин Ю.И., Розов В.А. // Успехи химии. 1971. Т. 40. Вып. 3. С. 369.
Geheb D., Kasanskaja H.F., Berezin J.V. // Ber. Bunsenges. Phys. Chem. 1972. Bd. 76. № 2. S. 160.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии