

Журнал физической химии, 2020, T. 94, № 3, стр. 418-423
Состояние поверхности катализаторов окисления СО, полученных нанесением платины на порошок плазмохимического нитрида титана
Е. Н. Кабачков a, b, Е. Н. Куркин a, b, Н. Н. Вершинин a, И. Л. Балихин a, b, В. И. Берестенко a, Ю. М. Шульга a, c, *
a Российская академия наук, Институт проблем химической физики
142432 Московская область, Черноголовка, Россия
b Российская академия наук, Научный центр
142432 Московская область, Черноголовка, Россия
c Национальный исследовательский технологический университет “МИСиС”
119049 Москва, Россия
* E-mail: yshulga@gmail.com
Поступила в редакцию 06.05.2019
После доработки 05.06.2019
Принята к публикации 18.06.2019
Аннотация
Осаждением кластеров платины на поверхность плазмохимического нитрида титана получены катализаторы низкотемпературного окисления монооксида углерода. Поверхность катализаторов изучена методом рентгеновской фотоэлектронной спектроскопии (РФЭС). Показано, что в исследуемых катализаторах платина контактирует не только с тонким слоем диоксида титана, но и, возможно, непосредственно с оксинитридом титана. Установлено, что обработка катализатора монооксидом углерода не приводит к полному восстановлению платины. При сравнении РФЭ-спектров исходных порошков нитрида титана и катализаторов показано также, что при синтезе катализатора толщина оксидной пленки на поверхности нитрида уменьшается.
Нитрид титана (TiN) нашел широкое применение благодаря своей твердости, коррозионной стойкости и высокой температуре плавления [1–3], а также декоративным свойствам, поскольку его спектр отражения очень похож на спектр отражения золота [4–7]. В последнее время TiN стали использовать также для получения допированного азотом высокоактивного анатаза [8].
В настоящей работе методом рентгеновской фотоэлектронной спектроскопии (РФЭС) исследованы катализаторы низкотемпературного окисления монооксида углерода в комбинированных системах фотокаталитической очистки воздуха. В качестве подложки при получении катализаторов использовали ультрадисперсный порошок TiN, полученного ранее путем восстановления тетрахлорида титана в потоке азотной плазмы СВЧ-разряда [9, 10]. Исследование свойств катализаторов на подложке из нитрида титана в реакции окисления СО в малых концентрациях (менее 100 мг/м3) при 295 К показало, что при содержании платины в катализаторе от 9 до 15 мас. % скорость окисления СО в 120 раз выше скорости окисления СО на платиновой черни с удельной поверхностью 30 м2/г. Скорость окисления СО на подложке из диоксида титана марки Hombikat (Germany) при содержании платины в катализаторе 12 мас. % ниже в 1.6 раза, по сравнению с нитридом титана, содержащим 12 мас. % платины. Более подробно результаты каталитических испытаний катализатора на основе нитрида титана представлены в [11].
Вследствие большого практического интереса к TiN в литературе можно найти много публикаций, посвященных его исследованию, в том числе и с использованием метода РФЭС [12–19]. Однако, единого подхода к интерпретации экспериментальных РФЭ-спектров нитрида титана до сих пор не существует. Особенно это относится к количественным оценкам, что связано как с разными способами вычитания фона, так и со сложностью самого объекта. Дело в том, что нитрид титана обладает широкой областью гомогенности и склонностью к окислению. Состав и структура окисленного слоя на поверхности нитрида титана зависят как от способа получения и условий хранения, так и от размера частиц.
Цель настоящей работы заключалась в определении электронного состояния атомов платины на поверхности катализатора, а также в аттестации поверхности подложки и выяснения изменений, которые могут происходить с подложкой в процессе синтеза катализатора.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез нитрида титана
Порошки нитрида титана получали водородным восстановлением тетрахлорида титана в потоке микроволновой плазмы азота при атмосферном давлении. Смесь паров тетрахлорида титана с водородом в необходимом соотношении вводили в плазменный поток азота со среднемассовой температурой около 3000 К, полученный в плазмотроне с помощью микроволнового генератора с частотой 2450 МГц и максимальной полезной мощностью 5 кВт. Дисперсностью получаемых порошков управляли изменением расхода TiCl4, который составлял 0.1 г/мин при получении порошка TiN со средним размером частиц 18 нм и 0.25 г/мин при получении порошка с размером частиц 36 нм, расход плазмообразующего азота в обоих случаях составлял 4 м3/ч, расход водорода – 0.5 м3/ч. Химическое взаимодействие реагентов и конденсация наночастиц нитрида титана происходили в трубчатом реакторе диаметром 50 мм и длиной 250 мм, внутренние стенки которого были футерованы кварцем. Образовавшиеся в реакторе частицы нитрида титана после охлаждения потока отделяли от газовой фазы фильтрацией на рукавном фильтре.
Синтез композита с платиной
Для получения катализатора на основе наночастиц нитрида титана использовали метод осаждения кластеров каталитического металла (Pt) на поверхность носителя TiN при восстановлении платинохлористоводородной кислоты H2PtCl6 · 6H2O формиатом лития LiCOOH в водном растворе, содержащем взвешенные частицы TiN, аналогичный методу, описанному нами ранее [20]. Катализатор получали следующим образом: при 20°С водный раствор H2PtCl6 · 6H2O (10‒2 моль/л) смешивали с водным раствором формиата лития (0.04–0.1 моль/л). Затем в нагретую до 60°С водную суспензию нитрида титана с концентрацией твердых частиц 0.8 г/л, полученную с помощью ультразвуковой обработки, вводили необходимое количество водного раствора H2PtCl6 · 6H2O и формиата лития. После периода индукции (8–15 мин) происходит осаждение кластеров платины на поверхности наночастиц нитрида титана. После выдержки раствора в течение 24 ч при комнатной температуре проводили промывку дистиллированной водой (5–6 раз) катализатора от продуктов реакции. Катализатор СО сушили при температуре 80°С в течение 24 ч. Затем проводили частичное восстановление кластеров платины в среде азота, с объемной долей СО 10% при температуре 90°С в течение 4 ч. Для исследования выбирали два катализатора К18 и К32 с содержанием платины 12 мас. %. Катализаторы К18 и К32 различались тем, что в качестве подложки в них использовали нитрид титана со средним размером частиц 18 и 32 нм соответственно. Средний размер частиц нитрида титана определяли из результатов измерения удельной поверхности порошков по низкотемпературной адсорбции молекулярного азота (метод БЭТ). Расчет размера частиц l проводили по формуле Sуд= = 6/lρ, где Sуд – удельная поверхность порошка, ρ – удельная плотность нитрида титана.
Метод исследования
РФЭ-спектры получали с использованием электронного спектрометра Specs PHOIBOS 150 MCD и рентгеновской трубки с магниевым анодом (hν = 1253.6 эВ). Вакуум в камере спектрометра не превышал 4 × 10–8 Па. Спектры регистрировали в режиме постоянной энергии пропускания (40 эВ для обзорных спектров и 10 эВ для отдельных линий). Обзорный спектр записывали с шагом 1.00 эВ, тогда как спектры отдельных линий –с шагом 0.03 эВ. Вычитание фона проводили по методу Ширли [21], декомпозицию спектров – по набору смешанных гауссовских/лоренцевых пиков в рамках программного обеспечения Casa XPS 2.3.19. Для количественных оценок использовали табличные значения удельных плотностей (4.24 г/см3 для TiO2 и 5.44 г/см3 для TiN), а также следующие значения глубин выхода фотоэлектронов [22]: λ1 = $\lambda _{{{\text{Ti}}2p}}^{{{\text{Ti}}{{{\text{O}}}_{2}}}}$ = 3.08 nm, λ2 = $\lambda _{{{\text{Ti}}2p}}^{{{\text{TiN}}}}$ = 1.73 nm.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Исходный нитрида титана
На рис. 1 представлен обзорный спектр одного из образцов плазмохимического нитрида титана. Заметим сразу, что спектры других образцов принципиально не отличаются от приведенного. В табл. 1 перечислены аналитические линии для исследуемого образца, их интенсивности и рассчитанные из этих интенсивностей содержания элементов (в атомных процентах) в слое образца, анализируемого методом РФЭС (2–4 нм). Видно, что частицы нитрида покрыты толстым слоем загрязнений, происхождение которых связано с высокой активностью наночастиц нитрида титана и условиями их достаточно длительного хранения на воздухе. Присутствие кремния и серы в образце мы связываем с особенностями технологии получения нитрида титана.

Таблица 1.
Состав (ат. %) исследуемых образцов, рассчитанных из интенсивностей аналитических линий РФЭ-спектров
| Образец | C | N | O | Pt | Si | Ti | S |
|---|---|---|---|---|---|---|---|
| TiN | 58.8 | 6.3 | 23.3 | – | 2.5 | 7.1 | 1.8 |
| К32 | 75.9 | 1.8 | 15.4 | 0.4 | 1.2 | 4.9 | >0.1 |
| К18 | 54.3 | 5.6 | 26.5 | 0.9 | 0.2 | 12.3 | >0.1 |
Проанализируем форму спектров Ti2p и N1s. Известно, что спектр Ti2p индивидуального соединения титана представляет собой спин-орбитальный дублет, который описывается двумя пиками (Ti2p1/2 и Ti2p3/2) с соотношением интенсивностей 1 : 2 и расстоянием между пиками 5.7 эВ [23]. Полученный нами экспериментальный спектр Ti2p плазмохимического нитрида титана хорошо описывается шестью пиками или тремя дублетами (рис. 2), соответствующими нитриду (1), оксинитриду (2) и оксиду (3). Положения и относительные интенсивности пиков Ti2p3/2 приведены в табл. 2.

Таблица 2.
Положения, полуширины и интенсивности пиков Ti2p3/2, полученные при декомпозиции спектров Ti2p исходного порошка плазмохимического нитрида титана и катализаторов на его основе
| Образец | Пик | Eb, эВ | FWHM, эВ | I, % | d, нм |
|---|---|---|---|---|---|
| TiN | 1 | 455.7 | 1.8 | 15.1 | |
| 2 | 457.3 | 1.7 | 6.9 | 3.7 | |
| 3 | 459.0 | 1.8 | 44.7 | ||
| K18 | 1 | 456.1 | 2.0 | 30.3 | |
| 2 | 457.6 | 1.6 | 11.2 | 1.6 | |
| 3 | 459.1 | 1.7 | 25.1 | ||
| K32 | 1 | 455.8 | 1.9 | 29.4 | |
| 2 | 457.4 | 1.7 | 13.1 | 1.5 | |
| 3 | 458.9 | 2.1 | 24.1 |
Оценка толщины оксидной пленки d, рассчитанная по формуле (см., например, [21]):
где I1 и I3 – интенсивности пиков в табл. 2, $\lambda _{{{\text{Ti}}2p}}^{{{\text{Ti}}{{{\text{O}}}_{{\text{2}}}}}}$ – глубина выхода фотоэлектронов Ti2p из оксидного слоя на поверхности нитрида, дает d = 3.7 нм. Формула (1) для мелкодисперсных частиц дает завышенное значение d, поскольку она выведена для плоского бесконечного образца, покрытого оксидной пленкой, и не учитывает вклады в интенсивность I3 оксидной пленки от боковых поверхностей наночастиц TiN.
Расчет отношения I3/I1, проведенный для наночастицы кубической формы в предположении, что эмиссия фотоэлектронов регистрируется в направлении, перпендикулярном одной из граней куба, дает следующее выражение:
(2)
$\begin{gathered} {{I}_{{\text{3}}}}{\text{/}}{{I}_{1}}\, = \,A{{l}^{2}}[1\,--\,(1\,--\,2d{\text{/}}l)\exp (--d{\text{/}}{{\lambda }_{1}})\left] / \right[{{(l\,--\,2d)}^{2}}\, \times \\ \times \;\exp (--d{\text{/}}{{\lambda }_{2}})], \\ \end{gathered} $Сравнивая значения d, полученные по формулам (1) и (2), можно считать, что толщина оксидной пленки на поверхности исследуемых наночастиц составляет приблизительно 2 нм. Это означает, что в случае частиц размером 16 нм объем оксидной пленки составляет 33% от всего объема частицы.
На спектре N1s (рис. 3) помимо основного пика, относящегося к азоту в решетке мононитрида (Есв = 397.1 эВ) (см., например, [12, 23]), можно выделить еще два пика с Есв = 399.2 и 401.6 эВ (табл. 3). Согласно данным [24, 25], пик с Есв = = 399.2 эВ можно связать с атомами азота в решетке оксинитрида Ti(N,O). Тем не менее, однозначно установленного соответствия между пиками 2 на спектрах N1s и Ti2p, по нашему мнению, пока нет. Как будет показано ниже, в оксидном слое также могут находиться атомы азота с Есв ~ 399.2 эВ. Что касается отнесения пика с Есв = 401.6 эВ, то в литературе этот пик часто приписывают молекулярному азоту [25–27], который образуется при окислении нитрида. Однако, если обратиться к РФЭ-спектрам индивидуальных комплексов переходных металлов с молекулярным азотом [28–32], то мы обнаружим что линия N1s расщепляется на два пика. Если предположить, что N2 координируется как в биядерном комплексе, то энергия связи должна быть такой же как у экзо-атома моноядерного комплекса, т.е. ниже указанной величины не менее, чем на 1 эВ. По нашему мнению, происхождение пика с Есв = = 401.6 эВ остается до сих пор под вопросом. Более того, многие авторы не отмечают этот пик. По нашему мнению, вклад в фотоэмиссию вблизи 401.6 эВ могут давать адсорбированные молекулы NO, атомы азота в решетке TiO2 и фотоэлектроны N1s от нитридного азота, которые потеряли часть своей энергии на возбуждение переходов электронов из зоны проводимости в свободную зону. Кстати, асимметрию фотоэлектронного пика можно также частично объяснить взаимодействием внутренней дырки с электронами проводимости [33, 34]. Таким образом, исходный порошок нитрида титана покрыт довольно толстой пленкой оксида титана, которая в своем составе содержит атомы азота. Между нитридом и оксидом на его поверхности находится тонкий слой оксинитрида.

Таблица 3.
Положения, полуширины и интенсивности пиков, полученные при декомпозиции спектров N 1s исходного порошка плазмохимического нитрида титана и катализаторов на его основе
| Образе | Пик | Eb, эВ | FWHM, эВ | I, % |
|---|---|---|---|---|
| TiN | 1 | 397.1 | 2.3 | 71.3 |
| 2 | 399.2 | 2.5 | 19.3 | |
| 3 | 401.6 | 2.8 | 9.2 | |
| K18 | 1 | 397.1 | 2.0 | 18.1 |
| 2 | 399.4 | 2.6 | 80.1 | |
| 3 | 401.6 | 1.9 | 1.7 | |
| K32 | 1 | 397.1 | 2.7 | 30.8 |
| 2 | 399.4 | 2.3 | 61.4 | |
| 3 | 401.6 | 2.7 | 7.7 |
Устойчивость объемных образцов нитрида титана в окислительной среде хорошо известна. Понятно, что плотность атомов титана в TiO2 заметно ниже, чем в TiN (данные по удельной плотности TiO2 и TiN приведены выше). Следовательно, TiO2 на поверхности TiN не может служить защитной пленкой, препятствующей диффузии кислорода к нитриду титана. Тем не менее, многие авторы утверждают, что оксидная пленка на поверхности нитрида титана состоит из чистого TiO2 [35, 36]. Данные, полученные нами ранее [37–39] и в настоящей работе, указывают на присутствие оксинитрида в качестве переходного слоя между нитридом и оксидом. Именно этот слой и является барьером, препятствующим окислению нитрида. Эти данные также совпадают с выводами работ [40].
Катализаторы
Содержание элементов (в ат. %) в приповерхностном слое катализаторов представлены в табл. 1. Видно, что, как и в исходном образце, на поверхности катализатора присутствует достаточно толстая углеводородная пленка (большое содержание углерода). Причина появления углеводородных загрязнений поверхности указаны выше. Можно отметить, что в процессе приготовления катализаторов отношение [N/Ti]at, рассчитанное из интегральных интенсивностей линий N1s и Ti2p, уменьшается в 2 раза.
Результаты декомпозиции РФЭ-спектров катализаторов Ti2p представлены в табл. 2. Видно, что в катализаторах отношение I3/I1 меньше такового в исходном нитриде титана. Следовательно, в катализаторе уменьшается толщина оксидной пленки на поверхности нитрида. Возникает вопрос, как это могло произойти? Можно полагать, что при восстановлении платины произошло также восстановление поверхностного оксида. Однако, тогда возникает второй вопрос – в каком виде присутствует в катализаторе восстановленная часть оксидной пленки? Очевидно, что, если произошло восстановление до металла, то контакт с воздухом снова приведет к окислению металла, и формально ничего не должно поменяться. Нам кажется, что к увеличению соотношения I3/I1 приводят ультразвуковое перемешивание и последующая промывка водой в процессе нанесения платины. При этих операциях верхний рыхлый слой оксидной пленки может механически разрушаться, и отделившиеся мелкие частицы оксида удаляются из образца при промывке. Анализируя данные табл. 2, следует также отметить, что интенсивность пика 2 (I2) в катализаторах выше, чем для исходного образца.
В спектре N1s (рис. 4) катализатора основным пиком стал пик с Есв = 399.2 эВ. Синхронное увеличение интенсивностей от оксинитридного слоя в спектрах N1s и Ti2p означает, что влияние этого приповерхностного слоя на электронные свойства поверхности существенно возросло. Нельзя исключать также непосредственного контакта платины с оксинитридом, поскольку оксидный слой необязательно должен быть сплошным. Следовательно, свойства контакта между каталитически активным металлом (Pt) и подложкой в исследуемом катализаторе существенным образом отличаются от контакта Pt/TiO2.

Спектр Pt4f хорошо описывается двумя дублетами Pt4f7/2 и Pt4f5/2 (рис. 5), один из которых с Есв(Pt4f7/2) = 71.4 эВ по своему положению соответствуют металлической платине, второй (с Есв(Pt4f7/2) = 74.3 эВ) – оксиду Pt4+. Следует отметить, что обработка катализатора монооксидом углерода не приводит к полному восстановлению платины.
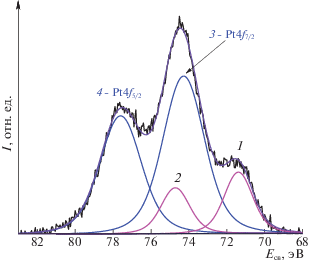
Отметим здесь также то, что интенсивность линии Pt4f заметно меньше, чем следовало бы ожидать в предположении о поверхностном распределении 12 мас. % платины (табл. 1). Если пересчитать приведенный в этой таблице поверхностный состав катализаторов из атомных процентов в массовые, то, конечно, доля платины увеличится, однако, для обоих исследованных катализаторов она останется меньше 12 мас. %. Причиной этого может быть то, что частицы платины соединяют между собой несколько частиц носителя. Такая конструкция естественным образом затрудняет выход Pt4f-фотоэлектронов. Для системы платина на TiO2 часто наблюдается такое явление, как сильное взаимодействие с носителем, когда металл закрывается оксидной пленкой [41, 42].
Таким образом, исследование методом РФЭС поверхности высокоактивных катализаторов окисления СО, полученных в результате обработки порошка плазмохимического нитрида титана платинохлористоводородной кислотой, показало, что: 1) в результате синтеза катализатора оксидная пленка на поверхности нитрида титана обогащается азотом, а ее толщина уменьшается; 2) поверхностное содержание Pt меньше объемного; 3) обработка катализатора монооксидом углерода не приводит к полному восстановлению платины.
Список литературы
Sundgren J.E., Johansson B.O., Karlsson S.E., Hentzell H.T.G. // Thin Solid Films. 1983. V. 105. P. 367.
Sproul W.D., Rudnik P.J., Gogol C.A. // Thin Solid Films. 1989. V. 171. P. 171.
Cheng H.-E., Wen Y.-W. // Surf. Coat. Technol. 2004. V. 179. P. 103.
Zega B., Kornmann M., Amiguet J. // Thin Solid Films. 1977. V. 45. P. 577.
Buhl R., Pulker H.K., Moll E. // Thin Solid Films. 1981. V. 80. P. 265.
Mumtaz A., Class W.H. // J. Vac. Sci. Technol. 1982. V. 20. P. 345.
Niyomsoan S., Grant W., Olson D.L., Mishra B. // Thin Solid Films. 2002. V. 415. P. 187.
Liu G., Yang H.G., Wang X. et al. // J. Am. Chem. Soc. 2009. V. 131. P. 12868.
Троицкий В.Н., Гуров С.В., Берестенко В.И. // Химия высоких энергий. 1979. № 13. С. 267.
Балихин И.Л., Берестенко В.И., Домашнев И.А. и др. Установка и способ получения нанодисперсных порошков в плазме СВЧ-разряда: Патент РФ № 2252817 зарегистрирован 27.05.2005 г.
Вершинин Н.Н., Берестенко В.И., Ефимов О.Н. и др. // Химия высоких энергий. 2019. № 4. С. 267.
Shulga Y.M., Troitskii V.N., Aivazov M.I., Borodko Y.G. // Zh. Neorg. Khim. 1976. V. 21. P. 2621.
Johansson L.I., Stefan P.M., Shek M.L., Christensen A.N. // Phys. Rev. 1980. V. B22. P. 1032.
Porte L., Roux L., Hanus J. // Phys. Rev. 1983. V. B28. P. 3214.
Bertoti I., Mohai M., Sullivan J.L., Saied S.O. // Appl. Surf. Sci. 1995. V. 84. P. 357.
Haasch R.T., Lee T.Y., Gall D. et al. // Surf. Sci. Spectra. 2000. V. 7. P. 193.
Bertoti I. // Surf. Coat. Technol. 2002. V. 151–152. P. 194.
Glaser A., Surnev S., Netzer F.P. et al. // Surf. Sci. 2007. V. 601. P. 1153.
Patscheider J., Hellgren N., Haasch R.T. et al. // Phys. Rev. 2011. V. B 83. P. 125124.
Вершинин Н.Н., Бакаев В.А., Берестенко В.И. и др. // Химия высоких энергий. 2017. Т. 51. С. 50.
Briggs D. and Seah M.P. (eds), Practical Surface Analysis by AES and XPS. Wiley, Chichester, 1983.
Seah M.P., Dench W.A. // Surf. Interface Anal. 1979. V. 1. P. 2.
Moulder J.F. et al. Handbook of X-ray Photoelectron Spectroscopy; Perkin-Elmer: Eden Prairie, MN, 1992.
Jiang N., Zhang H.J., Bao S.N. et al. // Physica 2004. V. B 352. P. 118.
Milosev I., Strehblow H.-H., Navinsek B., Metikos-Hukovic M. // Surf. Interface Anal. 1995. V. 23. P. 529.
Saha N.C., Tompkins H.G. // J. Appl. Phys. 1992. V. 72. P. 3072.
Esaka F., Furuya K., Shimada H. et al. // J. Vac. Sci. Technol. 1997. A. V. 15. 2521.
Leigh G.J., Murrell J.N., Bremser W., Proctor W.G. // Chem. Commun. 1970. P. 1661.
Leigh G.J., Bremser W. // J. Chem. Sot. Dalton Trans. 1973. P. 612.
Nefedov V.I., Lenenko V.S., Shur V.B. et al. // Inorg. Chim. Acta, 1973. V. 3. P. 499.
Chatt J., Elson C.M., Hooper N.E., Leigh G.J. // J. Chem. Sot. Dalton Trans. 1975. P. 2392.
Brant P., Feltham R.D. // J. Less-Comm. Metals. 1977. V. 54. P. 81.
Doniach S., Sunjic M. // J. Phys. C. 1970. V. 3. P. 285.
Islam M.N., Ghosh T.B., Chopra K.L. et al. // Thin Solid Films 1996. V. 280. P. 20.
Heide N., Schultze J.W. // Nucl. Instrum. Methods Phys. Res. 1993. V. B80/81. P. 467.
Francois J.C., Massiani Y., Gravier P. et al. // Thin Solid Films. 1993. V. 223. P.223.
Шульга Ю.М., Троицкий В.Н. // Порошковая металлургия. 1979. № 10. С. 1.
Шульга Ю.М., Клящицкий Г.Я., Рубцов В.И. и др. // Поверхность. 1991. № 9. С. 67.
Rubtsov V.I., Shulga Y.M., Troitski V.N. et al. // Phys. Low-Dim. Struct. 1995. № 12. P. 287.
Massiani Y., Medjahed A., Gravier P. et al. // Thin Solid Films. 1990. V. 191. P. 305.
Tauster S.J. // Accounts of Chemical Research. 1987. V. 20. P. 389.
Pan C.J. et al. // Journal of the Taiwan Institute of Chemical Engineers. 2017. V. 74. P. 154.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии