

Журнал физической химии, 2020, T. 94, № 3, стр. 393-403
Особенности адсорбции газов, паров и жидкостей микропористыми адсорбентами
А. А. Фомкин a, *, Г. А. Петухова a
a Российская академия наук, Институт физической химии и электрохимии им. А.Н. Фрумкина
119071 Москва, Россия
* E-mail: fomkinaa@mail.ru
Поступила в редакцию 14.06.2019
После доработки 08.08.2019
Принята к публикации 03.09.2019
Аннотация
Рассмотрены основные особенности адсорбции газов, паров и жидкостей в микропористых адсорбентах, адсорбционной деформации и термодинамики адсорбции. Представлены результаты расчетов адсорбции газов на микропористых адсорбентах в области сверхкритических температур по теории объемного заполнения микропор М.М. Дубинина и методом молекулярной динамики. Показано, что при переходе через область критических состояний газовой фазы адсорбат в микропорах не претерпевает экстремальных изменений. Установлено, что при адсорбции газов и паров адсорбционная деформация “жестких” адсорбентов не превосходит 1% и в значительной степени определяется химией поверхности и пористой структурой адсорбента.
Вне зависимости от химического состава, микропористые адсорбенты, представляют собой особый класс пористых твердых тел, в которых физическая адсорбция приводит к существенному изменению свойств адсорбированного вещества. В теории объемного заполнения микропор (ТОЗМ) [1], развитой в работах школы М.М. Дубинина впервые были сформулированы основные физико-химические свойства этих систем. В таких порах вещество целиком находится в перекрывающихся адсорбционных полях противоположных стенок микропор. Поэтому его свойства существенно отличаются от свойств вещества в равновесной объемной фазе. По классификации М.М. Дубинина [2], эффективные размеры микропор соизмеримы с эффективными размерами сорбирующихся молекул. Область размеров X (диаметров или ширины) микропор распределена на ультрамикропоры (собственно микропоры, X < < 1.2–1.4 нм) и супермикропоры (1.2–1.4 нм < X < < 3.0–3.2 нм). Поры с эффективными размерами в интервале: 3.0–3.2 нм < X < 100–200 нм выделены в класс мезопор. Адсорбция в мезопорах происходит по механизму послойного заполнения, приводящего к капиллярной конденсации. Размеры мезопор намного больше размеров адсорбированных молекул. Они являются переходными между микропорами и макропорами, имеющими размеры более 100–200 нм. Адсорбция в макропорах происходит по механизму послойного заполнения поверхности. Макропоры оказывают существенное влияние на кинетику процесса, выполняя, как и мезопоры, функцию транспортных пор для адсорбируемых молекул. Аналогичные принципы положены и в основу классификации пор по размерам, принятой YUPAC [3].
Потенциальные барьеры, на входах в микропоры, в значительной степени экранируют влияние равновесной фазы и молекул соседних полостей на адсорбированные молекулы в порах. Адсорбционное поле микропористого адсорбента навязывает адсорбированному веществу свою структуру и сильно диспергирует его. Например, в каждой полости активного угля или цеолита в среднем находится от нескольких молекул до нескольких десятков молекул [1, 4]. Эти особенности микропористой системы резко меняют свойства вещества, находящегося в адсорбированном состоянии.
В процессе адсорбции, микропористые адсорбенты являются не просто инертными носителями адсорбционного поля, они также активно участвуют в адсорбционном взаимодействии [5]. Внешне это проявляется в деформации адсорбента и изменении его упругопластических свойств в зависимости от а,р,Т-параметров состояния адсорбционной системы.
Распространение адсорбционных процессов в область высоких давлений привело к развитию термодинамики адсорбции, учитывающей не только неидеальность равновесной фазы, но и неинертность адсорбента [6, 7]. Анализ поведения термодинамических функций адсорбции в зависимости от а,р,Т-параметров системы позволяет сделать выводы о состоянии и структуре адсорбированного вещества в микропорах. Пористая структура твердого тела оказывает решающее влияние на адсорбцию газов, паров и жидкостей. Реальные адсорбенты, как правило, имеют более или менее широкое распределение пор по размерам [8–10].
Особенности свойств адсорбционных систем “микропористый адсорбент–адсорбат” особенно ярко проявляются в поведении изотерм и изостер адсорбции, адсорбционной деформации адсорбента, а также в поведении термодинамических функций адсорбционных систем, их зависимости от а–р–Т-параметров состояния.
Адсорбция газов и паров на микропристых адсорбентах
В промышленности наибольшее применение нашли адсорбенты с развитой микропористой структурой, обладающие, высокой адсорбционной активностью [8, 10]. К адсорбентам такого типа относятся большинство цеолитов, а также микропористые углеродные адсорбенты и силикагели. В цеолитах микропоры образуются в процессе формирования их кристаллической структуры [4]. Микропористые углеродные адсорбенты с узким распределением пор по размерам образуются при не очень высоких степенях обгара карбонизата [9]. Исследование адсорбции газов, паров и жидкостей широкого круга веществ разной природы: Хе, Кr, Аr, N2, О2, Н2, СН4, СО2, CF3Cl, н-С4Н10, н-С5Н12, н-С7Н16, н-С9Н20, H2O, C6H6, C2H5OH в интервалах давлений (0.1 Па–150 МПа) и температур (77–600 К) на адсорбентах с узким распределением пор по размерам: цеолитах NaХ, NaА; микропористых углеродных адсорбентах: ПАУ-10, АУК показало [4, 8, 11], что для всех этих систем адсорбция обратима и растет с ростом давления.
В качестве примера на рис. 1 и 2 представлены изотермы адсорбции Хе, H2O и C6H6 на цеолите NaX с объемом микропор Wo = 0.323 см3/г и радиусом α-полостей х = 0.62 нм [12]. Аналогичные зависимости адсорбции газов от давления получены и при адсорбции на микропористых активных углях. Как следует из рис. 1, изотермы адсорбции с ростом температуры почти симбатно смещаются в область повышенных давлений. Их ход не изменяется при переходе через критическую температуру Хе (Тc = 289.7 К), что свидетельствует об отсутствии конденсационных эффектов в адсорбате. Аналогичные зависимости обнаружены и для других адсорбционных систем [13–22].
Рис. 1.
Изотермы адсорбции Хе на цеолите NaX при различных температурах: 1 – 150, 2 – 165, 3 – 180, 4 – 210, 5 – 240, 6 – 280, 7 – 296, 8 – 310, 9 – 330, 10 – 350, 11 – 370, 12 – 500, 13 – 600 К.
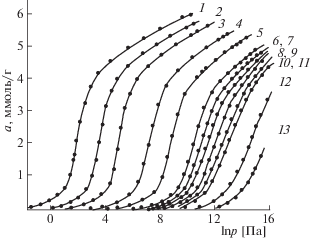
Рис. 2.
Изотермы адсорбции воды (1–3) и бензола (4, 5) на цеолите NaX при различных температурах: 1 – 303, 2 – 313, 3 – 323, 4 – 303, 5 – 323К; μ – химический потенциал равновесной фазы. Темные значки – адсорбция пара; полузатемненные значки – адсорбция на линии давления насыщенного пара; светлые значки – адсорбция жидкости при давлениях до 100 МПа.

Независимо от того, в каком состоянии находится адсорбтив, химические потенциалы μ адсорбтива и адсорбата в микропорах равны между собой. Это означает, что изотерма адсорбции а = = f(μ)T должна быть единой не только при адсорбции газа, но и при адсорбции пара и жидкости. Представленные на рис. 2 изотермы адсорбции H2O и C6H6 на цеолите NaX [23] в координатах а–μ, получены совмещением данных по адсорбции пара, жидкости, находящейся под давлением своего насыщенного пара, и жидкости при высоких гидростатических давлениях (до 100 МПа).
Химический потенциал сжатой жидкости при гидростатическом давлении P рассчитывали по уравнению:
где $P_{s}^{0}$ – давление насыщенного пара жидкости при избыточном гидростатическом давлении, равном нулю, $v\left( Р \right)$ – удельный объем сжатой жидкости. Химический потенциал насыщенного пара при избыточном гидростатическом давлении, равном нулю, ($\mu (P_{s}^{0})$ принимали за условный нуль отсчета при данной температуре.Как следует из рис. 2, изотермы адсорбции в микропористых адсорбентах не изменяют своего наклона при переходе из области пара в область сжатой жидкости через линию давления насыщенного пара. Отсутствие скачков на изотермах адсорбции, даже в области перехода изотермы в область жидкого состояния равновесной фазы, свидетельствует об отсутствии фазовых переходов первого рода в микропористых адсорбентах. Оценки числа сорбирующих полостей в цеолите [4] и активных углях [24, 25], дают величины ~1020–1021 на грамм адсорбента. Высокая дисперсность адсорбированного вещества, когда в каждой полости адсорбента может находиться в среднем до нескольких десятков молекул, мешает образованию зародышей новой фазы.
В термодинамике адсорбции важную роль играют изостеры адсорбции – кривые зависимости давления равновесной фазы от температуры при постоянной величине адсорбции. В литературе [4, 11, 26, 27] отмечается, что в координатах ln p – T–1 изостеры адсорбции в хорошем приближении остаются линейными в широких интервалах давлений и температур. На рис. 3 в качестве примера представлены изостеры адсорбции Хе на цеолите NaX.
Рис. 3.
Изостеры адсорбции Хе на цеолите NaX при различных величинах адсорбции: 1 – 0.1, 2 – 0.2, 3 – 0.4, 4 – 1.0, 5 – 2.5, 6 – 3.5, 7 – 4.0, 8 – 4.5, 9 – 4.7, 10 – 4.9, 11 – 5.15, 12 – 5.3, 13 – 5.5, 14 – 5.8 ммоль/г; lnеPs – линия давления насыщенного пара; ркр, Ткр, Ткип, Ттт – соответственно критические давление и температура, температуры кипения и тройной точки ксенона.

Из рис. 3 следует, что изостеры адсорбции Хе остаются линейными во всем интервале давлений и не изменяют своего хода при переходе через критическую температуру равновесной фазы. При высоких заполнениях объема микропор изостеры адсорбции, начинающиеся в области адсорбции пара, далее линейно продолжаются в закритической области. На рис. 3 пунктиром отмечен ход изостер в области, где равновесная фаза представляет собой сжатый жидкий ксенон. Возможность такого хода изостер адсорбции подтверждается исследованиями адсорбции воды и бензола в цеолите NaX, изученных при гидростатических давлениях до 100 МПа [23].
Описание адсорбционных равновесий на основе теории объемного заполнения микропор
Применение теории объемного заполнения микропор (ТОЗМ) для описания адсорбционных равновесий на основе структурно-энергетических характеристик адсорбента и физико-химических параметров адсорбируемых веществ общеизвестно. Теория позволяет также определить по изотерме адсорбции стандартного пара бензола структурно-энергетические характеристики адсорбента: удельный объем микропор Wo, эффективную полуширину х для щелевидных пор или радиус пор для модели цилиндрических пор и стандартную характеристическую энергию адсорбции Ео. В качестве термического уравнения адсорбции наибольшее распространение получило уравнение Дубинина–Радушкевича:
где а – адсорбция, определяемая как полное содержание адсорбата в микропорах; ао(Т) – предельная величина адсорбции данного вещества при давлении р, равном давлению насыщенного пара рs; А – дифференциальная мольной работа адсорбции; Е – характеристическая энергия адсорбции пара данного вещества. В общем случае константа в показателе степени может быть и не равна 2. Тогда уравнение носит название Дубинина–Астахова [1].При температурах Т от температуры Tкип до температуры тройной точки Ттт на линии насыщения считается, что плотность адсорбата ρad равна плотности жидкости ρl. Поэтому ао(Т) = = Woρl. Для более высоких температур, от Tкип до критической Ткр температурную зависимость предельной адсорбции рассчитывают по методу Дубинина–Николаева [1]:
(3)
${{а}_{о}}\left( Т \right) = {{а}_{о}}\left( {{{Т}_{{{\text{кип}}}}}} \right)\exp \left[ { - \alpha \left( {Т - {{Т}_{{{\text{кип}}}}}} \right)} \right],$Дифференциальная мольная работа адсорбции определяется как
где R – универсальная газовая постоянная, fs – летучесть насыщенного пара исследуемого вещества, f – летучесть равновесной фазы.В (1) характеристическая энергия адсорбции исследуемого вещества определяют как Е = βЕо, где β = Р/Ро – коэффициент аффинности, определяемый через отношение парахоров исследуемого пара Р и стандартного пара бензола Ро при температурах кипения. В ТОЗМ на основе рентгеновских исследований микропористых углеродных адсорбентов принята связь между эффективной полушириной микропор х0 и стандартной характеристической энергией адсорбции бензола Е0 в виде х0= 12/Еo.
Применение ТОЗМ в области сверхкритических температур обычно ограничено интервалом, распространяющимся на 100–150 К выше критической температуры. В этой области стандартные условия – давление ps и предельная адсорбция ао – определяются линейной экстраполяцией кривой давления насыщенного пара в закритическую область в координатах ln ps – T–1 и кривой предельной адсорбции в координатах ln ao – T по (2) [1]. Аналогичные подходы использовались и в других работах [28, 29]. Такие подходы имеют естественные ограничения, обусловленные заданием стандартных состояний ps и ao при температурах выше критической.
В [30] использован другой подход. В области температур, превышающих критическую, изотермы адсорбции газов были рассчитаны экстраполяцией, исходя из условия линейности изостер. При этом опорные изотермы адсорбции для температур кипения Ткип и критической Ткр, были рассчитаны по ТОЗМ. Целесообразность использования свойства линейности изостер адсорбции при температурах ниже и выше критической обусловлена не только экспериментальными фактами, но и близостью к линейности изостер адсорбции, рассчитанных по ТОЗМ в интервале температур Ткип ≤ T ≤ Ткр. Нелинейность изостер составляет ~10–3 1/К [31]. На основе приведенной методики в хорошем приближении были рассчитаны изотермы адсорбции СН4, Хе, Кr, Аr, СО2, N2 , O2 и др. газов. В качестве примера, на рис. 4 представлены результаты расчета адсорбции метана на микропористом углеродном адсорбенте ПАУ-10, полученном путем терморазложения поливинилиденхлорида в инертной среде. Адсорбент ПАУ-10 имеет узкое распределение пор по размерам (Wо = 0.47 см3/г; Ео = 30.0 кДж/моль; хо = 0.80 нм). На изотермах адсорбции отсутствуют гистерезис и характерный подъем, связанный с адсорбцией в мезопорах при приближении к ps.
Рис. 4.
Изотермы адсорбции СН4 на микропористом углеродном адсорбенте ПАУ-10 при различных температурах: 1 – 120, 2 – 140, 3 – 170, 4 – 190, 5 – 210, 6 – 240, 7 – 273, 8 – 303 К. Сплошные линии – расчет на основе ТОЗМ по термическому уравнению Дубинина–Радушкевича; символы – эксперимент.

Как следует из рис. 4, расчет на основе ТОЗМ, с использованием уравнения Дубинина–Радушкевича и свойства линейности изостер, удовлетворительно описывает ход экспериментальных изотерм адсорбции при давлениях до 12 МПа в докритической и сверхкритической областях температур. Наиболее существенные отклонения расчетных величин адсорбции от эксперимента наблюдаются при низких температурах в области малых заполнений. Такая ситуация не противоречит ТОЗМ [1], поскольку в ней область применимости ограничивается интервалом заполнений микропор от 0.25 до 0.95.
Адсорбция квантовых газов
Исследования адсорбции квантовых газов (гелий, водород) на микропористых адсорбентах показывают, что в области низких температур, меньших Ткр, изменение адсорбции с температурой не подчиняется обычным закономерностям, свойственным для неквантовых газов. При расчете адсорбционных равновесий по ТОЗМ это проявилось в том, что температурный коэффициент предельной адсорбции α, рассчитанный по методу Дубинина–Николаева [1], для водорода оказался отрицательным [32]. Такой результат расчета, вероятно, – следствие квантовых эффектов, отмечавшихся при адсорбции водорода в области температур ниже 50 К [33].
В связи с этим температурную зависимость предельной адсорбции водорода aо(Т) определяли с использованием корреляционной зависимости между дифференциальной мольной изостерической теплотой адсорбции среднего заполнения объема микропор адсорбента $\left\langle {{{q}_{{{\text{st}}}}}} \right\rangle $ и обратными величинами термических коэффициентов предельной адсорбции α = –(dln ao/dT)ps. На рис. 5 представлены эти характеристики для ряда систем, различающихся своими физико-химическими свойствами, в координатах $\left\langle {{{q}_{{{\text{st}}}}}} \right\rangle $ – (1/α). Сплошная кривая – аппроксимация полиномом.
Рис. 5.
Зависимость дифференциальной мольной изостерической теплоты адсорбции среднего заполнения микропор $\left\langle q \right\rangle $ различных веществ от обратной величины температурного коэффициета предельной адсорбции α для различных систем: 1 – (н-С8Н18–цеолит NaX), 2 – (Н2О – NaX), 3 – (С6Н6 – NaX), 4 – (н-С7Н16–NaX), 5 – (Н2О – NaX), 6 – (CF3C1 – NaX), 7 – (N2 – NaX), 8 – (CO2–NaX), 9 – (Ar–NaX), 10 – (N2 – активный уголь), 11 – (Кr–NaX), 12 – (CH4 – активный уголь ПАУ-10), 13 – (Xe–NaX), 14 – (CH4–NaX), 15 – точка, соответствующая адсорбции водорода. Сплошная кривая – аппроксимация.

Кривая должна исходить из нуля, поскольку теплоте адсорбции, равной нулю, соответствует нулевая энергия межмолекулярного взаимодействия “адсорбат–адсорбент” и “адсорбат–адсорбат”. Поэтому α = –(dln ao/dT)ps → ∞, а 1/α → 0. Поскольку дифференциальная теплота адсорбции водорода по данным [34, 35] составляет ~4 кДж/моль, то из рис. 5 следует, что α = 2.58 × × 10–3 К–1. Аналитически зависимость $\left\langle {{{q}_{{{\text{st}}}}}} \right\rangle $ = = f(1/α) хорошо аппроксимируется полиномом:
(5)
$\left\langle q \right\rangle = 58.131{{\alpha }^{{ - 3}}} - 2.237{{\alpha }^{{ - 2}}} + 2.8264{{\alpha }^{{ - 1}}}.$Как следует из табл. 1, к оптимальным показателям для аккумуляторов водорода (6.5 мас. %; 695 м3(Н2) м–3) [37] наиболее близки показатели адсорбции при температурах до 200 К.
Адсорбционная деформация микропористых адсорбентов
Неинертность адсорбента проявляется в виде изменений размеров и формы адсорбента при адсорбции, причем для адсорбентов, имеющих анизотропию объемных свойств, адсорбционная деформация анизотропна [38]. Энергетический вклад адсорбционной деформации может быть учтен через зависимость объема адсорбента от адсорбции.
Адсорбционная деформация микропористых адсорбентов в зависимости от а,р,Т-параметров адсорбционного равновесия может быть и положительной, и отрицательной. Для относительно “жестких” адсорбентов, таких как активные угли, цеолиты, она обычно не превышает процента. Поэтому до недавнего времени исследованию адсорбционной деформации адсорбентов уделялось мало внимания. Однако, расширение областей применения адсорбционных процессов, особенно в область высоких давлений требует уточнения вклада адсорбционной деформации в термодинамические характеристики адсорбции. Кроме того, адсорбционная деформация в значительной степени определяет механическую прочность и стабильность адсорбента в высокоскоростных процессах, таких как КБА-процессы, в которых число циклов “адсорбции–десорбции” увеличивается в тысячи раз. Теории адсорбционной деформации до недавнего времени не уделялось должного внимания. Феноменологические подходы [39–41] не смогли объяснить детали адсорбционных взаимодействий, приводящих к адсорбционной деформации, ее зависимости от температуры и заполнения микропор. В последние годы появились новые подходы, основанные на применении молекулярно-статистических методов к адсорбции в микропорах. В [42] для объяснения адсорбционной деформации нанопористых адсорбентов использована статистико-механическая теория молекулярной структуры поверхностных слоев. Показано, что наблюдаемые эффекты адсорбционного сжатия адсорбентов – следствие анизотропии тензора давления адсорбата в микропорах. В других работах использовали теорию функционала плотности [43] и молекулярно-статистические подходы [44]. Тем не менее, разработка теории адсорбционной деформации твердых тел пока находится в начальной стадии.
Зависимости адсорбционной деформации от величины адсорбции и температуры могут иметь области сжатия и расширения. На рис. 6 представлены зависимости относительной линейной адсорбционной деформации микропористого углеродного адсорбента АУК от величины адсорбции метана в области сверхкритических температур 243–393 К при давлениях до ~10 МПа.
Рис. 6.
Зависимости адсорбционно-стимулированной деформации микропористого углеродного адсорбента АУК от адсорбции СН4 и среднего количества молекул метана в единичной микропоре, при различных температурах: 1 – 177.65, 2 – 216.2, 3 – 243.3, 4 – 273.15, 5 – 313, 6 – 333, 7 – 393 К. Символы – экспериментальные данные, линии – аппроксимация.

Как следует из рис. 6, при адсорбции метана на активном угле АУК кривые адсорбционной деформации адсорбента имеют область сжатия только при относительно низких температурах (177.65–313 К). Максимальная адсорбционная деформация расширения адсорбента достигает ~0.3%; деформация сжатия не превышает 0.03%. В области более высоких температур (313–393 К) область сжатия отсутствует. Сжатие твердого тела – следствие проявления сил взаимодействия адсорбированных молекул СН4 с противоположными стенками микропор. В данном случае диаметр молекул метана 0.42 нм соизмерим с эффективной шириной пор адсорбента АУК, равной 0.82 нм. Однако с ростом заполнения объема микропор уменьшается среднее расстояние между адсорбированными молекулами СН4, а также молекулами и стенками пор, нарастают силы отталкивания, что приводит к значительному расширению адсорбента АУК. При 177.65 К и 6 МПа максимальное расширение адсорбента АУК в ~10 раз превосходит первоначальное сжатие.
С ростом температуры область начального сжатия адсорбента постепенно сужается и при ~333 К пропадает. При 393 К в начале изотермы наблюдается небольшое резкое расширение адсорбента до 0.002%, сменяющееся далее плавным ростом с увеличением адсорбции СН4. Аналогичные зависимости получены также при адсорбции СО2, Аr, Хе, Кr, СН4, N2, О2.
Причиной начального расширения адсорбента АУК не может быть только увеличение кинетической энергии адсорбированных молекул, поскольку величины адсорбции малы, а дифференциальная теплота адсорбции СН4 достаточно велика (~22 кДж/моль). Такое поведение адсорбционной деформации, вероятно, вызвано анизотропией упругих свойств углеродного адсорбента с температурой и, в частности, ослаблением связей между гексагональными слоями углерода в стенках щелевидных пор адсорбента с ростом температуры. В результате первые молекулы, попадая в пору, “стягивают” ближайшие слои углерода, что приводит к нарушению баланса сил в твердом теле, и увеличению расстояния между последующими слоями. В макроскопическом плане это приводит к расширению твердого тела. Дальнейшее расширение адсорбента с ростом адсорбции, вероятно, – следствие изменения баланса сил, связанных с увеличением внутреннего давления в адсорбате с ростом адсорбции в микропорах и увеличением сжимаемости пористого твердого тела с ростом температуры.
Адсорбция молекул, имеющих эффективные размеры, близкие к размерам микропор адсорбента имеют свои особенности. Изучена зависимость адсорбционной деформации адсорбента АУК при адсорбции н-пентана, н-гептана, н-октана при температурах 243.3, 273.15, 293, 313, 353 и 393 К. В качестве примера, на рис. 7 представлены типичные зависимости относительной линейной деформации адсорбента АУК от величины адсорбции для н-пентана. Как следует из рис. 7, адсорбционная деформация адсорбента АУК в начальной области заполнений микропор практически отсутствует. Для самой низкой температуры 243.3 К при адсорбции 4.8 ммоль/г наблюдается небольшая область сжатия до 0.01%, а затем адсорбционная деформация переходит в область расширения и стремительно растет до 0.7% при адсорбции 5.4 ммоль/г. При более высоких температурах адсорбционная деформация переходит в область расширения при меньших величинах адсорбции.
Рис. 7.
Зависимости адсорбционно-стимулированной деформации микропористого углеродного адсорбента АУК от адсорбции н-С5Н12, и среднего количества молекул н-пентана в единичной микропоре при различных температурах: 1 – 243.3, 2 – 273.15, 3 – 293, 4 – 313, 5 – 353, 6 – 393 К. Символы – экспериментальные данные, линии – сглаживающие кривые.

Причиной, вызывающей такое поведение деформационных кривых, вероятно, являются стерические соотношения и специфичность взаимодействия молекул с поверхностью стенок микропор. В области начальных заполнений, в силу близости размеров молекул и размеров микропор, энергия взаимодействия молекул со стенками пор находится в минимуме, и сила близка к нулю. С ростом заполнения микропор, нарастают силы отталкивания между всеми участниками взаимодействия, и адсорбент расширяется. Влияние стерических факторов при адсорбции, в частности, проявляется в замедленой кинетике адсорбции. Время установления равновесия при малых давлениях доходило до 5–8 ч.
Энергетика адсорбции
Энергетика адсорбции газов и паров в микропористых адсорбентах существенно отличается от адсорбции на открытой поверхности непористого твердого тела. В первом случае адсорбция происходит по механизму объемного заполнения микропор, а во втором – адсорбат образует на поверхности твердого тела отдельную фазу. Определение объема твердого тела вместе с микропорами, в общем случае достаточно сложная задача. Для кристаллических цеолитов это можно сделать на основе данных рентгеноструктурного и химического анализа [4]. В общем случае для целиком микропористых адсорбентов эта задача была решена в [45]. Адсорбция, определяемая таким образом, является полным содержанием или абсолютной адсорбцией, а не избытком, в объеме микропор.
По определению, дифференциальная изостерическая теплота адсорбции является разностью между мольной энтальпией газовой фазы hg и дифференциальной энтальпией адсорбционной системы h1 = (dH1/dа)T:
(7)
${{q}_{{{\text{st}}}}} = {{h}_{{\text{g}}}} - {{(d{{H}_{1}}{\text{/}}d{{N}_{{\text{a}}}})}_{T}} = {{h}_{{\text{g}}}} - {{h}_{1}}.$В [6, 7] показано, что дифференциальную изостерическую теплоту адсорбции можно рассчитать на основе равновесных адсорбционных и деформационных данных по уравнению:
(8)
$\begin{gathered} {{q}_{{{\text{st}}}}} = - Rz{{(d\ln P{\text{/}}d{{T}^{{ - 1}}})}_{a}}[1 - {{(d{{{v}}_{1}}{\text{/}}da)}_{T}}{\text{/}}{{V}_{{\text{g}}}}] - \\ - \;{{(dP{\text{/}}da)}_{T}}[{{{v}}_{1}} - T{{(d{{{v}}_{1}}{\text{/}}dT)}_{a}}], \\ \end{gathered} $Если давления малы, z = 1 и адсорбент не деформируется при адсорбции ${{v}_{1}} \ne {{v}_{1}}(a,T)$, то
(9)
${{q}_{{{\text{st}}}}} = - R{{(d\ln р{\text{/}}d{{T}^{{ - 1}}})}_{a}} + {{{v}}_{1}}{{\left( {dP{\text{/}}d{{a}_{a}}} \right)}_{T}}.$На рис. 8 представлены зависимости дифференциальной мольной изостерической теплоты адсорбции метана от величины адсорбции на микропористом углеродном адсорбенте ПАУ-10 при давлениях до 12 МПа. Как следует из рис. 8, учет всех поправок, в соответствии с (8), приводит к существенной температурной зависимости теплоты адсорбции. Неидеальность газовой фазы и адсорбционная деформация адсорбента практически не влияют на теплоту адсорбции при температуре 120 К. В области начальных заполнений микропор до 3 ммоль/г теплота адсорбции быстро падает с 24 до 19 кДж/моль за счет заполнения молекулами метана наиболее активных адсорбционных центров. Дальнейшее объемное заполнение микропор до 9.5 ммоль/г происходит при теплоте, близкой к ~18–19 кДж/моль. При высоких заполнениях (~10 ммоль/г) на кривой теплоты адсорбции появляется слабо выраженный максимум (~20 кДж/моль) и затем резкое падение до ~15 кДж/моль при адсорбции ~11.5 ммоль/г.
Рис. 8.
Зависимости дифференциальной мольной изостерической теплоты адсорбции СН4 на микропористом углеродном адсорбенте ПАУ-10 от адсорбции при различных температурах: 1 – 120, 2 – 150, 3 – 170, 4 – 190, 5 – 273, 6 – 370, 7 – 410, 8 – 500, 9 – 600 К.

Максимум на кривой теплоты адсорбции СН4 на ПАУ-10 появляется вследствие роста теплоты адсорбции за счет увеличения энергии притяжения “адсорбат–адсорбат” и падения теплоты за счет нарастания энергии отталкивания на малых расстояниях. Как следует из рис. 7, учет всех поправок, в соответствии с (8), приводит к существенной температурной зависимости теплоты адсорбции.
Такие эффекты часто наблюдаются при адсорбции паров на цеолитах [4].
В [46] это было подтверждено с учетом адсорбционной деформации цеолита прямым экспериментальным калориметрическим исследованием адсорбции Хе на цеолите NaX при давлениях до 20 МПа. На активных углях максимумы на кривой теплоты адсорбции обнаруживаются значительно реже и, в основном, при адсорбции на адсорбентах с узким распределением пор по размерам. Адсорбенты АУК и ПАУ-10 отвечают этим требованиям.
Анализ зависимостей изменения дифференциальной энтропии и изостерической теплоемкости адсорбционной системы показывает, что при высоких заполнениях микропор возможно образование адсорбционных ассоциатов за счет энергии притяжения между адсорбированными молекулами. Адсорбат в микропорах настолько сильно диспергирован, что образование жидкой фазы становится невозможным, но предвестники фазы – молекулярные ассоциаты – образуются. Образование ассоциатов в микропорах объясняет появление максимумов на кривых зависимости дифференциальной мольной изостерической теплоемкости адсорбционной системы Са от температуры с ростом заполнения объема микропор адсорбированными молекулами. На рис. 9 показано, что с ростом адсорбции метана, максимумы на кривых теплоемкости становятся все более выраженными.
Рис. 9.
Температурные зависимости дифференциальной мольной изостерической теплоемкости адсорбционной системы “метан – микропористый углеродный адсорбент ПАУ-10” при различных величинах адсорбции: 1 – 1.0, 2 – 3.0, 3 – 4.0, 4 – 5.0, 5 – 6.0, 6– 8,0, 7 – 9.0, 8 – 9.5, 9 – 10.0, 10 – 10.3, 11 – 10.5 ммоль/г.

Это положение проверено с использованием численного моделирования адсорбционной системы методом молекулярной динамики.
Численное моделирование адсорбции в щелевидных микропорах
В расчетах методом молекулярной динамики (МД) [25], применительно к адсорбции в модельной поре были использованы атом-атомные потенциалы в виде универсальных силовых полей при помощи пакета молекулярно-динамических программ TINKER [47] с силовыми полями OPLS-AA [48] и MM3 [49]. В качестве модели рассматривали микропору в виде “шайбы” с открытыми боковыми стенками. Адсорбирующие поверхности представляли собой графеновые однослойные плоскости. Модель микропоры диаметром 38.8 нм и шириной щели (расстоянием между центрами атомов углерода, образующих адсорбирующие поверхности) d = 0.82 нм, помещалась в центр программно задаваемой ячейки в форме параллелепипеда 8 × 8 × d нм c периодическими граничными условиями. Пространство за пределами собственно микропоры моделирует объемную фазу. В ходе численного эксперимента молекулы адсорбата могли находиться в адсорбционной, поверхностной и паровой фазах. В такую систему при постоянной температуре помещали определенное количество молекул, и проводили расчет их траекторий.
На рис. 10 и 11 представлены результаты МД-моделирования адсорбции метана в модельной щелевидной микропоре при заполнениях 0.1 и 0.9 от максимального. Как следует из рисунков, ассоциирование метана происходит в центральной части поры. Большая часть адсорбированных молекул находится в ассоциате. С ростом заполнения ассоциаты метана увеличиваются. Характерно, что структура ассоциатов двухслойная. Слои располагаются параллельно графеновым плоскостям.
Рис. 10.
Мгновенный снимок МД-траектории 20 молекул СН4 в щелевидной поре с графеновыми стенками при 273 К. Зачерненные кружки – атомы углерода.

Рис. 11.
Мгновенный снимок МД-траектории 160 молекул СН4 в щелевидной поре с графеновыми стенками при 273 К.

Проведены расчеты изотерм адсорбции метана (в широком интервале температур – ниже и выше критической), этана, пропана, бутана, этилена пропилена, бензола и этанола, а также изотерм компонентов жидкой смеси бензол–этанол и смеси газов пропан–бутан. Проведен анализ молекулярных наноструктур адсорбированных метана, бензола и этанола, а также количественный расчет концентраций и изомерного состава ассоциатов адсорбированного и жидкого этанола [50, 51].
ЗАКЛЮЧЕНИЕ
Микропористые адсорбенты, представляют собой особый класс пористых твердых тел, в которых физическая адсорбция приводит к существенному изменению свойств адсорбированного вещества. Вещество целиком находится в перекрывающихся адсорбционных полях противоположных стенок микропор. Поэтому его свойства существенно отличаются от свойств вещества в равновесной объемной фазе. Адсорбционное поле микропористого адсорбента навязывает адсорбированному веществу свою структуру и сильно диспергирует его. Адсорбенты активно участвуют в адсорбционном взаимодействии. Внешне это проявляется в деформации адсорбента и изменении его упругопластических свойств в зависимости от а,р,Т-параметров состояния адсорбционной системы. Особенности свойств адсорбционных систем “микропористый адсорбент – адсорбат” особенно ярко проявляются в поведении изотерм и изостер адсорбции, адсорбционной деформации адсорбента, а также в поведении термодинамических функций адсорбционных систем, их зависимости от а-р-Т-параметров состояния. Независимо от того, в каком состоянии находится адсорбтив, химические потенциалы μ адсорбтива и адсорбата в микропорах равны между собой. Это означает, что изотерма адсорбции а = f(μ)T должна быть единой не только при адсорбции газа, но и при адсорбции пара и жидкости. Отсутствие скачков на изотермах адсорбции, даже в области перехода изотермы в область жидкого состояния равновесной фазы, свидетельствует об отсутствии фазовых переходов первого рода в микропористых адсорбентах. В координатах ln p – T–1 изостеры адсорбции в хорошем приближении остаются линейными в широких интервалах давлений и температур. При высоких заполнениях объема микропор изостеры адсорбции, начинающиеся в области адсорбции пара, далее линейно продолжаются в закритической области. Расчет на основе ТОЗМ с использованием уравнения Дубинина–Радушкевича и свойства линейности изостер удовлетворительно описывает ход экспериментальных изотерм адсорбции при давлениях до 12 МПа в докритической и сверхкритической областях температур. Расширение областей применения адсорбционных процессов, особенно в область высоких давлений требует уточнения вклада адсорбционной деформации в термодинамические характеристики адсорбции. Кроме того, адсорбционная деформация в значительной степени определяет механическую прочность и стабильность адсорбента в высокоскоростных процессах разделения смесей газов. Зависимости адсорбционной деформации от величины адсорбции и температуры могут иметь области сжатия и расширения. Энергетика адсорбции газов и паров в микропористых адсорбентах существенно зависит от давления равновесной фазы и адсорбционной деформации адсорбента.
Анализ зависимостей изменения дифференциальной энтропии и изостерической теплоемкости адсорбционной системы показывает, что при высоких заполнениях микропор возможно образование адсорбционных ассоциатов за счет энергии притяжения между адсорбированными молекулами. Адсорбат в микропорах настолько сильно диспергирован, что образование жидкой фазы становится невозможным, но предвестники фазы – молекулярные ассоциаты – образуются. Образование ассоциатов в микропорах объясняет появление максимумов на кривых зависимости дифференциальной мольной изостерической теплоемкости адсорбционной системы Са от температуры с ростом заполнения объема микропор адсорбированными молекулами при заполнениях 0.1 и 0.9 от максимального. Структура адсорбционных ассоциатов и деформация микропористого адсорбента существенно зависят от энергии адсорбции, соотношения размеров пор и молекул адсорбата, а также от степени заполнения объема порового пространства.
Список литературы
Дубинин М.М. Адсорбция и пористость. М.: ВАХЗ, 1972. 126 с.
Дубинин М.М. // Изв. АН СССР. Сер. хим. 1974. С. 996.
YUPAC Manual of Symbols and Terminology, Appendix 2. Pt. I // Colloid and Surface Chemistry. Pure Appl. Chem. 1972. V. 31. P. 578.
Брек Д. Цеолитовые молекулярные сита /Пер. с англ. М.: Мир. 1976. 781 с.
Беринг Б.П., Майерс А., Серпинский В.В. // Докл. АН СССР. 1970. 193. 119.
Бакаев В.А. Молекулярная теория физической адсорбции: Дис. … д-ра физ.-мат. наук. М.: МГУ, 1989. 348 с.
Fomkin A.A. // Adsorption, 2005. V. 11. № 3/4. P. 425.
Поляков Н.С., Петухова Г.А. // Росс. хим. журн.1995. Т. 39. № 6. С. 7.
Петухова Г.А., Поляков Н.С., Лупашку Ф.Г., Монахова Л.И. // Изв. АН. Сер. хим. 1995. № 10. С. 1934.
Петухова Г.А. // Защита металлов. 2008. Т. 44. № 2. С. 170
Dubinin M.M., Stoeckli H.F. // J. Colloid Interface Sci. 1980. V. 75. № 1. P. 34.
Фомкин А.А., Серпинский В.В., Фидлер К. // Изв. АН СССР. Сер. хим. 1982. № 6. С. 1207.
Фомкин А.А., Серпинский В.В. // Там же. 1974. № 9. 2108.
Беринг Б.П., Селиверстова И.И., Серпинский В.В., Фомкин А.А. // Там же. С. 2816.
Чхаидзе Э.В., Фомкин А.А., Серпинский В.В., Цицишвили Г.В. // Там же. 1985. № 5. С. 974.
Чхаидзе Э.В., Фомкин А.А., Серпинский В.В. и др. // Там же. 1986. № 4. С. 929.
Харитонов А.Б., Синицын В.А., Фомкин А.А. // Там же. 2000. № 6. С. 1018.
Fomkin A.A. // Adsorption. 2005. V. 11. № 3/4. P. 425.
Школин А.В., Фомкин А.А., Яковлев В.Ю. // Изв. АН. Сер. хим. 2007. № 3. С. 382.
Фомкин А.А. // Физикохимия поверхности и защита материалов. 2009. Т. 45. № 2. С. 133.
Потапов С.В., Фомкин А.А., Синицын В.А. // Изв. АН. Сер. хим. 2009. № 4. С. 717.
Потапов С.В., Фомкин А.А., Синицын В.А., Школин А.В., Пулин А.Л. // Физикохимия поверхности и защита материалов. 2010. Т. 46. № 5. С. 445.
Фомкин А.А., Авраменко В.А., Селиверстова И.И., Серпинский В.В. // Докл. АН СССР. 1986. Т. 288. № 3. С. 678.
Дубинин М.М. В сб. Углеродные адсорбенты и их применение в промышленности. М.: Наука. 1983. С. 100.
Школин А.В. Адсорбционно-стимулированная деформация микропористого углеродного адсорбента с узким распределением пор по размерам при адсорбции углеводородов в широких интервалах давлений и температур: Дис…канд. наук. М.: ИФХЭ РАН, 2008. 263 с.
Bülov M., Shen D., Jale S. // Applid Surface Science. 2002. V. 196. P. 157.
Barrer R.M., Papadopoulos R. // Proc. Roy. Soc. London. 1972. V. A326. 1566. P. 315.
Дубинин М.М., Кадлец О., Серпинский В.В. // Изв. АН СССР. Сер. хим. 1984. С. 502.
Овчаренко И.Е., Толмачев А.М., Фомкин А.А. // Изв. РАН. Сер. хим. 1993.Т. 42. № 9. С. 1668.
Фомкин А.А., Чхаидзе Э.В., Серпинский В.В., Цицишвили Г.В., Дубинин М.М. // Изв. АН СССР. Сер. хим. 1989. № 1. С. 43.
Школин А.В., Фомкин А.А., Яковлев В.Ю. // Изв. АН. Сер. хим. 2007. № 3. С. 382.
Фомкин А.А., Синицын В.А. // Физикохимия поверхности и защита материалов. 2008. Т. 44. № 2. С. 163.
Stan G., Cole M.W. // J. Low Temp. Phys. 1998. V. 110. P. 539.
Коган В.С., Бреславец К.Г. В сб. Адсорбция и пористость / Под ред. М.М. Дубинина, В.В. Серпинского. М.: Наука, 1976. С. 270.
Бреславец К.Г., Коган В.С., Миленко Ю.Я. и др. // В сб. Вопросы атомной науки и техники, Харьков: Изд-во ФТИ АН УССР. 1972. Вып. 1 (2). С. 12.
Фомкин А.А., Синицын В.А. // Физикохимия поверхности и защита материалов. 2008. Т. 44. № 2. С. 163.
Dillon A.C., Jones K.M., Bekkedahl T.A. et al. // Nature. 1997. № 386. P. 377.
Твардовский А.В., Фомкин А.А., Тарасович Ю.И. и др. // Изв. РАН. Сер. хим. 1992. № 6. С. 1270.
Flood E.A. // Can. J. Chem. 1957. V. 35. P. 48.
Беринг Б.П., Красильникова O.K., Серпинский В.В. // Докл. АН СССР. 1976. Т. 231. № 2. С. 373.
Серпинский В.В., Якубов Т.С. // Изв. АН СССР. Сер. хим. 1981. № 1. С. 71
Русанов А.И. // Коллоидн. журн. 2007. Т. 69. С. 861.
Ustinov E.A., Do D.D. // Carbon . 2006. V. 44. P. 2652.
Kovalczyk P., Ciach A., Neimark A.V. // Langmuir. 2008. V. 24. P. 6603.
Фомкин А.А., Селиверстова И.И., Серпинский В.В. // Изв. АН СССР. Сер. хим. 1986. № 2. С. 280.
Gusev V.Y., Fomkin A.A. // Adsorption Science and Technology. 1991. V. 8. № 2. P. 75.
http: // dasher.wustl.edu/tinker/
Jorgensen W.L., Maxwell D.S., Tirado-Rives J. // J. Am. Chem. Soc. 1996. V. 118. P. 11225.
Allinger N.L., Yuh Y.H., Lii J.-H. // Ibid. 1989. V. 111. P. 8551.
Толмачев А.М., Анучин К.М., Крюченкова Н.Г., Фомкин А.А. // Физикохимия поверхности и защита материалов. 2011. Т. 47. № 2. С. 124.
Толмачев А.М., Фирсов Д.А., Анучин К.М., Кузнецова Т.А. // Там же. 2009. Т. 45. № 2. С. 177.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии