

Журнал физической химии, 2020, T. 94, № 5, стр. 657-671
Теоретические и экспериментальные исследования субнаноразмерных молекул и кластеров
О. П. Чаркин a, *, Н. М. Клименко b
a Российская академия наук, Институт проблем химической физики
Черноголовка, Россия
b Московская государственная академия тонкой химической технологии им. М.В. Ломоносова
Москва, Россия
* E-mail: charkin@icp.ac.ru
Поступила в редакцию 09.09.2019
После доработки 09.09.2019
Принята к публикации 17.09.2019
Аннотация
Дан краткий обзор теоретических и экспериментальных исследований серии субнаноразмерных молекул и кластеров, выполненных авторами совместно с сотрудниками Института атомных и молекулярных наук и Центра геномики Академии Синика (Тайпей, Тайвань) с использованием квантово-химических расчетов и экспериментальных методов лазерной спектроскопии и масс-спектрометрии в газовой фазе. Рассмотрены системы: а) фрагментация и ассоциация гема, гемина и бета-гематина; б) структура, стабильность и хиральность изомеров протонированного октамера серина; в) стабильность и структура Li- и Na-замещенных сахаридов, фуллеролов и фуллеролоподобных полигидроксильных кластеров; г) теоретическое предсказание и экспериментальный синтез и исследования эндоэдрального метан-фуллеренового кластера CH4@C60.
Наши совместные исследования с Академией Синика (Тайвань) выполнялись в основном в рамках тайваньского национального проекта, созвучного с российским проектом “Здоровье нации”. В нем, помимо прикладных направлений, значительное внимание уделялось фундаментальным исследованиям структуры, свойств и механизмов элементарных реакций биологически активных соединений, включая теоретические и экспериментальные исследования свободных (изолированных) молекул и ионов, выполняемым на молекулярном уровне.
Экспериментальные исследования изолированных биологических молекул и ионов в газовой фазе проводились в Институте атомных и молекулярных наук и в Центре геномики Академии Синика (Тайпей) с помощью масс-спектрометрических и спектроскопических методов с использованием лазерного облучения, электроспрея, столкновительной ионизационной диссоциации (CID), МАLDI и других техник.
Квантово-химические расчеты выполнялись нами (О.П.Ч. и Н.М.К.) в рамках метода функционала плотности (DFT) в приближении Бекке–Ли–Янга–Парра (B3LYP) с базисами типа 6-31G* и 6-311++G**, которые, как правило, удовлетворительно воспроизводят экспериментальные значения геометрических параметров и (при использовании масштабных множителей) частот нормальных колебаний. Погрешности расчетов абсолютных значений энергий связей более значительны и могут достигать 5 и более ккал/моль. Поэтому при обсуждении результатов расчетов энергетических характеристик основное внимание концентрировалось не столько на абсолютных величинах, сколько на их относительных изменениях и на тенденциях поведения относительных энергий в рядах родственных молекул. Поскольку все члены каждого такого ряда рассчитывались в одинаковом приближении, точность у рассчитанных относительных величин обычно оказывается выше, чем у абсолютных значений.
Следует отметить, что экспериментальные подходы в исследованиях субнаноразмерных частиц в газовой фазе на молекулярном уровне нередко сталкиваются с серьезными трудностями, поскольку такие объекты уже слишком сложны для спектроскопических методов и одновременно недостаточно велики для дифракционных измерений. Сочетание расчетов с экспериментом в этой области обещает быть особенно плодотворным. Наше сотрудничество, помимо получения биологически значимых результатов, казалось нам методически интересным в двух аспектах: а) сопоставление наших расчетов с экспериментальными данными на уровне свободных молекул и ионов не осложняется сольватацией, ассоциациями и другими эффектами внешней среды, существенными для исследований в конденсированных средах, и может считаться наиболее адекватным, и б) все рассмотренные системы относятся к классу структурно-нежестких соединений, представляющих особый интерес для структурной химии.
Наше сотрудничество с Академией Синика протекало не всегда гладко из-за случаев несоответствия результатов расчетов и измерений. Такие случаи представляли особый интерес, поскольку они заставляли “теоретиков – точнее считать, а экспериментаторов – точнее мерить” и обоюдно уточнять результаты и их интерпретации для достижения адекватного соответствия. В настоящей работе мы проиллюстрируем некоторые из полученных результатов, а также сложности в спорах теоретиков с экспериментаторами, которые нередко возникают в совместных исследованиях структурно-нежестких молекул.
ГЕМ, ГЕМИН, ГЕМАТИН. СТРУКТУРА, СТАБИЛЬНОСТЬ, ФРАГМЕНТАЦИЯ, АССОЦИАЦИЯ
Молекула гема FeC34H32N4O4 имеет строение порфиринового кольца с трехвалентным атомом железа в центре с близкими триплетным и квинтетным состояниях и восемью периферийными группами, в том числе двумя пропионатными (Pr), двумя винильными (Vy) и четырьмя метильными (Me). Гем является ключевым сайтом гемоглобина и миоглобина, ответственным за трансформацию кислорода и процессы дыхания в живых телах. Исследования его структуры, стабильности и механизмов реакций с участием гема, его положительного иона гемина и их производных, включая различные каналы фрагментации и ассоциации, важны для понимания молекулярных механизмов трансформации кислорода в живых организмах и для направленных поисков лекарств против малярии и других болезней крови (см., например, многотомное издание [1] и обширную библиографию в нем).
При исследовании масс-спектра гема в Институте молекулярных и атомных наук была обнаружена группа линий, отвечающих фрагментации гемина с последовательным отрывом периферийных групп. На рис. 1 прослеживается эволюция масс-спектра гема в зависимости от времени его лазерного облучения [2–4]. Стартовый пик 616 отвечает исходному иону гемина FeC34H32N4O$_{4}^{ + }$. Через 0.2 с после начала облучения появляется пик 557, отвечающий отрыву карбоксометильного хвоста (Сm) от первой пропионатной группы, причем его интенсивность возрастает в первые две секунды, далее монотонно уменьшается и через минуту спадает до нуля. С запозданием на 0.2 с появляется пик 498, отвечающий отрыву второй карбоксометильной группы, а еще через 2 с появляются четыре пика, отвечающие быстрому последовательному отрыву метильных групп и т.д. На этом основании сделан качественный вывод, что при фрагментации гемина первыми отрываются наиболее слабо связанные карбоксометильные, следующими – метильные и последними – наиболее сильно связанные винильные группы. В ранней масс-спектрометрической работе [5] были измерены энергии последовательного отрыва карбоксометильных групп гемина и найдено, что у первой группы энергия D1 (Cm) составляет 2.5 ± 0.3 эВ, а у второй группы энергия D2 (Cm) оказалась намного меньше, чем D1 (Cm), причем D2 (Cm) из-за своей малости не могла быть измерена количественно и оценена лишь в несколько десятых долей эВ (см. табл. 1). С нашей точки зрения, результаты [5] представлялись неожиданными, поскольку было непонятно, какие резкие перестройки химических взаимодействий могли бы ослабить вторую связь на порядок по сравнению с первой.
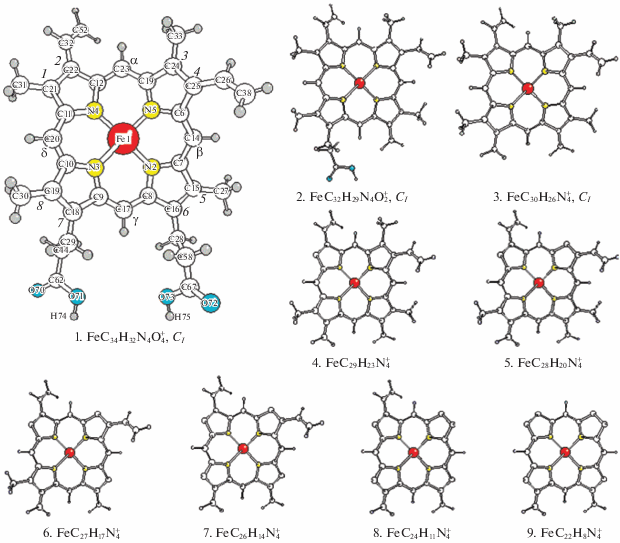
Таблица 1.
Энергии фрагментации гема и гемина с последовательным отрывом периферийных групп CMe, Me и Vy (B3LYP/6-311++G**), эВ
| Фрагментация | Расчет | Эксп. |
|---|---|---|
| FeC34H32N4O4 → FeC32H29N4O2 + CMe – $D_{1}^{ \circ }$(CMe) | 2.70 | |
| FeC32H29N4O2 → FeC30H26N4 + CMe – $D_{2}^{ \circ }$(CMe) | 2.49 | |
| FeC34H32N4${\text{O}}_{4}^{ + }$ → FeC32H29N4${\text{O}}_{2}^{ + }$ + CMe – $D_{1}^{ + }$(CMe) | 2.16 | 2.5 ± 0.3; 1.9 ± 0.3 |
| FeC32H29N4${\text{O}}_{2}^{ + }$ → FeC30H26${\text{N}}_{4}^{ + }$ + CMe – $D_{2}^{ + }$(CMe) | 2.47 | ≪D1; 2.4 ± 0.3 |
| FeC32H29${\text{N}}_{4}^{ + }$ → FeC29H23${\text{N}}_{4}^{ + }$ + Me – $D_{1}^{ + }$(Me) | 4.64 | |
| FeC29H23${\text{N}}_{4}^{ + }$ → FeC28H20${\text{N}}_{4}^{ + }$ + Me – $D_{2}^{ + }$(Me) | 4.46 | |
| FeC28H20${\text{N}}_{4}^{ + }$ → FeC27H17${\text{N}}_{4}^{ + }$ + Me – $D_{3}^{ + }$(Me) | 4.87 | |
| FeC27H17${\text{N}}_{4}^{ + }$ → FeC26H14${\text{N}}_{4}^{ + }$ + Me – $D_{4}^{ + }$(Me) | 4.63 | |
| FeC26H14${\text{N}}_{4}^{ + }$ → FeC24H11${\text{N}}_{4}^{ + }$ + Vy – $D_{1}^{ + }$(Vy) | 5.31 | |
| FeC24H11${\text{N}}_{4}^{ + }$ → FeC22H8N+ + Vy – $D_{2}^{ + }$(Vy) | 5.64 |
В рамках приближения B3LYP мы рассчитали равновесную геометрию, частоты нормальных колебаний, характеристики распределения электронной плотности гемина и продуктов его ступенчатой фрагментации. На рис. 2 изображены их равновесные структуры 1 и 2–9 соответственно. Расчеты энергий отрыва карбоксометильных групп (см. табл. 1) дали близки значения D1 (Cm) = = 2.2 и D2 (Cm) = 2.3 эВ соответственно, из которых первая оказалась близкой к экспериментальному значению 2.5 ± 0.3 эВ [5], в то время как вторая находится в резком несоответствии с оценкой в нескольких десятых эВ [5].
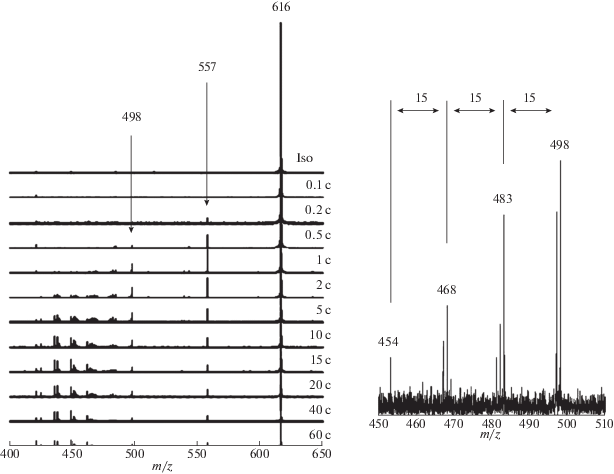
Первой реакцией наших коллег было пожелание “считать лучше”. Однако наши повторные расчеты с более полными и гибкими базисными наборами слабо повлияли на теоретические результаты, в то время как при внимательном рассмотрении экспериментальных данных возникли вопросы. Анализ рассчитанных геометрических параметров, частот нормальных колебаний и характеристик распределения электронной плотности у гемина и его фрагментов не обнаружил ощутимой перестройки или изменений этих параметров при отрыве как первой, так и второй групп Cm. В ряду структур FeC34H32N4O$_{4}^{ + }$ (1) – FeC32H29N4O$_{2}^{ + }$ (2) – FeC30H26N$_{4}^{ + }$ (3) эти параметры, в том числе параметры эквивалентных внешних групп и связей внешних групп с порфириновым кольцом, меняются в обычных пределах 0.01–0.02 Å для межъядерных расстояний, нескольких градусов для валентных углов и нескольких десятков см–1 для частот валентных колебаний. В свете этих данных, экспериментальная оценка энергии D2 отрыва второй группы Cm, предложенная в [5], представляется явно заниженной и требующей уточнения. После известной настойчивости с нашей стороны китайские коллеги повторили масс-спектроскопические измерения и нашли новые значения D1 (Cm) = 1.9 ± 0.2 и D2 (Cm) = 2.4 ± 0.3 эВ (см. [2–4]) в удовлетворительном полуколичественном соответствии с данными нашего расчета для обеих связей.
Подобная ситуация неполного соответствия расчетов с экспериментом повторилась при рассмотрении геометрического строения β-гематина. Эта проблема возникла в связи с тем, что в масс-спектре гема был обнаружен пик с массой 1232, который мог быть отнесен к положительному иону димера гема (C34H32N4O4)$_{2}^{ + }$. Вопросы о возможности существования димера гема, о его структуре, стабильности и взаимодействии с молекулой О2 представляют значительный интерес для более полного понимания механизмов активации кислорода в клетках на молекулярном уровне. Было интересно выяснить, как образуется ион с массой 1232: при ионизации нейтральной молекулы димера гема или путем ассоциации мономерных молекул гема и гемина, и т.д.
В качестве предварительного шага в поисках структуры димера гема мы оптимизировали близкий к ней молекулярный фрагмент, вырезанный из структуры кристаллического β-гематина, являющегося синтетическим и структурным аналогом малярийного пигмента гемазоина, который был исследован рентгенографическим методом в [6] (рис. 3). Структура гематина содержит двухслойные цепочки из тех же феррипорфириновых колец, в которых внешние винильные и метильные группы остаются свободными, а среди пропианатных групп их внутренняя пара связывает кольца друг с другом посредством внутренних ковалентных связей Fe–О.
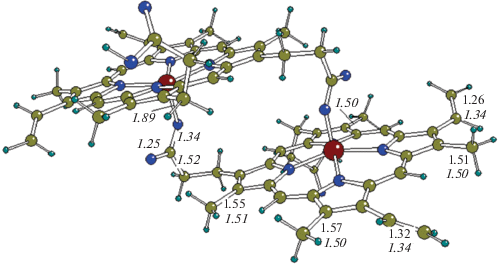
Мы рассчитали колебательные частоты гематина в двух вариантах: 1) при фиксированных рентгенографических координатах атомов, взятых из [6], и 2) после оптимизации геометрии в приближении B3LYP, где рентгенографические координаты использовались в качестве стартовых и далее оптимизировались. Результат получился неожиданным. В первом варианте получено несколько больших мнимых частот. Во втором варианте оптимизация геометрии гематина приводит к локальному минимуму ППЭ (все колебательные частоты действительны) и сопровождается понижением полной энергии системы на ∼2.5 эВ. Иными словами, расчеты свидетельствовали в пользу того, что рентгенографическая структура существенно менее выгодна по сравнению с оптимизированной и не соответствует особой точке поверхности потенциальной энергии. Причина столь значительного расхождения вряд ли могла быть приписана влиянию решетки кристаллического гематина и оставалась непонятной.
В этом случае реакцией наших коллег также было традиционное пожелание “считать лучше”, однако при внимательном рассмотрении экспериментальных значений структурных параметров к ним также возникли вопросы. Сравнение рентгенографических и оптимизированных характеристик показало, что в подавляющем большинстве случаев, включая параметры порфиринового кольца и большинства периферийных групп, рассчитанные и экспериментальные значения удовлетворительно согласуются в пределах обычных нескольких сотых долей ангстрема для межъядерных расстояний и нескольких градусов для валентных углов. Вместе с тем, часть данных [6], которые касаются длин связей периферийных групп с кольцом и количественных различий параметров у эквивалентных групп, представляются спорными. Как уже отмечалось выше, рассчитанные геометрические параметры эквивалентных групп остаются консервативными и, в частности, длины одинарных связей С–С всех четырех метильных групп с кольцом лежат в узких пределах 1.50–1.51 Å. Напротив, значения этих длин, приведенные в [6], варьируют в широком интервале от 1.50 до 1.57 Å. В расчетах длины двойных С=С связей обеих винильных групп практически совпадают (1.34 Å), тогда как в [6] для них даются разные значения 1.26 и 1.32 Å, причем первая (короткая) связь, если судить по ее длине, должна быть промежуточной между двойной и тройной. На фоне хорошей переносимости структурных параметров, предсказанной в расчетах, остается непонятным, какие физические причины могли бы привести к столь сильным структурным различиям параметров эквивалентных групп, приведенных в [6], и столь большому выигрышу энергии при оптимизации. В работах [2–4] сделан вывод о необходимости уточнения экспериментальной структуры гематина и предположено, что возможная причина расхождений лежит в недостаточной точности схемы Ритвельда при обработке результатов рентгеновских измерений для соединений, содержащих тяжелые атомы в сочетании с удаленными группами легких атомов. Аналогичные искажения с недооценкой одних типов расстояний и переоценкой других известны для других комплексных соединений тяжелых металлов.
Следует отметить еще, что, согласно расчетам, для колебательных спектров гема, гемина и особенно димера гема и гематина характерно наличие большого числа низких деформационных частот в интервале 20–100 см–1 с большими амплитудами колебаний. Для более адекватного описания подобных структурно-нежестких частиц необходимо их рассмотрение в рамках динамических подходов.
Что касается стабильности димера гема и его положительного иона, то их расчеты с использованием различных стартовых структур свидетельствуют, что при оптимизации нейтрального димера во всех рассмотренных случаях мономеры раздвигаются друг от друга на расстояние ∼4–5 Å, так что нейтральный димер, скорее всего, может считаться мало стабильным к диссоциации на мономеры. Напротив, положительный ион димера гема на ∼8–10 ккал/моль выгоднее системы изолированных мономеров гема и гемина и может считаться достаточно стабильным и ответственным за пик 1232 в масс-спектре.
DFT-ПРЕДСКАЗАНИЕ, ЭКСПЕРИМЕНТАЛЬНЫЙ СИНТЕЗ И ИССЛЕДОВАНИЕ ЭНДОЭДРАЛЬНЫЕ ФУЛЛЕРЕНА СН4@C60
В наших работах [9, 10] была рассчитана серия эндоэдральных фуллеренов с небольшими молекулами (тетраэдрических гидридов МH4, ацетилена и других) внутри клеток С60, С80, С84 и трубок С120 и С240. На основании этих расчетов была предсказана возможность существования эндоэдрального метан-фуллеренового кластера СН4@С60 (см. левую часть рис. 4), у которого рассчитанная затрата энергии, необходимая для внедрения молекулы метана, была оценена ∼15–20 ккал/моль, перенос заряда между гостем и хозяином малоощутим (не более 0.01 е), фуллереновый каркас С60 слабо реагирует на внедрение с малыми изменениями своего радиуса и химических сдвигов ЯМР (в пределах ∼0.002–0.004 Å и ∼1 ppm), а частота его симметричного “дыхательного” колебания Ag остается практически неизменной (в пределах нескольких см–1). Напротив, изменения свойств гостя по сравнению со свойствами свободной молекулы СН4 выражены вполне отчетливо. При ее внедрении связи С–Н укорачиваются на ∼0.010 Å, константы магнитного экранирования на атомах С и Н возрастают на ∼6.5 и ∼7 ppm соответственно. Частоты валентных колебаний a и t увеличиваются на ∼120 см–1, а деформационные частоты е и t – на ∼10–12 см–1, при этом интенсивности, отвечающие этим колебаниям, уменьшаются в несколько раз. Рассчитанная потенциальная поверхность в окрестности минимума оказалась столь пологой, что при центральном положении атома С в [9–12] не удалось локализовать наиболее выгодную относительную ориентацию гостя и клетки, и мы ограничились выводом, что при нормальных условиях молекула метана может квазисвободно вращаться во внутренней полости фуллерена.
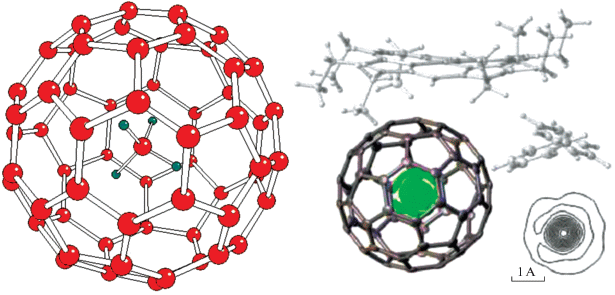
Наша статья с предсказанием существования и расчетами свойств СН4@С60 в период 2002–2003 гг. была направлена в ряд зарубежных журналов, но, встретив настороженный прием с возражениями типа “расчеты слишком опережают эксперимент” или “авторы не предлагают метода получения предсказанного кластера”, была принята и опубликована в “Журнале неорганической химии” в 2004 г.
В 2019 г., т.е. 15 лет спустя, в работе Витби с сотр. [11] впервые получен кластер СН4@C60, который был охарактеризован методами Х-Ray, масс-спектрометрии и ЯМР-спектроскопии высокого разрешения (см. правую часть рис. 4. В ее нижнем правом углу приведена карта распределения электронной плотности во внутренней полости каркаса С60).
Сравнение данных [9, 10] и [11] показывает, что экспериментальные результаты находятся в хорошем соответствии с теоретическими предсказаниями. Так, измеренный ЯМР химический сдвиг на каркасном атоме углерода эндоэдрального кластера (относительно свободной молекулы С60) очень мал и составляет 0.53 ppm. Измеренные химические сдвиги на атомах Н (7.87 pm) и С (4.98 ppm) метана относительно свободной молекулы СН4 также неплохо коррелируют с предсказанными значениями ∼7 и ∼6.5 ppm. Времена протонной спин-решеточной релаксации свидетельствуют, что метан не имеет фиксированной ориентации, а свободно вращается во внутренней сфере фуллеренового каркаса и структура кластера СН4@C60 должна описываться в рамках динамической модели, совместимой с “делокализованным квантовым состоянием”. Для полноты картины было бы интересным сравнить еще изменения длин связей С–Н и частот колебаний внедренного метана, и, как сообщают авторы [11], ими планируются дальнейшие ИК-спектроскопический и криогенный ЯМР-эксперименты. Можно надеяться, что синтез кластера СН4@С60 послужит “первой ласточкой”, за которой последуют синтезы новых эндоэдральных фуллеренов с небольшими внедренными молекулами типа NH3, СО2, Н2СО и др. Вместе с тем, каркас С60 имеет слишком тесную внутреннюю полость, что сильно ограничивает круг потенциальных гостей. На основании нашего расчетного опыта (в обзоре [10] была предсказана возможность существования нескольких десятков эндофуллеренов с более сложными гостевыми молекулами) новых интересных результатов можно ожидать для кластеров с более просторными клетками типа С80, С84 и т.д.
На наш взгляд, описанная картина служит иллюстрацией того, насколько долгим и сложным может быть путь от теоретического предсказания до экспериментальной проверки. Расчеты СН4@C60 на компьютере [9] заняли не более нескольких дней, в то время как эксперимент [11] потребовал виртуозного исполнения сложного многоступенчатого синтеза, включающего функциализацию фуллерена и открытие в нем 14‑атомного “окна”, внедрение молекулы СН4 через это окно в атмосфере метана при давлении 1500 атм, последовательное “зашивание” окна, восстановление замкнутого шарообразного каркаса С60 и, наконец, очистку каркаса от поверхностных функциональных групп с последующей сложной техникой измерений. Можно добавить еще одно общее замечание, что теоретик, делающий предсказания, должен быть готов жить долго, чтобы успеть узнать их судьбу еще на этом свете.
СТРУКТУРА, СТАБИЛЬНОСТЬ, СПЕКТРЫ И ГОМОХИРАЛЬНОСТЬ ПРОТОНИРОВАННОГО ОКТАМЕРА СЕРИНА Ser8Н+
Серин Ser = NH2CН(CH2OH)COOH – простейшая хиральная аминокислота, в молекуле которой хиральный атом углерода окружен аминной NH2, карбоксильной COOH и гидроксометильной CH2OH группами и которая занимает особое положение среди других аминокислот. Во-первых, в масс-спектрах его протонированных форм те пики, которые отвечают ионам октамера Ser8Н+ и его ассоциатам, кратным октамеру, на порядок интенсивнее, чем у промежуточных аналогов, и считаются наиболее стабильными. Во-вторых, протонированный октамер серина гомохирален: его реакции газофазного дейтерообмена, в которых смешивались октамер L8-формы с дейтерированным октамером D8-формы, обнаружили в продуктах не статистическое смешение L и D, а явную предпочтительность L‑формы. В-третьих, гомохиральный октамер способен обменивать часть своих молекул Ser на другие аминокислоты и встраивать в себя только один тип гостевого энантиомера с той же фиксированной хиральностью.
Для фармакологии энантиомерная чистота лекарств очень важна. В качестве примера можно напомнить талидомид, который в 1960-х годах в США предписывался беременным женщинам как мягкое седативное средство против тошноты и недомоганий. После этого начали рождаться младенцы с недоразвитыми конечностями и другими следами физического уродства. Выяснилось, что у талидомида один энантиомер действительно снимает недомогания, но второй калечит эмбрион. Вопросы, почему два зеркальных отражения выполняют столь разные биологические функции и как получать не рацемат, а один требуемый энантиомер достаточной чистоты, имеют давний интерес. Возникла идея получить гомохиральные макрокристаллы серина, построенные из октамеров Ser8 как из стабильных строительных блоков и способные выполнять функции “реакторов асимметричного синтеза” энантиомеров различных аминокислот с фиксированной хиральностью в макроколичествах, что представило бы несомненный интерес для фармакологии и медицины. Кроме того, следы серина регулярно находят в метеоритах, и в литературе есть предположения о том, что гомохиральный серин может иметь непосредственное отношение к проблеме зарождения жизни на земле.
Исследованиям структуры и стабильности протонированного октамера серина на молекулярном уровне было посвящено большое число работ (см., например, [12–16] и библиографию в них), в которых выяснилось, что как расчеты, так и экспериментальные измерения приводили к разноречивым результатам. Так, сечения ионной мобильности свидетельствуют в пользу компактной структуры, в которой все ОН-группы связаны в водородные мостики, и в работах [12, 13] на основании DFT-расчетов с базисом 6-31G была предложена предпочтительная кубоподобная цвиттер-ионная структура (на рис. 5 обозначена как I). Вместе с тем, кинетика реакции газового дейтерообмена свидетельствует о присутствии, по крайней мере, двух изомеров – компактного и “рыхлого”, которые реагируют с разной скоростью. На основании расчетов с перебором более 3000 возможных конформеров по методу Монте-Карло в [15] в качестве предпочтительной была предложена структура с одной свободной гидроксильной группой. В Институте атомных и молекулярных наук с помощью перестраиваемого лазера был измерен колебательный спектр иона Ser8H+ и найден интенсивный пик в области 3680 см–1 (изображен в верхней половине рис. 5), свидетельствующий о наличии в структуре Ser8H+ нескольких свободных ОН-групп. На основании анализа перечисленных структур можно было бы заключить, что каждая из них объясняла свою часть опытных данных, но не могла объяснить другие результаты.
Рис. 5.
CID-IR-спектр протонированного октамера серина Ser8H+ (верхний рисунок) и оптимизированные структуры альтернативных изомеров (нижний рисунок) вместе с их рассчитанными ИК-спектрами [16].
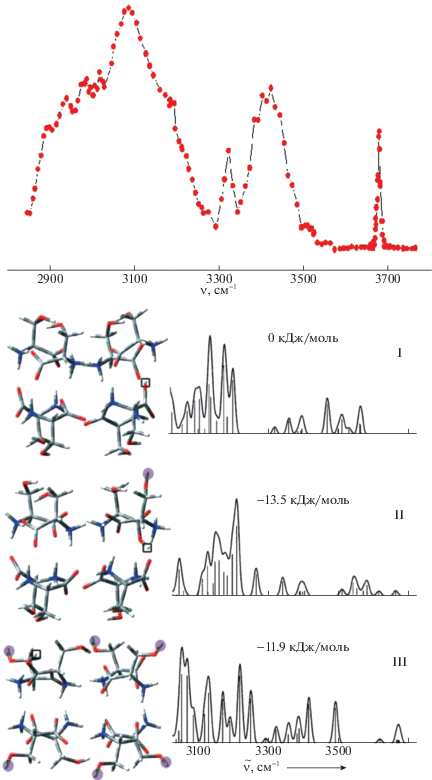
В наших расчетах [16], выполненных в рамках приближения B3LYP/6-311++G**, помимо перечисленных выше структур, были рассмотрены более 10 новых гомохиральных и смешанных конфигураций октамера с разным соотношением свободных и мостиковых ОН-групп. Заметим, что расчеты подобных ассоциатов, которые связаны густой сеткой лабильных водородных связей и которые имеют не менее 10–15 низких колебательных частот в интервале 15–50 см–1, связаны с очень большими затратами машинного времени. Согласно расчетам, иону октамера Ser8Н+ отвечает совокупность, по крайней мере, из 5–6 близколежащих предпочтительных конформеров, имеющих от 2 до 6 свободных ОН-групп, у которых рассчитанная полоса ν(ОН) в области ∼3650–3670 см–1 лучше других соответствует экспериментальному ИК-спектру как по частоте, так и по интенсивности. При этом наилучшее соответствие получено для структуры с шестью ОН-группами (на рис. 5 обозначена как III). Предпочтительные конформеры расположены тесной группой в пределах нескольких ккал/моль и могут одновременно присутствовать в условиях обычных температур. Предложенные ранее структуры менее предпочтительны. Компактная структура [12], содержащая только мостиковые ОН-группы, не отвечает стационарной точке ППЭ. Структура [15] с одной свободной ОН-группой лежит на несколько ккал/моль выше, чем структура [12], и в ней недооценивается интенсивность полосы ν(ОН) ∼ 3650–3670 см–1. Ковалентная структура [13] лежит еще на 25–30 ккал/моль выше и представляется невыгодной.
Ни одна из рассмотренных структур, старых и новых, не дает убедительного объяснения гомохиральности Ser8H+. Наши расчеты наиболее выгодного конформера с гомохиральной структурой L8 и со смешанными структурами вплоть до L4D4 показали, что каждая единичная замена L- на D-форму требует небольших затрат энергии (∼1.0–1.5 ккал/моль) и энергетический фактор может играть определенную роль. Вследствие пологости ППЭ цвиттер-ионных конформеров расчеты их активационных барьеров затруднены, но можно полагать, что эти конформеры могут трансформироваться друг в друга при небольших изменениях энергии. При этом переход каждой отдельно взятой гидроксильной группы из мостикового состояния в свободное может сопровождаться кооперативным перераспределением всей лабильной системы водородных мостиков, которая может с легкостью переключать мостики с одних пар атомов N и O на другие. Подобные лабильные ассоциаты деформируются столь легко, что статический подход с фиксированными координатами ядер для их описания становится неадекватным. Необходимы динамические подходы, включающие одновременное рассмотрение всего ансамбля низколежащих изомеров.
СТРУКТУРА И СТАБИЛЬНОСТЬ Li- И Na-ЗАМЕЩЕННЫХ ПОЛИСАХАРИДОВ И ФУЛЛЕРОЛОВ В ИХ MALDI-СПЕКТРАХ
В масс-спектрометрических исследованиях фрагментации пептидов, олигосахаридов и карбогидратов, помимо протонированных форм, часто используются их ион-молекулярные комплексы с метками катионов щелочных металлов. В масс-спектрах карбогидратов типа сахарозы (Suc) помимо обычных ионов типа Suc · Na+ нередко встречаются серии пиков, отвечающих ионам типа (Suc – nH + nNa) · Na+, в которых несколько гидроксидных атомов Н замещены атомами натрия (H/Na-замещения) или иного электроположительного металла. Так, в масс-спектре глюкозы с матрицей тригидроксоацетофенона найдены пики, соответствующие одно-, двух- и трехкратно H/Na-замещенным производным [17]. При исследованиях мономеров, димеров и тримеров динуклеотидов, содержащих фосфатную группу и различные комбинации из аденина, цистеиана, гуанина или тимина, показано [18], что атомы Na могут замещать до шести атомов Н в димерах, до девяти – в тримерах и т.д. Дополнительные атомы щелочных металлов быстро увеличивают отрицательный заряд на углеродном скелете пропорционально числу замещений и могут сильно влиять как на структуру замещенных олигосахаридов, так и на характер их фрагментации. Для интерпретации сложных масс-спектров крупных олигомеров важны знание “слабых звеньев” в структуре карбогидратов, оценка энергий, требуемых для замещения атомов Н на атомы Li, Na или K, и выявление тенденций в изменениях структуры, стабильности и спектроскопических характеристик замещенных сахаридов с увеличением числа замещений. Для исследования этих вопросов нами были выполнены BLYP/6-311+G**-расчеты последовательно H/Li- и Na/H-замещенных молекул глюкозы, сахарозы и мелизитозы, а китайскими коллегами измерены их MALDI-спектры с ацетатом натрия в матрице тригидроксоацетофенона. Здесь мы вкратце рассмотрим результаты на примере производных глюкозы [17], альтернативные структуры которых и MALDI-спектр приведены соответственно на рис. 6 и 7.
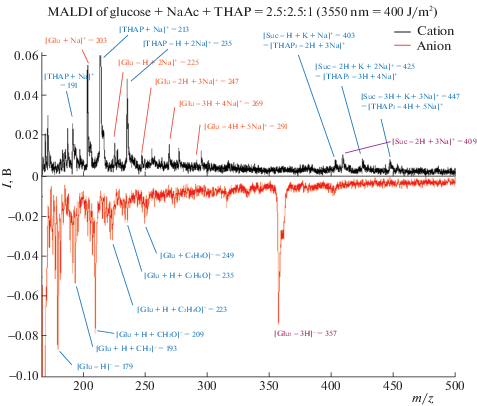
Рис. 7.
Оптимизированные структуры изомеров Na-замещенных молекулы глюкозы (1–6) и ее ион-молекулярного комплекса (7–12) [17].
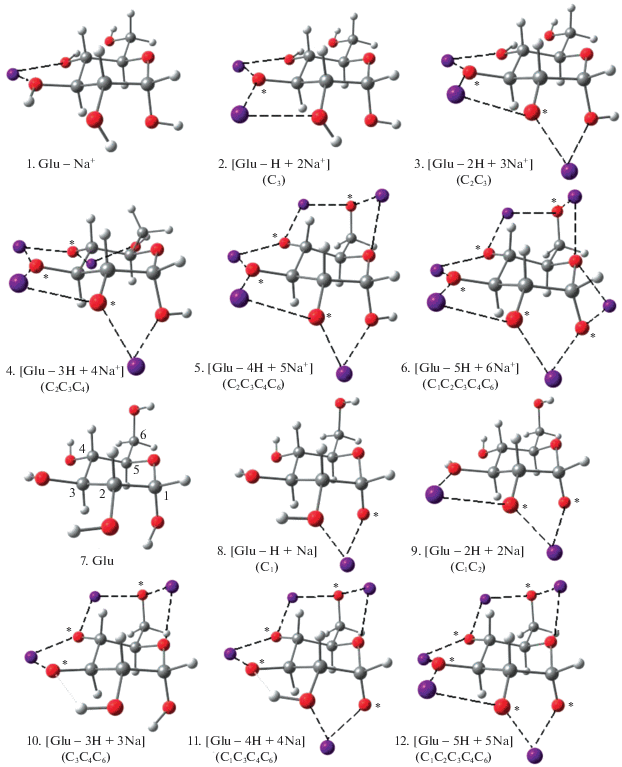
Согласно расчетам, по мере увеличения числа замещений n кольцевой каркас ощутимо уплощается, атомы Na приближаются к кольцу на ∼0.10 Å, терминальные связи С–Н и С–С удлиняются на 0.01–0.02 и 0.02–0.04 Å , а связь С–О укорачивается на 0.01 Å. Частоты валентных колебаний сдвигаются на ∼200 и ∼100 см–1 в длинноволновую область для связей С–Н и С–С и на ∼100–150 см–1 в коротковолновую область для карбонильной группы. Эти сдвиги значительны и могут быть прослежены методами ИК-спектроскопии. Наибольшие изменения претерпевают связи Ос–С между внутрициклическим атомом Ос и соседними атомами углерода, особенно с атомом С1, связанным с аномерным гидроксильным атомом кислорода. При увеличении n до 5 связь Ос–С удлиняется на ∼0.11 Å, а ее частота νвал(OcC1) уменьшается на ∼300 см–1, что свидетельствует о значительном ослаблении этой связи и повышении вероятности ее разрыва при фрагментации глюкозы.
В рамках анализа заселенностей, при увеличении n отрицательный заряд на кольце возрастает почти на 2 е. Соответственно рассчитанное сродство нейтральных молекул (Suc – nH + nNa) к присоединению катиона Na+ быстро увеличивается от ∼50 ккал/моль у свободной до 83 ккал/моль у пятикратно замещенной глюкозы. Энергии ступенчатого замещения ΔE(H/Na) свидетельствуют, что первое замещение у иона Glu · Na+ слабоэкзотермично (+4 ккал/моль), а все последующие эндотермичны (в пределах –10 ккал/моль, см. табл. 2). У нейтральных молекул все замещения, в том числе первое, эндотермичны и требуют затрат энергии в тех же пределах –10 ккал/моль. Эти затраты не слишком велики и также могут быть компенсированы при лазерном облучении в условиях MALDI-эксперимента. Такое же соответствие получено при сравнении наших расчетов и масс-спектров сахарозы и мелизитозы. Это дает основание надеяться, что расчеты будут столь же эффективно дополнять и уточнять результаты измерений для более сложных карбогидратов.
Таблица 2.
Энергии E(H/Na) последовательных H/Na-замещений в гидроксогруппах катионов и нейтральных молекул глюкозы и THAP, в ккал/моль (расчет B3LYP/6-311++H** [17])
| Катионы | |||
|---|---|---|---|
| [Glu – (n – 1)H + nNa]+ + NaAc → → [Glu – nH + (n + 1)Na]+ + Hac | E(H/Na) | [THAP – (n – 1)H + n Na]+ + NaAc → → [THAP – nH + (n + 1)Na]+ + HAc | E(H/Na) |
| Glu.Na+ | +3.7 | THAP.Na+ | +5.5 |
| [Glu – H + 2Na]+ | –3.6 | [THAP – H + 2Na]+ | –3.0 |
| [Glu – 2H + 3Na]+ | –8.6 | [THAP – 2H + 3Na] + | –10.0 |
| [Glu – 3H + 4Na]+ | –10.0 | ||
| [Glu – 4H + 5Na]+ | –5.8 | ||
| Нейтральные молекулы | |||
| [Glu – n H + nNa] + NaAc → → [Glu – (n + 1)H + (n + 1)Na] + HAc | E(H/Na) | [THAP – n H + nNa] +NaAc → → [THAP – (n + 1)H + (n + 1)Na] + NaAc | E(H/Na) |
| Glu | –9.0 | THAP | –4.3 |
| [Glu – H + Na] | –11.7 | [THAP – H + Na] | –16.6 |
| [Glu – 2H + 2Na] | –11.6 | [THAP – H + 2Na] | –20.3 |
| [Glu – 3H + 3Na] | –10.3 | ||
| [Glu – 4H + 4Na] | –11.4 | ||
На следующем этапе мы распространили данный подход на соединения с большим числом гидроксильных групп и выполнили совместные MALDI-исследования системы фуллерол C60(ОH)24 + + ацетат натрия и DFT-расчеты серии ионов Na-замещенных фуллеренола C60(ОH)nNa24–n · Na+ с разным числом H/Li- и H/Na-замещений n [19, 20]. Как известно, фуллеренолы растворяются в воде и могут представлять интерес для фармакологии, использоваться в качестве лекарственных средств после соответствующей функционализации.
Оптимизация структуры с последовательными замещениями H/Li и H/Na проводилась сначала над одной и далее над всеми четырьмя шестиатомными гранями одновременно (см. рис. 8 и 9).
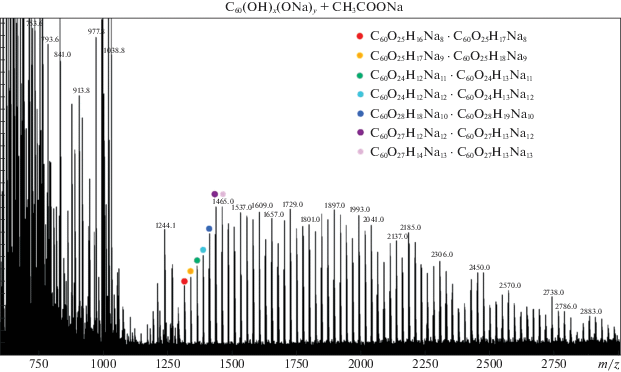
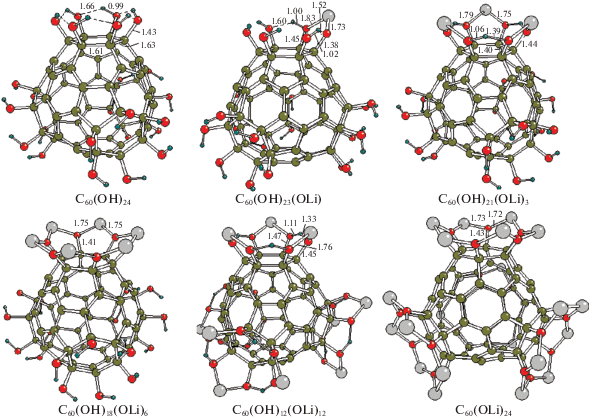
В табл. 3 приведены три типа энергий замещений H/Na: суммарные энергии E(n), отвечающие полной совокупности n замещений; парциальные энергии ΔE(n/(n – 1)) единичных последовательных замещений с изменением n на единицу и сродство к присоединению катиона Na+ при разных значениях n. Нетрудно видеть, что в случае нейтральных молекул первые четыре замещения, локализованные при общей шестиугольной грани, практически не требуют ощутимых затрат энергии (в пределах 1 ккал/моль) и могут практически свободно реализоваться уже при обычных условиях в виде молекул “солей” многозарядных фуллеренольных анионов. В случаях замещений при четырех гранях одновременно парциальные энергии ΔE(n/(n – 1)), приходящиеся на каждое “коллективное” H/Li-замещение (от n – 1 к n), немного возрастают в пределах ∼1–2 ккал/моль. Можно полагать, что в условиях умеренно повышенных температур суммарное число замещений у фуллеренола C60(ОH)24 может достигать 16. Энергии, необходимые для пятого и шестого замещений при одной грани увеличиваются до ∼4 и ∼8 ккал/моль соответственно, но эти величины также остаются не слишком большими и могут компенсироваться при лазерном возбуждении в MALDI-эксперименте. Но при пятом и тем более шестом коллективных замещениях одновременно при четырех гранях суммарные затраты энергии резко возрастают, так что экспериментальное определение числа дополнительных H/Na-замещений сверх 16 оказываются сложной задачей, поскольку результаты могут зависеть от энергетической подпитки и других факторов в условиях эксперимента. На качественном уровне выполненные расчеты и MALDI-эксперимент свидетельствуют в пользу вывода о возможности существования газообразных молекул и ионов фуллеренолов C60(ОH)nNa24 – n и C60(ОH)nNa24 – n · Na+ с большим числом замещений, но для более детальных исследований подобных систем метод MALDI, вероятно, оказывается слишком “жестким”. Интерпретация их масс-спектров затрудняется вследствие перекрывания слабых сигналов, отвечающих H/Na-замещениям, с интенсивными сигналами, которые соответствуют фрагментации каркаса С60 со ступенчатым отрывом двухатомных молекул С2.
Таблица 3.
Суммарные энергии Е(n) одновременных n замещений H/Li, парциальные энергии ΔЕ(n/(n – 1)) последовательных однократных H/Li-замещений и сродство к катиону лития A(Li+) у производных фуллеренола, в ккал/моль (расчет B3LYP/6-31G* [19, 20])
| m | n | C60(OH)m(OLi)n | C60(OH)m(OLi)n · Li+ | C60(OH)m(OLi)n | ||
|---|---|---|---|---|---|---|
| E(n) | ΔЕ(n/(n – 1)) | E(n) | ΔЕ(n/(n – 1)) | A(Li+) | ||
| Однократные H/L-замещения (в одном С6-кольце) | ||||||
| 23 | 1 | –0.2 | –0.2 | 66.8 | ||
| 22 | 2 | –0.6 | –0.4 | |||
| 21 | 3 | –1.0 | –0.4 | +10.0 | +3.3 | 75.6 |
| 20 | 4 | –1.2 | –0.2 | |||
| 19 | 5 | –3.8 | –2.6 | |||
| 18 | 6 | –8.2 | –4.4 | +17.8 | +2.6* | 93.1 |
| Четырехкратные Н/L-замещения (в четырех С6-кольцах одновременно) | ||||||
| 20 | 4 | –0.6 | 0.6* | |||
| 16 | 8 | –11.6 | –2.3* | |||
| 12 | 12 | –17.1 | –1.4* | +14.0 | –0.6* | 97.6 |
| 8 | 16 | –27.2 | –2.6* | ∼0 | –3.5* | 90.2 |
| 4 | 20 | –69.5 | –10.6* | –11.3 | –3.0* | 123.7 |
| 0 | 24 | –107.4 | –9.2* | –32.6 | –5.3* | 136.0 |
ЗАКЛЮЧЕНИЕ
Наше рассмотрение концентрировалось в основном на структурно-нежестких молекулах и кластерах, которые содержат 100 и более атомов и для которых (при всех различиях их химического состава и природы межатомных взаимодействий) характерны большое число низких частот деформационных колебаний, пологие поверхности потенциальной энергии с мелкими локальными минимумами и склонность к протеканию низкобарьерных внутримолекулярных перегруппировок, как локальных (свободное вращение молекулы СН4 внутри кластера СН4@С60), так и кооперативных (миграция катионов щелочных металлов в кластерах C60(OH)nNa$_{{25 - n}}^{ + }$, перестройки лабильной сетки водородных мостиков в ионе октамера серина Ser8H+ и т.д.). Поскольку и расчеты, и эксперименты выполнялись на уровне изолированных молекул, сопоставление их результатов не требовало учета влияния окружающей среды. Вместе с тем, их газофазные исследования на молекулярном уровне и интерпретация результатов измерений часто связаны с преодолением специфических трудностей технического и принципиального характера из-за низкой симметрии, структурной нежесткости и других особенностей наноразмерных частиц, которые велики и сложны для измерений и интерпретации в рамках спектроскопических подходов и одновременно недостаточно велики для использования дифракционных методов. При определении структуры таких систем статический подход нередко оказывается недостаточным. Рассчитанные поверхности потенциальной энергии, геометрическое и электронное строение локальных минимумов и переходных состояний, ИК- и ЯМР-спектры и другие молекулярные характеристики могут быть особенно полезны в случаях, когда разные экспериментальные методы приводят к разноречивым результатам и когда для более полного и адекватного описания необходимо использование динамических подходов.
Авторы благодарят проф. А.М. Мебеля, Ш.‑Х. Лина, Ч.-В. Чена и И.-С. Ванга за приглашение к сотрудничеству и Вычислительный центр и Центр геномики Академии Синика (Тайпей, Тайвань) за предоставление машинного времени и помощь в проведении расчетов.
Список литературы
The Porphyrin Handbook / Ed. by K.M. Kadish, K.M. Smith, R. Guilard. New York: World Science, 2011.
Чаркин O.П., Клименко Н.M., Нгуен T.П. и др. // Журн. неорган. химии. 2005. Т. 50. № 9. С. 1504.
Charkin O.P., Klimenko N.M., Nguyen P.T. et al. // Chem. Phys. Letters. 2005. V. 415. P 362.
Чаркин O.П., Клименко Н.M., Нгуен T.П. и др. // Журн. неорган. химии. 2006. Т. 51. № 10. С. 1714.
Charkin O.P., Klimenko N.M., Charkin D.O. et al. // J. Phys. Chem. A. 2007. V. 111. P. 9207.
Nonose S., Tanaka H., Okai N. et al. // Eur. Phys. J. D. 2002. V. 20. P. 619.
Pagola S., Stephens P.W., Bohle D.S. et al. // Nature. 2000. V. 404. P. 307.
Чаркин О.П. // Журн. неорган. химии. 2009. Т. 54. № 4. С. 665. https://doi.org/10.1134/S0036023609040172
Чаркин О.П., Клименко Н.М., Чаркин Д.О., Мебель А.М. // Журн. неорган. химии. 2004. Т. 49. С. 792.
Charkin O.P., Klimenko N.M., Charkin D.O. // Adv. Quantum Chem. 2009. V. 58. Chapt. 3. P. 69. https://doi.org/10.1016/S0065-3276(09)00707-2
Woodworth S., Sitinova G., Alom S. et al. // Angew. Chem. Int. Ed. 2019. V. 58. Iss. 15. P. 5038. https://doi.org/10.1002/anie.201900983
Julian R.R., Hodyss R., Kinnear B. et al. // J. Phys. Chem. B. 2002. V. 106. P. 1219.
Cook R.G., Zhang D., Koch K.J. et al. // Anal. Chem. 2001. V. 73. P. 3646.
Kong X., Tsai I.-A., Sabu S. et al. // Angew. Chem. Int. Ed. 2006. V. 45. P. 4130.
Schalley C.A., Weis P. // Int. J. Mass Spectrom. 2002. V. 221. P. 9.
Kong K., Lin C., Oh H.-B. et al. //Chem. Phys. Chem. 2009. V. 10. P. 603.
Чаркин О.П., Клименко Н.М., Чен Б.Г. и др. // Журн. неорган. химии. 2011. Т. 56. № 1. С. 65. https://doi.org/10.1134/S0036023611010074
Baker E.S., Gidden J., Fersoco A., Bowers M.T. // Phys. Chem. Chem. Phys. 2004. V. 6. P. 2786.
Чаркин О.П., Клименко Н.М., Ванг И.-С. // Журн. неорган. химии. 2012. Т. 57. № 7. С. 1044. https://doi.org/10.1134/S0036023612070091
Charkin O.P., Klimenko Y.M., Wang Y.-S. // In Handbook on Fullerene: Synthesis, Properties and Applications. Eds: R. F. Verner, C. Benvegn. New York: Nova Science Publishers, Inc., 2011. Ch. 6. P. 313.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии