

Журнал физической химии, 2020, T. 94, № 3, стр. 381-392
Физико-химические аспекты ингибирования коррозии металлов
Ю. И. Кузнецов a, *, Н. Н. Андреев a, А. И. Маршаков a
a Российская академия наук, Институт физической химии и электрохимии им. А.Н. Фрумкина
Москва, Россия
* E-mail: kuznetsov@ipc.rssi.ru
Поступила в редакцию 19.06.2019
После доработки 19.06.2019
Принята к публикации 03.09.2019
Аннотация
Рассмотрены основные достижения сотрудников ИФХЭ РАН в развитии фундаментальных научных основ пассивации и ингибирования коррозии металлов и сплавов.
Систематические физико-химические исследования коррозии металлов в Академии наук СССР были начаты в 1929 г. выдающимся ученым академиком Владимиром Александровичем Кистяковским (1865–1952), основавшем коллоидно-электрохимическую Лабораторию (ЛАКЭ) (г. Ленинград). Борьба с коррозией металлов привлекала внимание В.А. Кистяковского еще в начале 20-го века. В 1907 г. он указал на возможность замедления коррозии магния введением в агрессивную среду щелочи. Он также предложил использовать замедлители для защиты нефтеналивных судов от коррозии.
В 1934 г. Академия наук была переведена в Москву, где ЛАКЭ преобразовали в Коллоидо-электрохимический институт АН СССР (КЭИН), в состав которого входил сформированный ранее в ЛАКЭ Отдел коррозии. Первым директором КЭИН также был В.А. Кистяковский, который оставался им до 1939 г. Под его руководством в 1934 г. была проведена I Всесоюзная конференция по коррозии металлов, а в 1938 г. – совещание по вопросам коррозии и борьбы с ней. Представленные на них научные труды были опубликованы. Несмотря на широкую область научной деятельности В.А. Кистяковского, следует заметить, что значительную ее часть он посвятил фильмовой (пленочной) теории пассивности металлов и разработке методов противокоррозионной защиты (ПКЗ).
В 1939 г. директором КЭИНа стал выдающийся физикохимик академик Александр Наумович Фрумкин (1895–1976), работы которого сыграли важную роль в развитии не только электрохимии, но и коррозии металлов. В 1945 г. КЭИН был переименован в Институт физической химии РАН. Его директором до 1949 г. оставался академик А.Н. Фрумкин.
В 1939 г. в КЭИН пришел другой выдающийся ученый в области физической химии и металловедения член-корреспондент АН СССР Георгий Владимирович Акимов (1901–1953), который реорганизовал Отдел защиты металлов и сыграл решающую роль в формировании отечественной школы специалистов в области коррозии и защиты металлов. Развернутые им широким фронтом работы фундаментального и прикладного характера сделали Институт головным в Академии наук по исследованию проблем коррозионного разрушения металлов и сплавов и ПКЗ.
Г.В. Акимов теоретически рассмотрел коррозию металлов в присутствии различных замедлителей, анализируя их влияние на этот процесс с помощью поляризационных диаграмм. Георгий Владимирович всесторонне поддерживал начатые во второй половине 1940-х гг. его учеником И.Л. Розенфельдом (1914–1981) широкие исследования ингибирования коррозии металлов неорганическими соединениями. Важным итогом этих исследований явилась публикация в 1953 г. первой в мире книги, полностью посвященной ингибиторам коррозии металлов (ИК) [1]11. С 1953 г. исследования ингибиторов коррозии интенсивно развивались в новой лаборатории коррозии и защиты металлических сооружений, возглавляемой И.Л. Розенфельдом. Об их важности свидетельствует и тот факт, что уже в начале 1960-х гг. она была переименована в лабораторию ИК.
Важнейшим событием явилась организация Г.В. Акимовым сети коррозионных станций как испытательного полигона для изучения не только коррозионной стойкости металлов и сплавов, но и средств их ПКЗ, в первую очередь покрытии и ИК. Г.В. Акимов еще 15.11.1945 направил Президенту АН С.И. Вавилову письмо-обоснование о необходимости создания в нашей стране сети коррозионных станций. Его инициатива была поддержана Президиумом АН СССР, и первые в стране станции начали работать уже в 1946 г., а к 1948 г. в ИФХ СССР начала функционировать их сеть: Московская промышленная, Звенигородская сельская (60 км от Москвы), Северная приморская (пос. Дальние Зеленцы на побережье Баренцева моря) и Южная приморская (г. Батуми). С 1956 г. к ним присоединились Геленджикская морская коррозионная станция, а в 1973 г. – Дальневосточная приморская (г. Владивосток). Именно с началом работы сети станций в ИФХ АН СССР стали интенсивно развиваться исследования коррозионного поведения материалов в атмосфере, почве и в море.
Закономерности атмосферной коррозии металлов (АКМ) и в настоящее время остаются важнейшими в исследованиях коррозии металлов в природных средах, поскольку она до сих пор наносит, вероятно, наибольший экономический ущерб. Весомый вклад в теорию и практику защиты от атмосферной коррозии металлов был сделан Н.Д. Томашовым, И.Л. Розенфельдом и Ю.Н. Михайловским [4], которые эффективно руководили в Институте научными коллективами сотрудников, работавшими в этой области. И.Л. Розенфельд, обобщая результаты исследований АКМ, уже в 1960 г. опубликовал, по-видимому, первую в мире монографию по этому вопросу, ценность которой не утрачена и в наши дни [5]. Более поздние достижения в изучении механизма АКМ, описании и прогнозировании скоростей коррозии металлов в разных атмосферных условиях суммированы в монографиях [6, 7]. Неудивительно, что такие исследования вызвали большой интерес к возможностям ИК, среди которых видное место заняли летучие ИК (ЛИК).
ЛЕТУЧИЕ ИНГИБИТОРЫ КОРРОЗИИ МЕТАЛЛОВ
Атмосферная коррозия. Особенность действия ЛИК – способность испаряться, в виде паров достигать поверхности металла и, взаимодействуя с ней, обеспечивать надежную защиту от коррозии. Пары ЛИК проникают в щели и зазоры, часто недоступные другим средствам защиты, обеспечивают торможение коррозии и под слоями продуктов коррозии и отложений. В ИФХ АН СССР исследования и разработка эффективных ЛИК атмосферной коррозии велись с середины 1950-х годов прошлого столетия. И.Л. Розенфельд с сотр. [8] исследовали способность к парофазной защите стали широкого круга неорганических солей, эфиров органических кислот, амидов, аминов и их солей (преимущественно бензоатов). Ими разработан комплекс ускоренных лабораторных испытаний, имитирующих естественные условия, приборы, позволяющие оценивать защитные свойства ЛИК под тонкими пленками электролита [9], а также новые методы исследования коррозионно-электрохимического поведения металлов в атмосфере ЛИК.
К середине 1960-х годов исследования ЛИК приобретают новую направленность и ориентируются на создание ЛИК, способных:
– замедлять одновременно коррозию разных металлов;
– прочно адсорбироваться на поверхности металлов, обеспечивая эффект антикоррозионного последействия;
– иметь давление насыщенных паров ЛИК (po) не ниже 10–5–10–6 мм рт. ст.
Этим требованиям удовлетворяют два класса ЛИК: соли нитробензойных кислот с аминами и диалкиламинокетоны. При разработке первого класса ЛИК, являющихся сильными окислителями, реализовался новый для того времени принцип ингибирования коррозии – перевод металлов в пассивное состояние за счет ускорения ими катодной реакции [8, 10]. Известный ЛИК этого класса – м-нитробензоат гексаметиленимина– и ингибированная им бумага (МБГИ) нашли применение для консервации сложных приборов и механизмов в разных отраслях промышленности.
Второй класс ЛИК, разработанных в конце 1960-х годах в ИФХ АН СССР, – диалкиламинокетоны – не обладал окислительными свойствами, но лучший из них – 1-диэтиламино-2-метил-бутанон-3, получивший промышленное название ИФХАН-1, отличался высокой эффективностью защиты черных и широкого набора цветных металлов. Как показано в [11], его защитные свойства связаны с необратимой адсорбцией не только самого ЛИК, но и продуктов его окисления и их димеризации на поверхности металла.
Многие органические ЛИК класса низших гетероалкилированных аминов, например, диалкиламинокетоны, являются жидкостью с высоким значением p°. Это неудобно для практики, поэтому И.Л. Розенфельд и Ю.И. Кузнецов предложили использовать в качестве носителей ЛИК силикагели и цеолиты. Данный способ защиты металлов от атмосферной коррозии с помощью ЛИК был запатентован в СССР, США, Японии, а также нескольких странах Европы и продолжает использоваться в настоящее время.
В начале 1990-х годов промышленный выпуск ЛИК, в том числе разработанных в ИФХ АН СССР, был прекращен, а сырьевая база для их производства разрушена. Кроме того, многие из ЛИК перестали отвечать ужесточившимся экологическим требованиям. Решение этой проблемы требовало развития представлений о механизмах парофазной защиты металлов и методов направленного синтеза ЛИК с заданными свойствами. Важно было разработать количественную оценку давления насыщенных паров органических соединений (р°). В связи с этим, анализируя влияние структуры органических соединений на давление их паров, Н.Н. Андреев и Ю.И. Кузнецов [12] пришли к выводу о линейности изменений стандартной свободной энергии испарения ($\Delta G_{{{\text{исп}}}}^{{^{ \circ }}}$) при варьировании заместителя R в ароматических соединениях. Оказалось, что изменения величин ln p°, функционально связанных с $\Delta G_{{{\text{исп}}}}^{{^{ \circ }}}$: ln p° = $ - \Delta G_{{{\text{исп}}}}^{{^{ \circ }}}{\text{/}}RT$, при варьировании R пропорциональны для жидких монозамещенных нафталинов, тиофенов и бензолов. По данным о p° бензолов, принятых за стандартную серию соединений, были рассчитаны и табулированы ξR – константы летучести R:
(1)
${{\xi }_{{\text{R}}}} = \lg p{{_{{(293\,{\text{К}})}}^{{{\text{o,Ph}} - {\text{R}}}}}^{{}}}--\lg p_{{(293\,{\text{К}})}}^{{{\text{o,Ph}} - {\text{H}}}},$(2)
$\lg {{p}^{{{\text{o,X}} - {\text{R}}}}} = {{\alpha }_{{\text{X}}}}~ + {\text{ }}{{\kappa }_{{\text{X}}}}{{\xi }_{{{\text{R}}.}}}$Рис. 1.
Зависимости $\lg p_{{{\text{(293 К)}}}}^{{^{ \circ }}}$ для жидких монозамещенных (а – нафталинов (1) и тиофенов (2); б – гексанов (3) и циклогексанов (4)) от ξR.
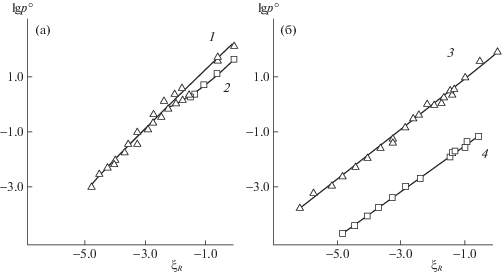
Было установлено, что вклады пространственно разделенных R в $\Delta G_{{{\text{исп}}}}^{{^{ \circ }}}$ органических веществ независимы и аддитивны. Это позволяло использовать для количественной оценки p° жидких полизамещенных соединений корреляционные уравнения вида:
(3)
$\lg {{p}^{{{\text{o,X}} - {{{\text{R}}}_{1}} \ldots {{{\text{R}}}_{n}}}}} = \alpha + {{\kappa }_{{\text{X}}}}\Sigma {{\xi }_{{\text{R}}}}.$Величины p° соединений с близко расположенными R, например о-замещенных ароматических ЛИК, выше расчетных, из-за внутримолекулярных взаимодействий и экранирования R от межмолекулярных сил. Гибкость развиваемому методу придали исследования [14], в которых установлена возможность оценки p° жидких органических ЛИК в широком диапазоне температур корреляционных зависимостей типа:
(4)
$\lg p_{{(T)}}^{{{\text{o}},{\text{X}} - {\text{R}}}} = a + b{\text{/}}T + c{{\xi }_{{\text{R}}}} + d{{\xi }_{{\text{R}}}}{\text{/}}T.$Дальнейшее развитие этого направления исследований связано с разработкой способов приближенной оценки величин ξR и методов реконструкции опорных зависимостей по косвенным данным [15]. Хотя такая система оценки летучести органических соединений существенно помогала при разработке новых ЛИК, она эффективна для оценки летучести лишь в отсутствие в соединениях ионных связей. Между тем, расчет lg p° для солей органических кислот и оснований также важен для солевых ЛИК. В связи с этим в [16] показано, что основу для таких расчетов дает рассмотрение системы “соль–ее насыщенный водный раствор–пар”. Состав и давление паров компонентов солей для веществ, не взаимодействующих с водой необратимо, идентичны таковым для индивидуальных соединений. Анализ равновесий в системе и оценка в приближениях Рауля–Генри давлений паров неассоциированных кислоты и основания в ее паровой фазе привел авторов к выводу о диссоциативном механизме парообразования солей “ониевого” типа и определил возможность расчета их p°.
В 1990-х годах в ИФХ АН СССР продолжались исследования формирования ЛИК тончайших защитных слоев и их способности к “защитному последействию”. Ю.И. Кузнецов с соавт. [17, 18], используя эллипсометрию, РФЭ- и ИК-спектроскопию, изучили наноразмерные слои, сформированные из паровой фазы на окисленном железе и порошке γ-Fe2O3 N,N-диэтиламинопропионитрилом (ДЭАПН). Оказалось, что, хотя ДЭАПН имеет высокую летучесть, его десорбция с защищаемой поверхности в атмосферу не завершается и через 8 мес. В этом важную роль играет не только хемосорбция ЛИК, но и его физическая адсорбция, которая формирует “несвязанный” и ассоциированный слои, медленно разрушающиеся даже в вакууме. Убедительные доказательства частичной необратимости адсорбции этаноламинов на железе и низкоуглеродистой стали получены в [19] эллипсометрическим методом и коррозионными испытаниями на “защитное последействие”.
В начале 2000-х годов в Институте вновь интенсифицировались исследования механизма возникновения коррозии в системах, защищенных ЛИК. Получила развитие модель, в соответствии с которой, металл не подвержен коррозии, пока концентрация ЛИК в поверхностной влаге превышает защитную. Для изолированных от окружающей среды систем защита определяется начальным периодом совместной экспозиции металла и ЛИК. Если его пары, диффундируя от источника, создают в поверхностной влаге на металле защитную концентрацию раньше, чем на нем возникнут очаги коррозии, система будет защищена длительное время. Это условие защиты положено в основу разработки модели парофазной защиты металлов над водными растворами ЛИК [20]. При этом сделан важный вывод: в тонких пленках поверхностной влаги на металле сохраняются те же закономерности влияния химической структуры ЛИК на их защитные свойства, что и в объеме раствора.
Согласно [21], причины нарушения парофазной защиты металла в герметичных объемах заключаются в перераспределении ЛИК в системе под действием теплообменных процессов, аналогичном происходящему при дистилляции растворов. Локальное обеднение поверхностного электролита ЛИК до уровня ниже защитного приводит со временем к возникновению на металле очагов коррозии. Учет этого факта привел к созданию нового способа парофазной защиты герметичных систем, основанного на использовании ЛИК, образующих с водой азеотропные смеси. При использовании таких ЛИК в дозировке, несколько превышающей его концентрацию в азеотропе, которая в свою очередь должна быть выше концентрации в поверхностной пленке, необходимой для полной защиты металла во всем возможном интервале температур среды, нельзя подобрать режим теплообмена, инициирующий коррозию. Способ защиты сталей с применением азеотропных с водой ЛИК, реализован в товарном продукте ИФХАН-8, предназначенном для защиты их в герметичных объемах.
В [22] установлено, что в неполностью герметичных объемах снижение поверхностной концентрации ЛИК и инициирование коррозии происходят за счет испарения его из системы и диффузии агрессивных компонентов атмосферы, в том числе воды, внутрь ее. Последний процесс часто определяет сроки парофазной защиты металла. Эффективность парофазной защиты металлов в таких системах может быть повышена рациональным подбором изолирующих материалов и совместным использованием ЛИК и осушителей.
Исследования защитного последействия ЛИК привели к созданию сотрудниками ИФХЭ РАН нового класса средств парофазной защиты металлов – камерных ингибиторов коррозии (КИН). Их использование подразумевает кратковременную обработку металлов парами малолетучих ИК в камерах при повышенных температурах, когда их значение р° резко возрастает. Обработанные таким образом металлы приобретают повышенную стойкость к атмосферной коррозии за счет образования наноразмерных адсорбционных пленок. Необратимость адсорбции в этом случае достигается в числе прочего за счет крайне медленного испарения КИН с поверхности. При рациональном подборе ИК камерная обработка металлов, может замедлять инициирование атмосферной коррозии в сотни и даже тысячи раз [23–27].
Углекислотная и сероводородная коррозия (СВК). ИК сталей в средах СО2 и Н2S востребованы для нефтегазовой промышленности, и неудивительно, что соответствующие исследования проводились в ИФХ неоднократно. Среди широкоизвестных достижений следует отметить разработки и исследования механизма действия диалкиламинонитрилов, проводившиеся в 1970‑х годах под руководством И.Л. Розенфельда, Л.В. Фроловой, В.М. Брусникиной и Л.П. Казанским [28].
Позднее, в середине 1990-х годов, Ю.И. Кузнецовым с сотр. начались исследования особенностей действия и разработка ЛИК для борьбы с СО2-коррозией стального оборудования и трубопроводов, продиктованные крупным заказом фирмы Phillips Petrolrum in Norway. Для борьбы с этим видом коррозии сталей традиционно для того времени основное внимание уделялось подщелачивающему действию реагентов. Однако в [29] авторами доказано, что, помимо повышения pH, важными факторами защитного действия ЛИК являются их адсорбция и влияние на формирование защитных карбонатных пленок. Убедительным доводом в пользу возможности эффективной защиты стали от СО2-коррозии не за счет изменения рН среды, служит подавление коррозии пеларгоновой или лауриновой кислотой, которые обладают неплохой летучестью.
Ю.И. Кузнецов с соавт. [30] изучили влияние различных классов органических соединений на коррозионное поведение стали в условиях, моделирующих работу систем транспортировки влажного газа, и доказали принципиальную возможность применения экологически безопасных водорастворимых ЛИК для защиты таких систем. Ю.И. Кузнецов, Р.К. Вагапов [31, 32] в это время изучали и возможность использования ЛИК и в Н2S-содержащих средах. Они впервые исследовали влияние химической структуры оснований Шиффа (ОШ) на защиту ими стали от СВК в водной и паровой фазах при рН раствора 2.5–8.4. Ими установлена линейная зависимость логарифма защитного эффекта ОШ от σ-констант Вепстера заместителей R в их молекулах и оптимум гидрофобности ОШ, превышение которого снижает степень защиты стали от СВК, в том числе и в присутствии СО2. Выявлено влияние ОШ на кинетику электрохимических реакций на стали при СВК и способность некоторых из них эффективно подавлять обе электродные реакции, предотвращая не только СВК, но и наводораживание сталей.
Позднее Р.В. Кашковский с соавт. [33, 34] продолжили исследования ЛИК при защите стали от СВК и нашли, что вторичные и третичные амины превосходят в эффективности первичные, особенно в газопаровой фазе. Их защитная способность увеличивается с ростом гидрофобности молекул, характеризуемой логарифмом коэффициента распределения амина в системе октанол–вода (lg P). Введение карбоксилатов в ингибиторы на основе летучих аминов может увеличить их защитное действие в газовой фазе, содержащей H2S. Они также доказали важную роль поверхностной пленки сульфидов железа при защите сталей такими ЛИК. Оказывается, что адсорбция ЛИК аминного типа из газовой фазы на окисленной воздухом стали протекает намного медленнее и более обратимо, чем адсорбция на сульфиде железа. Летучий амин прочно адсорбируется на сульфидной поверхности стали и даже внедряется в структуру растущего сульфида, образуя плотный защитный барьер, замедляющий образование и рост трещин.
ПАССИВАЦИЯ МЕТАЛЛОВ В ВОДНЫХ РАСТВОРАХ ИНГИБИТОРОВ
Влияние ИК на пассивацию металлов всегда было и остается одним из важнейших аспектов их действия, по крайней мере, в нейтральных средах (рН 5–9). Среди неорганических соединений хорошо известны как пассиваторы нитриты, хроматы и фосфаты, механизм действия которых И.Л. Розенфельд начал изучать еще в конце 1940‑х гг. Его книга [1] написана до появления потенциостатов и многих физико-химических методов анализа поверхности металла. Однако уже в ней предположено, что даже для ИК анодного типа, обладающих окислительными свойствами, эффективность защиты от коррозии не всегда связана с их восстановлением, т.е. окислением ими поверхности. Позднее И.Л. Розенфельд снова обратился к изучению пассивации металла ИК [35] и объяснял пассивацию стали нитритом тем, что его адсорбция уменьшает концентрацию свободных электронов в оксидной пленке, препятствуя переносу через нее катионов железа в раствор. И.Л. Розенфельд исследовал механизм защитного действия и других неорганических анионов ${\text{МеО}}_{4}^{{n - }}$, среди которых ведущее место занимают хроматы. Специфическое действие этих ИК он связывал с тем, что, адсорбируясь на поверхности металла, они понижают общую свободную энергию системы и повышают стабильность оксидных пленок. В области потенциала полной пассивации (Епп) формирование пассивных слоев берет на себя кислород воды для этих ИК. Термодинамический расчет показал, что Епп соответствует потенциалу превращения оксида Fе3О4, формирующегося при более отрицательных потенциалах в оксид, близкий по составу к γ-Fе2О3.
И.Л. Розенфельд с сотр. в 1970-х годах начали исследования гетерополисоединений, способных проявлять сильные окислительные свойства в качестве ИК металлов в воде. Они оказались весьма эффективными при высоких температурах для защиты алюминиевых сплавов (t < 300°С). Эти исследования, продолженные в 1980–1990-х годах С.В. Олейником с сотр. [36, 37] показали, что фрагменты ИК, внедряясь в растущую оксидную пленку, модифицируют ее и придают ей не только антикоррозионные, но и противонакипные свойства. Позднее авторы [38], подробно исследовав ингибирование коррозии стали в 65% LiBr фосформолибдатом при t = 80–230°C, выявили пути его оптимального использования.
В середине 1970-х годов в лаборатории И.Л. Розенфельда, Ю.И. Кузнецов с сотр. приступил к широкому изучению органических ИК металлов для нейтральных сред. В 1976 г. ими показано, что при изменении химической структуры ИК его влияние на плотность тока пассивации (iп) железа можно количественно описать с помощью принципа ЛСЭ [39, 40]22. За критерий эффективности ИК здесь принимался логарифм коэффициента торможения растворения металла lg γан, где γан = iп,фон/iп,ин, а характеристикой полярности R его служит σ-константа Гамета или Тафта (для орто-замещенных соединений). Оказалось, что угловой коэффициент в уравнении Гамета–Тафта ρ = 0.57, характеризующий чувствительность реакционной серии к введению R, одинаков для акцепторных и донорных заместителей, хотя каждый из них образует отдельную ветвь V-образной зависимости33:
где знаки “+” и “–” указывают соответственно на донорную или акцепторную природу R. Этот подход дал ценную информацию о механизме действия ИК. Из многих систем, к которым он применялся, заслуживает внимания случай пассивации Cu и Zn в 0.5 М буферном растворе NaH2PO4 + NaOH (рН 11.7), исследованном в начале 1980‑х гг. Ю.И. Кузнецовым и Л.П. Подгорновой [40]. На примере 19 двузамещенных бензимидазола (БИ) они показали, что введение в БИ одинаковых полярных групп противоположным образом влияет на эффективность защиты меди: и цинка: где σI – индукционная константа R в ароматических соединениях, учитывающая его влияние на электронную плотность реакционного центра только по индукционному механизму. Аналогичные зависимости получены и при испытаниях, в которых определяли минимальную величину Син, при которой пассивируется металл. Противоположный характер влияния R на эффективность ИК объяснен различной способностью исследуемых металлов к образованию σ- и π-связей в их комплексах. В первом случае введение электроноакцепторных R в ИК облегчает пассивацию Сu, а во втором – введение электронодонорных R упрочняет σ-связь ИК с металлом и улучшает защиту Zn. Такие же закономерности наблюдали позднее при защите этих металлов двузамещенными бензтиазолов и 5(6)-нитро-БИ [41]. РФЭС-анализ поверхности пассивных пленок, образованных на Cu и Zn, подтвердил важную роль формирования комплексов этих органических ИК с металлом.В середине 1970-х гг. Ю.И. Кузнецов с соавт. [40], используя кулонометрический метод анализа пассивных пленок, впервые доказали возможность пассивации железа в нейтральных растворах за счет лишь адсорбции органических соединений (фенилантранилатом натрия, ФАН или диметилсульфоксидом). При этом пассивирование металла сопровождалось подавлением и его окисления, поэтому его иногда называют “безоксидной пассивацией”. Для подтверждения этого вывода были использованы различные методы анализа поверхности железа: эллипсометрический, электронография, вторичная ионная масс-спектрометрия и РФЭС.
В связи с важностью оценки взаимодействия ИК с металлом большое значение приобрело изучение формирования его первого адсорбционного слоя на металле методом in situ. Н.П. Андреева и Ю.И. Кузнецов в качестве такого метода широко использовали отражательную эллипсометрию. Важное преимущество ее применения в водных растворах – возможность сочетания этих измерений с электрохимическими.
Эллипсометрия может быть использована для изучения адсорбции органических ИК на поверхности электрода только при условии, что измеряемые углы Δ и Ψ при заданном электродном потенциале в исходном растворе не меняются во времени [40, 42]. Только в этом случае, различия в значениях Δ и Ψ при добавлении ИК, введенного в электрохимическую ячейку, служат мерой его адсорбции на электроде. Наилучшие условия для таких исследований получаются на электродах при потенциалах, соответствующих отсутствию интенсивного катодного выделения Н2 или анодного растворения металла. Зависимость изменения эллипсометрического угла (δΔ = Δ – Δ0, где значение Δ0 измерено на электроде перед добавлением изучаемого адсорбата) от концентрации адсорбата (Cин) может быть использована для определения типа изотермы адсорбции, а ее уравнение – для расчета констант адсорбции. В случае окисленного электрода следует полагать, что изменение концентрации электронов в поверхностном слое вследствие адсорбции несущественно влияет на величину Δ. Это утверждение основано на результатах работы [43]. Поскольку величина δΔ при адсорбции органического соединения, как правило, во много раз выше (иногда на несколько порядков), они были связаны в наших работах исключительно с изменением количества адсорбированного вещества или иона.
Еще в наших первых работах при изучении адсорбции ФАН в боратном буфере с рН 7.4, были выявлены особенности взаимодействия ИК с восстановленной и окисленной поверхностью чистого железа. Эллипсометрические изотермы согласуются с результатами исследования адсорбции методом СЭИ. Параметры изотерм, полученных с помощью двух независимых методов адсорбции ФАН на железном электроде, были очень близки. Несмотря на низкую величину ($ - \Delta G_{A}^{0}$) (<20 кДж/моль), доказано, что адсорбция ФАН может быть частично необратимой, и возможна пассивация металла даже в отсутствие оксидов железа. Аналогичные исследования, проводившиеся в нашем Институте уже более четверти века не только на Fe и сталях, но и на Сu, ее сплаве, Zn, Ni, Mg и сплаве Д 16, часто совместно с РФЭС-исследованиями, позволили получить ценную информацию о механизме адсорбции и защитного действия ИК, принадлежащих к различным классам органических соединений. Их подробный анализ выходит за рамки настоящей статьи, что отчасти можно найти в монографиях [7, 40] и обзорах [41, 42, 44–47].
Здесь же в качестве примера отметим работу [48], в которой изучена адсорбция БТА и нескольких его замещенных на Сu, а также их влияние на анодное растворение этого металла. Авторы, изучая адсорбцию этих ИК на Сu, адекватно описали ее полной изотермой Темкина:
(6)
${{\Theta }_{i}} = \frac{1}{{{{f}_{i}}}}\ln \frac{{1 + {{n}^{{ - 1}}}{{B}_{{i,\max }}}({{C}_{i}} - {{C}_{{i0}}})}}{{1 + {{n}^{{ - 1}}}{{B}_{{i,\min }}}({{C}_{i}} - {{C}_{{i0}}})}},$Они нашли, что величины ($ - \Delta G_{{a,\max }}^{0}$) линейно увеличиваются с ростом логарифма коэффициента распределения (lg P), характеризующего гидрофобность ИК в ряду БТА < 5-СН3-БТА < 5-Сl-БТА < 5-C5H11-БТА. Логарифм минимальной концентрации БТА и его производных, достаточной для самопроизвольной пассивации меди в боратном буфере с рН 7.4, содержащем 10 ммоль/л NaCl, а также величина защитного эффекта, хотя и не столь в явном виде, как ($ - \Delta G_{{a,{\text{max}}}}^{0}$), также линейно коррелирует с величиной lg P.
ИНГИБИРОВАНИЕ КИСЛОТНОЙ КОРРОЗИИ МЕТАЛЛОВ
Первые в ИФХ РАН исследования ИК в растворе кислот выполнялось в конце 1960-х годов И.Л. Розенфельдом и Д.М. Крамаренко, которые нашли, что из 40 изученных органических ИК наиболее эффективными оказались сульфоксиды, среди которых выделялись ди-н-бутилсульфоксид и ди-н-гексилсульфоксид (Z = 90–99%). Их защитный эффект вначале растет с увеличением алкила, а затем падает вследствие уменьшения растворимости высших гомологов в кислоте [49]. В лаборатории И.Л. Розенфельда эпизодически выполнялись и другие работы, связанные с ингибированием кислотной коррозии сталей, но систематические исследования по этой тематике стали проводится позднее.
В 1980-х гг. М.В. Поспелов с сотр., исследуя защиту стали от коррозии в 1.0 М Н2SO4 алифатическими аминами, нашли, что увеличение их эффективности продолжается с ростом концентрации Син вплоть до критической концентрации мицелообразования (ККМ) в этой среде. Описав зависимость скорости коррозии стали от Син уравнением изотермы Фрумкина и выявив сильное притяжение между адсорбирующимися частицами ИК, они пришли к выводу о важности гидрофобного взаимодействия алкилов. В этих работах проводилась параллель между поверхностной активностью аминов на границе раствор/воздух и их ингибирующим действием. Развивая эти исследования и расширяя круг органических ИК сталей (амины, диамины, ЧАС с С6–С16 алкилами или перфторалкилами) М.В. Поспелов с сотр. пришли к выводу (1990 г.), что адсорбция ингибирующих катионов и образование плотного защитного слоя обусловлены силами гидрофобного взаимодействия их неполярного радикала. При достаточно длинной неполярной цепи структура полярной группы и заряд на азоте существенно не влияют на эффективность ингибиторов этих классов. Для всех их алкил с С12 обеспечивает максимальную эффективность защиты, она уменьшается в ряду: алкилдиэтиламины > соли алкилпиридиния > алкилдиамины > > первичные алкиламины.
Начиная с 1980-х годов, в Институте развивается новое научное направление: изучение закономерностей взаимосвязи парциальных электродных реакций, протекающих при коррозии металлов. Были обоснованы представления об ингибирующем действии промежуточных продуктов восстановления кислородсодержащих окислителей (Ох), в частности, ионов ОН-, на коррозию металла [50–53].
Электровосстановление Ох в нейтральных средах может быть представлено в общем виде:
(7)
${\text{Ох}} + m{\text{e}} + p{{{\text{H}}}_{{\text{2}}}}{\text{O}} \to {\text{Red}} + n{\text{O}}{{{\text{H}}}^{ - }},$(8)
$\lg \left( {\frac{{{{i}_{{\text{a}}}}}}{{i_{{\text{a}}}^{{\text{0}}}}}} \right) \approx - \frac{{{\gamma }{{С}_{{{\text{O}}x}}}}}{{A\sqrt С {\text{ }}}},$Уравнение (8) показывает связь между защитной концентрацией окислителя при постоянном коэффициенте торможения растворения металла, например, при $\lg \left( {\frac{{{{i}_{{\text{a}}}}}}{{i_{{\text{a}}}^{{\text{0}}}}}} \right) = - 1$, и величиной γ. Эффективность оксоанионов как ИК железа в нейтральном сульфатном растворе растет в ряду: ${\text{NO}}_{2}^{ - }$ (γ = 0.5) < ${\text{CrO}}_{4}^{{2 - }}$ (γ = 0.66) < ${\text{MoO}}_{4}^{{2 - }}$ (γ = 1) < ${\text{WO}}_{4}^{{2 - }}$ (γ = 2) [53], что подтверждается линейной зависимостью защитной концентрации окислителей от величины 1/γ в соответствии с (8) (рис. 2).
Рис. 2.
Зависимость защитной концентрации ингибитора от 1/γ при коррозии железа в 0.05 М растворе Na2SO4 (рН 7). Окислители: NaNO2 (1), K2CrO4 (2), Na2MoO4 (3), K2WO4 (4).

Эти представления использованы для объяснения влияния органических окислителей (нитробензоата натрия, 2,4-динитробензоата натрия, нитрофенола, толухинона) на процесс анодного растворения железа [54]. Отмечено, что продуктами восстановления органических окислителей, кроме поверхностных ионов ОН-, являются органические катионы, которые должны образовывать запирающий слой, адсорбируясь на отрицательно заряженной поверхности металла. Данное предположение подтверждено при изучении совместного действия неорганических Ох и органических аминов, которые образуют органические катионы в результате протонирования. Показано, что в присутствии таких смесей степень торможения коррозии железа пропорциональна коэффициенту γ, который характеризует количество “избыточных” поверхностных ионов OH-, образующихся при восстановлении Ох [55].
Таким образом, на основании представлений о влиянии продуктов реакции восстановления кислородсодержащих окислителей на кинетику сопряженного анодного процесса удалось получить количественную зависимость между степенью торможения растворения металла, концентрацией и природой ингибитора окислительного типа.
В лаборатории Ю.И. Кузнецова систематические исследования ИК металлов в водных растворах кислот начали проводиться с 2006 г. (совместно с доцентом Калужского педагогического университета Я.Г. Авдеевым, впоследствии докторантом, а затем ведущим научным сотрудником этой лаборатории). Важную роль в становлении этого научного направления сыграла разработка и всестороннее изучение высокоэффективного ИК для растворов различных кислот и СВК производного аминотриазола, ИФХАН-92 [56–60]. На его примере были изучены особенности механизма защитного действия ИК в условиях высоких температур (до 200°С). Доказано, что такие ИК должны быть термостабильны, а их защитное действие обусловлено хемосорбцией с последующим формированием полимолекулярной пленки сложного состава.
Другое важное достижение теории ингибирования кислотной коррозии сталей – развитие физико-химических подходов к созданию на основе фосфорнокислых сред ингибированных растворов кислот, устойчивых к накоплению катионов Fe(III) [61–65]. Давно известно, что присутствие Fe(III) в кислотных растворах обычно существенно снижает защитное действие ИК, вводимых в травильный раствор. Среди причин этого негативного явления называют выведение ИК из раствора в результате коагуляции и слабое торможение им восстановления Fe(III), протекающего в диффузионном режиме. Однако фосфорная кислота имеет ряд преимуществ перед другими растворами минеральных кислот, поэтому разностороннее изучение эффективности ИК в ее растворах позволило установить состав, структуру и защитное последействие полислоев ИФХАН-92, формирующихся на поверхности стали в растворах H3PO4. Разработаны новые композиции на основе производных триазолов для защиты низкоуглеродистой стали в растворах H3PO4. Доказана уникальная способность композиции ИФХАН-92 с KCNS сохранять замедляющее действие на коррозию стали в фосфорнокислых растворах в присутствии катионов Fe(II) и Fe(III), что важно при использовании ИК в производственных условиях.
Этот факт позволил авторам, исследовавшим методом потенциометрии электрохимическое поведение катионов Fe на платиновом электроде в системах H2SO4(НСl, HClO4)–H3PO4–H2O (25–95°C) [65], предложить использовать в качестве травильных растворов такие смеси, как H3PO4 с H2SO4. Они показали, что окислительная способность 2.0 M H2SO4 + H3PO4, содержащей 0.10 М Fe(III), снижается по мере увеличения в ней мольной доли H3PO4. Этот эффект авторы объяснили формированием комплексов Fe(III) с фосфат-анионами, которые являются более слабыми окислителями в сравнении с его гидратными и сульфатными комплексами. Повышение температуры вызывает увеличение окислительной способности системы H2SO4–H3PO4–H2O–Fe(III). Однако это наименее выражено в системе H3PO4–H2O–Fe(III). Добавки H3PO4 к растворам H2SO4 переводят катионы Fe(III) из гидратных и сульфатных комплексов в фосфатные комплексы, что приводит к уменьшению их подвижности в водных растворах кислот.
ИНГИБИТОРЫ КОРРОЗИОННОГО РАСТРЕСКИВАНИЯ СТАЛЕЙ
В 2014–2017 гг. в ИФХЭ РАН совместно с ООО “Газпром-ВНИИГАЗ” проведен цикл исследований влияния ИК, относящихся к разным классам органических веществ, на процесс коррозионного растрескивания под напряжением (КРН) трубной стали в растворах, моделирующих грунтовый (подпленочный) электролит [66–70]. Дефекты КРН – наиболее опасный вид подземной коррозии газопроводов высокого давления. Коррозионные трещины на внешней стороне трубы появляются под отслоившимся изоляционным покрытием, поэтому электрохимическая защита трубопроводов не может предупредить возникновение и развитие этих дефектов [71, 72]. В этой связи весьма перспективна задача создания антикоррозионных защитных покрытий нового типа, которые должны не только изолировать металл от воздействия агрессивной среды, но и предупреждать развитие КРН.
Установлено, что диметилалкилбензиламмоний хлорид (Катамин АБ), алкилкарбоксилат натрия (АКН), N-содержащие соединения на основе кислот талового масла (ИФХАН-29), композиция на основе фосформолибденовой кислоты (ИФХАН-П-3) и ряд других ИК могут замедлить рост коррозионной трещины при статических механических напряжениях трубной стали Х70 в цитратном буфере (рН 5.5), моделирующем слабокислый грунтовый электролит (рис. 3) [66]. При этом наблюдается корреляция между скоростью роста коррозионной трещины и скоростью анодного растворения стали в электролитах, содержащих ИК. Следовательно, ведущим механизмом роста трещин в трубной стали в слабокислых электролитах является локальное анодное растворение металла [66, 67].
Рис. 3.
Скорости роста коррозионной трещины (${v}$, мм/ч) в трубной стали Х70 в цитратном буфере (рН 5.5) с добавкой 1 мМ Na2S (фон) и в присутствии различных ингибиторов коррозии: 1 – АКН, 2 – катамин АБ , 3 – АКН + катамин АБ (1 : 1), 4 – ИФХАН-П-3, 5 – ИФХАН-29.

Методом медленного растяжения образца с постоянной скоростью было изучено влияние ИК на трещиностойкость трубной стали в рН-нейтральном подпленочном электролите NS4 [73] и определены наиболее эффективные в данных условиях типы органических ингибиторов [69, 70]. В присутствии ионов сульфида процесс КРН интенсивно развивается, но ИК повышают стойкость трубной стали к КРН.
В результате разработана ингибиторная композиция КР-60, содержащая в своем составе продукты реакции жирных аминов с различными смесями высших ненасыщенных карбоновых кислот. Эффективность указанной композиции экспериментально доказана по результатам опытно-промышленных испытаний на участках магистральных газопроводов [74].
МИГРИРУЮЩИЕ ИНГИБИТОРЫ КОРРОЗИИ
Еще одна область применения ИК – защита стальной арматуры железобетонных конструкций. Срок их службы исчисляется многими десятилетиями. Однако агрессивное воздействие промышленной атмосферы, осадков, добавок, вводимых в бетон при его затворении, могут приводить к коррозии арматуры, к разрушению конструкций, иногда катастрофическому. ИК стальной арматуры железобетонных конструкций делятся на контактные и мигрирующие (МИК). Первые вводятся в бетон на стадии его затворения. Их исследованию посвящено большое количество статей и монографий. МИК наносятся на поверхность уже готовых железобетонных конструкций. Они мигрируют внутрь бетонного камня и, достигая стальной арматуры, тормозят ее разрушение. Их применение, обеспечивающее защиту конструкций, уже находящихся в эксплуатации и подверженных коррозии, весьма перспективно с экономической точки зрения.
Теория действия и методология создания МИК в значительной степени связаны с исследованиями специалистов ИФХЭ РАН. До их работ было принято считать, что проникновение МИК в бетонный камень связано с диффузией их паров. В этой связи в качестве МИК использовали летучие соединения, в первую очередь амины и аминоспирты. В работах сотрудников ИФХЭ РАН установлено, что способность МИК проникать в бетонный камень связана с капиллярными явлениями и может регулироваться подбором ПАВ [75]. Этот факт позволил отказаться от использования летучих аминов, защитная способность которых была обусловлена исключительно их способностью подщелачивать поровую жидкость бетона [76]. Вместо них были предложены МИК на основе водных растворов карбоксилатных ингибиторов и их смесей с традиционными для строительной области нитритами, модифицированных добавками ПАВ. Это оказало значительное влияние на практику противокоррозионной защиты железобетонных конструкций, повысило глубину защиты МИК и улучшило их технологические характеристики [77, 78].
Список литературы
Розенфельд И.Л. // Замедлители коррозии в нейтральных средах. М.: Изд-во АН СССР, 1953. 247 с.
Standard ISO 8044-1989
Химическая энциклопедия, М.: Сов. Энциклопедия, 1990. Т. 2. С. 434–436.
Стрекалов П.В., Маршаков А.И., Михайлов А.А. и др. // Коррозия: материалы, защита. 2005. № 12. С. 2.
Розенфельд. И.Л. // Атмосферная коррозия металлов. М.: Изд-во АН СССР, 1960. 372 с.
Михайловский Ю.Н. // Атмосферная коррозия металлов и методы их защиты. М.: Мир, 1989. 103 с.
Михайлов А.А., Панченко Ю.М., Кузнецов Ю.И. // Атмосферная коррозия и защита металлов. Монография. Тамбов: Изд. Першина, 2016. 555 с.
Розенфельд И.Л., Персианцева В.П. // Ингибиторы атмосферной коррозии. 1985. М.: Наука, 278 с.
Розенфельд И.Л., Жигалова К.А. // Ускоренные методы коррозионных испытаний металлов. (Теория и практика.). М.: Металлургия, 1966. 347 с.
Розенфельд И.Л., Кузнецов Ю.И., Кербелева И.Я. и др. // 3ащита металлов, 1975. Т.11. № 5. С. 612.
Овчинникова Н.С., Кузнецов Ю.И., Персианцева В.П. и др. // Коллоидн. журн. 1975. Т. 37. № 5. С. 991.
Андреев Н.Н., Кузнецов Ю.И. // Журн. физ. химии, 1993. Т. 67. № 9. С. 1912.
Andreev N.N., Kuznetsov Yu.I., Storozhenko T.Yu. // Mendeleev Comm. 1994. № 5. C. 173.
Андреев Н.Н., Кузнецов Ю.И., Беликова Т.В. // Журн. физ. химии. 1993. Т. 67. № 11. С. 2258.
Андреев Н.Н. // Защита металлов. 1998. Т. 34. № 2. С. 123.
Андреев Н.Н., Ибатуллин К.А. // Там же. 2002. Т. 38. № 1. С. 18.
Булгакова Р.А., Дорфман А.М., Кузнецов Ю.И. и др. // Там же. 1996. Т. 32. № 1. С. 48.
Андреева Н.П., Дорфман А.М., Кузнецов Ю.И. и др. // Там же. 1996. Т. 32. № 4. С. 437.
Андреев Н.Н., Андреева Н.П., Вартапетян Р.Ш. и др. // Там же. 1997. Т. 33. № 5. С. 521.
Андреев Н.Н., Кузнецов Ю.И., Федотова Т.В. // Там же. 2001. Т. 37. № 1. С. 5.
Андреев Н.Н., Кузнецов Ю.И. // Там же. 2002. Т. 38. № 5. С. 470.
Андреев Н.Н., Кузнецов Ю.И. // Коррозия: материалы, защита, 2004. № 3. С. 26.
Лучкин А.Ю., Гончарова О.А., Андреев Н.Н. и др. // Там же. 2017. № 11. С. 25.
Goncharova O.A., Luchkin A.Yu., Andreev N.N. et al. // Int. J. Corros. Scale Inhib. 2018. №. 4. P. 657.
Goncharova O.A., Kuznetsov Yu. I., Andreev N.N. et al. // Ibid. 2018. № 3. P. 340.
Goncharova O.A., Andreev N.N. Luchkin A.Yu. et al. // Materials and Corrosion, 2019. № 1. P. 161.
Goncharova O.A., Kuznetsov D.S., Andreev N.N. et al. // Int. J. Corros. Scale Inhib. 2019. № 2. P. 257.
Rozenfel’d I.L., Frolova L.V., Brusnikina V.M. // Soviet Scientific Reviews, Section B: Chemistry Reviews, 1987. V. 8. P. 115.
Кузнецов Ю.И., Андреев Н.Н., Ибатуллин К.А. // Защита металлов, 1999. № 6. С. 586.
Кузнецов Ю.И., Андреев Н.Н., Ибатуллин К.А. и др. // Там же. 2002. № 4. С. 368.
Кузнецов Ю.И., Вагапов Р.К. // Там же. 2001. № 5. С. 520.
Кузнецов Ю.И., Вагапов Р.К. // Там же. 2000. № 3. С. 238.
Кашковский Р.В., Кузнецов Ю.И., Вагапов Р.К. // Коррозия: материалы, защита, 2010. № 4. С. 13.
Кашковский Р.В., Кузнецов Ю.И., Вагапов Р.К. // Там же. 2011. № 1. С. 28.
Розенфельд И.Л. // Ингибиторы коррозии. М.: Химия, 1977. 352 с.
Ломакина С.В., Казанский Л.П., Шатова Т.С., Целых О.Г. // Защита металлов. 1997. № 6. С. 621.
Oleynik S.V., Anufriev N.G., Lomakina S.V. et al. // Proc. 8th Europ. Symp. on Corr. Inhhib., 1995. V. 2. P. 827.
Олейник С.В., Кузнецов Ю.И., Вартапетян А.Р. // Защита металлов, 2003. № 1. С. 16.
Жданов Ю.А., Минкин В.И. // Корреляционный анализ в органической химии. Ростов-на-Дону: Изд. РГУ, 1966. 470 с.
Kuznetsov Yu. I. // Organic Inhibitors of Corrosion of Metals. N.Y.: Plenum Press, 1996. 283 p.
Kuznetsov Yu.I. // Int. J. Corros. Scale Inhib. 2012. № 1. P. 3.
Kuznetsov Yu.I. // Ibid. 2015. № 4. P. 284.
Habraken F.H.P., Gijzeman O.L.I., Bootsma G.A. // Surface Science, 1980. № 1. P. 482.
Kuznetsov Yu.I. // Int. J. Corros. Scale Inhib. 2017. № 1. P. 1.
Kuznetsov Yu.I. // Ibid. 2016. № 4. P. 282.
Kuznetsov Yu.I. // Ibid. 2017. № 3. P. 209.
Kuznetsov Yu.I. // Ibid. 2017. № 4. P. 384.
Агафонкина М.О., Андреева Н.П., Кузнецов Ю.И. и др. // Журн. физ. химии. 2017. № 8. С. 1294.
Фролова Л.В., Кузнецов Ю.И., Вагапов Р.К. // Коррозия: материалы, защита. 2008. № 8. С. 43.
Спицын В.И., Михайловский Ю.Н. // Докл. АН СССР. 1982. Т. 266. № 5. С. 1184.
Михайловский Ю.Н. // Защита металлов. 1984. № 2. С. 179.
Михайловский Ю.Н., Попова В.М. // Там же. 1984. Т. 20. № 2. С. 204.
Михайловский Ю.Н., Засульская М.Н., Назаров А.П. и др. // Там же. 1985. № 3. С. 353.
Михайловский Ю.Н., Лукина Н.Б. // Там же. 1989. № 2. С. 197.
Лукина Н.Б., Маршаков А.И., Михайловский Ю.Н. // Там же. 1990. № 2. С. 333.
Авдеев Я.Г., Белинский П.А., Кузнецов Ю.И. и др. // Коррозия: материалы, защита, 2008. № 8. С. 16.
Авдеев Я.Г., Кузнецов Ю.И. // Там же. 2011. № 5. С. 30.
Авдеев Я.Г., Лучкин А.Ю., Кузнецов Ю.И. и др. // Там же. 2011. № 8. С. 20.
Авдеев Я.Г., Лучкин А.Ю., Кузнецов Ю.И. и др. // Там же. 2011. № 10. С. 26.
Авдеев Я.Г., Лучкин А.Ю., Кузнецов Ю.И. и др. // Там же. 2012. № 11. С. 20.
Авдеев Я.Г., Кузнецов Ю.И., Тюрина М.В. // Там же. 2012. № 5. С. 22.
Авдеев Я.Г., Тюрина М.В., Кузнецов Ю.И. и др. // Там же. 2013. № 6. С. 17.
Авдеев Я.Г., Тюрина М.В., Кузнецов Ю.И. // Там же. 2014. № 1. С. 18.
Avdeev Ya.G., Kireeva O.A., Kuznetsov D.S. et al. The Influence of // Protection of Metals and Physical Chemistry of Surfaces, 2018. № 7. P. 1298.
Avdeev Ya.G., Andreeva T.E., Panova A.V. et al. // Int. J. Corros. Scale Inhib., 2019. № 1. P. 139.
Игнатенко В.Э., Тьен Во, Маршаков А.И. и др. // Коррозия: материалы, защита. 2016. № 11. С. 27.
Marshakov A.I., Tien Vo, Ignatenko V.E. et al. // Int. J. Corros. Scale Inhib., 2017. № 2. P. 151.
Маршаков А.И., Ряховских И.В., Игнатенко В.Э. и др. // Научно-технический сборник “Вести газовой науки”. 2016. Т. 27. № 3. С. 48.
Малеева М.А., Редькина Г.В., Богданов Р.И. и др. // Коррозия. Территории Нефтегаз, 2015. № 2. С. 24.
Maleeva M.A., Ignatenko V.E., Shapagin A.V. et al. // Intern. J. Corros. and Scale Inhib. 2015. № 3. P. 226.
Арабей А.Б., Кношински З. // Коррозионное растрескивание под напряжением труб магистральных газопроводов. М.: Атлас, 2006. 106 с.
Marshakov A.I., Ignatenko V.E., Bogdanov R.I. et al. // Corrosion Sci.2014. № 3. P. 209.
Parkins R.N., Blanchard W.K., Delanty B.S. // Corrosion. 1994. V. 50. P. 394.
Арабей А.Б., Ряховских И.В., Мельникова А.В. и др. // Наука и техника в газовой промышленности. 2017. № 3. С. 3.
Старовойтова Е.В., Андреев Н.Н., Гедвилло И.А. и др. // Коррозия: материалы, защита. 2008. № 10. С. 22.
Андреев Н.Н., Пичугина Е.В., Лебедева Н.А. // Там же. 2005. № 7. С. 21.
Андреев Н.Н., Старовойтова Е.В., Лебедева Н.А. // Там же. 2007. № 5. С. 29.
Старовойтова Е.В., Гедвилло И.А., Жмакина А.С. и др. // Там же. 2009. № 6. С. 30.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии