

Журнал физической химии, 2020, T. 94, № 5, стр. 672-679
Моделирование влияния количества диметилсульфоксида на энергию взаимодействия ионов в нафион-подобных мембранах
А. С. Зюбин a, *, Т. С. Зюбина a, Е. А. Сангинов a, Р. Р. Каюмов a, Л. В. Шмыглева a, Ю. А. Добровольский a
a Российская академия наук, Институт проблем химической физики
Московская обл., Черноголовка, Россия
* E-mail: zyubin@icp.ac.ru
Поступила в редакцию 17.07.2019
После доработки 17.07.2019
Принята к публикации 17.09.2019
Аннотация
В рамках кластерного подхода с использованием функционала плотности B3LYP и базиса 6-31G* выполнено квантово-химическое моделирование удаления катионов лития и аммония от функциональной группы –O–CF2–CF2–SO$_{3}^{ - }$, характерной для Нафион-подобных полимерных мембран, в присутствии разного количества молекул диметилсульфоксида. Найдено, что в рассмотренных системах при малом количестве молекул пластификатора (n ≤ 4) катион достаточно жестко связан с анионом. При увеличении n не входящие в первую координационную сферу молекулы растворителя могут исполнять роль поляризуемой диэлектрической прослойки между катионом и анионом, что при умеренных затратах энергии (∼0.3 эВ) позволяет увеличивать расстояние между ними до 7–14 Å.
Одной из ключевых задач создания недорогих, долговечных и энергоемких электрохимических устройств (низкотемпературных топливных элементов, электрохимических сенсоров, металл-ионных батарей и аккумуляторов) является поиск новых электролитов. Они должны выдерживать зарядные напряжения до 5 В, быть устойчивыми к сильным восстановителям и окислителям, безопасными и недорогими. Использование водных или спиртовых растворов для металл-ионных систем исключено, так как неизбежна реакция вытеснения водорода металлом [1–7]. В этой области весьма перспективными представляются электролиты на основе пластифицированных мембран типа Нафион и Aquivion, которые содержат в своем составе перфторсульфонатные анионные функциональные группы (–O–CF2–CF2–SO$_{3}^{ - }$), в связи с их высокими механическими свойствами, химической стойкостью, униполярной проводимостью и широким диапазоном рабочих температур. К достоинствам этих материалов относится также то, что они легко отщепляют катионы, обеспечивая тем самым высокую концентрацию подвижных ионов в неводных средах, в частности в ДМСО, что делает их весьма перспективными для использования в металл-ионных аккумуляторах [6–15].
Определяющее влияние на транспортные свойства электролитов оказывают ассоциация и сольватация ионов, и исследование этих процессов является важной задачей в области ионики. Большинство публикаций в этой области посвящено изучению литий-ионного транспорта в подобных материалах, тогда как подвижность других ионов в данных системах изучена слабо. Поэтому научный интерес представляет исследование пластифицированных мембран Нафион в аммонийной форме, проводимость которых сравнима с протонной и литий-ионной формами мембран и значительно превышает проводимость по ионам остальных щелочных металлов [16–22]. В [23] при изучении свойств полимерного электролита на основе мембраны Нафион-115 в аммонийной форме, пластифицированной диметилсульфоксидом (ДМСО), была обнаружена необычная зависимость ионной проводимости и энергии активации проводимости в этом композите от содержания пластификатора. При n < 6 (n – количество молекул ДМСО на одну сульфогруппу) энергии активации проводимости полимерного электролита оказываются значительными (0.8–0.4 эВ), а проводимость –низкой. При n = 6–12 барьеры снижаются до 0.3–0.1 эВ (1 эВ = = 96.5 кДж/моль), а проводимость существенно возрастает. Дальнейшее увеличение содержания ДМСО не приводит к заметным изменениям параметров проводимости.
Весьма вероятно, что основную роль в формировании такой зависимости играет изменение характера сольватации ионной пары и потенциала взаимодействия катион–анион при изменении количества молекул растворителя. Однако не исключено, что эти особенности определяет строение катиона аммония и его взаимодействие с ДМСО с возможной передачей протона. Поэтому целью данной работы является изучение с помощью квантово-химического моделирования влияния количества молекул ДМСО на отделение двух разных катионов от сульфогруппы Нафиона – простого и компактного Li+ и более объемного и сложного NH$_{4}^{ + }$.
МЕТОДИКА РАСЧЕТОВ
Квантово-химическое моделирование было выполнено в кластерном приближении с использованием хорошо зарекомендовавшего себя в молекулярных расчетах гибридного функционала плотности B3LYP [24, 25] с валентно-двухэкспонентным базисом 6-31G*, включающим поляризационные функции, с помощью программного комплекса GAUSSIAN [26]. В качестве модельных систем были взяты содержащий сульфогруппу фрагмент цепочки Нафиона (С5OF11SO3) и от трех до 12-ти молекул ДМСО. Расстояние между атомом серы SO3-группы и катионом сканировалось от минимального, определяемого оптимизацией, и до такой величины, когда относительная энергия системы возрастает до 0.4–0.5 эВ относительно минимума.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Системы с катионом лития
При наличии трех молекул ДМСО (рис. 1) все они координируются к катиону, который при этом связан с атомом кислорода сульфогруппы. При увеличении расстояния Li–S молекулы ДМСО остаются связанными с катионом, а их положительно заряженные СН3-фрагменты разворачиваются к сульфогруппе. Потенциальная энергия системы при этом быстро растет (рис. 1). Четыре ДМСО могут формировать две близких по энергии структуры, в одной из которых катион связан с сульфогруппой и тремя ДМСО, а в другой – с четырьмя, которые могут легко переходить друг в друга. При близком контакте катион-анион реализуется первый вариант (a), а при увеличении расстояния Li–S – второй (c). Для перехода между ними требуется преодоление небольшого барьера (конфигурация b). Потенциальная энергия системы медленно меняется в интервале расстояний Li–S 3 – 6.5 Å, но затем начинает быстро расти (рис. 1).
Рис. 1.
Системы с тремя, четырьмя и шестью молекулами ДМСО вокруг фрагмента –SO$_{3}^{ - }$-Li+ (А) и их потенциальные кривые (Б) по расстоянию Li–S. Горизонтальные линии – начала отсчета энергии для каждой из них. Остальные обозначения см. текст.
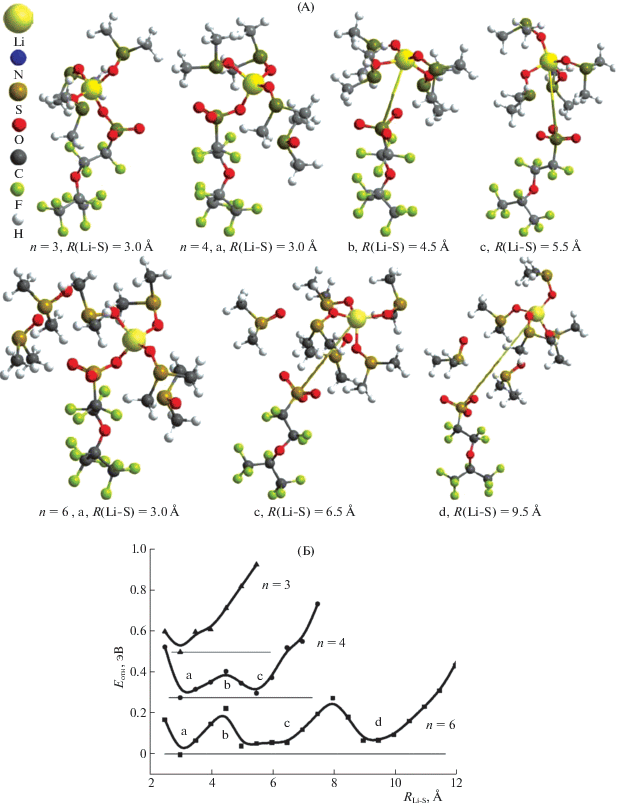
При наличии шести молекул ДМСО вокруг ионной пары (n = 6) ситуация оказывается похожей. При близком расположении (конфигурация a, контактная ионная пара) катион связан с сульфогруппой и тремя ДМСО, остальные молекулы пластификатора вытесняются во вторую координационную сферу. Переход к конфигурации (c, сольватированная ионная пара) с четырехкратной координацией катиона по ДМСО связан с преодолением почти такого же барьера, как и в предыдущем случае (конфигурация b). При возрастании расстояния Li–S молекулы ДМСО из второй координационной сферы меняют свое положение и формируют мостик между катионом и анионом (конфигурация d, разделенная ионная пара), что тоже требует преодоления небольшого энергетического барьера (рис. 1). Быстрый рост потенциальной энергии при n = 6 начинается с расстояния ∼11 Å.
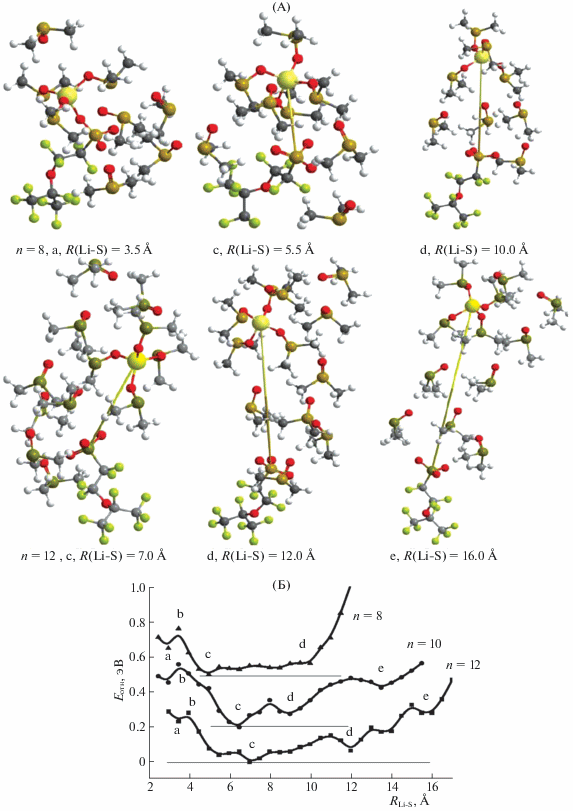
При дальнейшем увеличении количества молекул ДМСО вокруг ионной пары (n = 8, 10, 12) характер потенциальной кривой немного меняется: конфигурация a становится менее выгодной, чем с, в которой молекулы растворителя окружают оба иона. По мере роста расстояния Li–S не участвующие в формировании первой координационной сферы вокруг катиона молекулы растворителя создают дополнительную прослойку между катионом и анионом, что существенно увеличивает диапазон изменений этого расстояния: от 3 Å при n = 4 до 8–16 Å для n = 6–12 при умеренных затратах энергии (∼0.3 эВ), что дает возможность перемещения катиона от одной сульфогруппы к другой. Необходимость реорганизации “облака” ДМСО при изменении расстояния Li–S приводит к появлению небольших (∼0.2 эВ) потенциальных барьеров (рис. 2).
Системы с катионом аммония
Как и для Li+, для NH$_{4}^{ + }$ характерным координационным числом в данных системах является 4, но вследствие наличия связей N–H расстояния N–O в первой координационной сфере аммония существенно больше, чем Li–O в похожих конфигурациях (∼1.9 и 2.8 Å соответственно). Вследствие этого первая координационная сфера аммония имеет больший размер, чем у лития, и является менее плотной. В результате еще одна молекула ДМСО тоже может подходить к аммонию достаточно близко, на расстояние N–O ∼ ∼ 3.1–3.2 Å.
При наличии четырех молекул ДМСО аммоний может уходить от сульфогруппы при умеренных затратах энергии (∼0.3 эВ) на расстояние не более 7 Å. При увеличении количества молекул ДМСО в системе (n = 6, 8, 10 и 12) ситуация в качественном плане оказывается примерно такой же, как и для систем с литием: не участвующие в формировании первой координационной сферы вокруг катиона молекулы растворителя по мере увеличения расстояния катион–анион создают между ними дополнительную прослойку, что существенно увеличивает диапазон изменений этого расстояния (рис. 3, 4).
Рис. 3.
Системы с шестью и восемью молекулами ДМСО вокруг –SO$_{3}^{ - }$–NH$_{4}^{ + }$ (А) и их потенциальные кривые (Б) по расстоянию N–S. Горизонтальные линии – начала отсчета энергии для каждой из них. Обозначения см. текст.
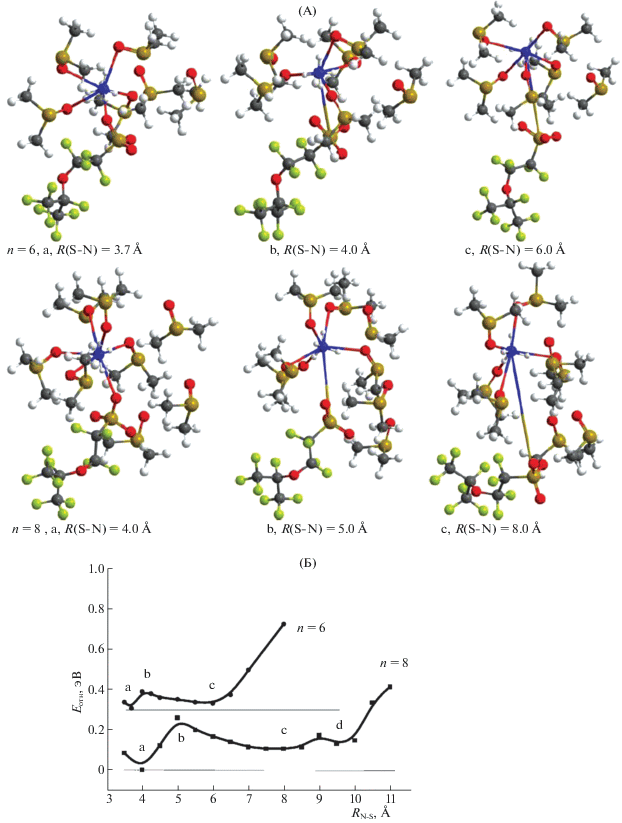
Рис. 4.
Системы с десятью и двенадцатью молекулами ДМСО вокруг –SO$_{3}^{ - }$–NH$_{4}^{ + }$ (А) и их потенциальные кривые (Б). Горизонтальные линии – начала отсчета энергии для каждой из них. Обозначения см. текст.
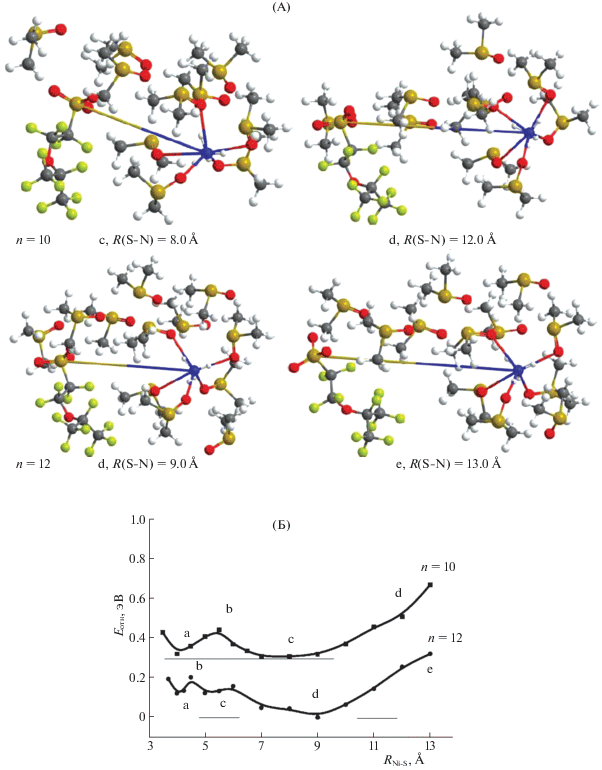
При n = 6 наиболее выгодна контактная ионная пара (структура а) с четырехкратной координацией катиона по ДМСО, по мере увеличения расстояния N–S-система проходит через небольшой барьер (b) и попадает в другой минимум (с), в котором с катионом связаны пять молекул растворителя, и лишь одна остается во второй координационной сфере. Этого недостаточно для формирования прослойки между ионами, и при дальнейшем увеличении расстояния S–N энергия начинает быстро расти. При добавлении двух молекул растворителя (n = 8) в качественном плане картина не меняется, но второй минимум (с) смещается на расстояние ∼8 Å, а существенный рост потенциальной энергии начинается лишь с 10 Å (рис. 3). Добавленные молекулы на малых расстояниях катион–анион формируют вокруг них вторую координационную сферу (конфигурации а и b), а на больших – создают прослойку между ними (конфигурации c и d). При дальнейшем увеличении количества молекул пластификатора (n = 10 и 12, рис. 4) качественная картина в основном сохраняется, но существенный рост потенциальной энергии начинается с расстояний S–N ∼ 12–13 Å.
Резюмируя полученные в данной работе результаты, можно сказать, что в рассмотренных системах с малым количеством ДМСО (n ≤ 4) катион достаточно жестко связан с анионом и не может удаляться от него на большие расстояния. При увеличении n не входящие в первую координационную сферу молекулы растворителя могут выполнять роль поляризуемой диэлектрической прослойки между катионом и анионом, что при затратах энергии ∼0.3 эВ позволяет увеличивать расстояние между ними при n = 6 до ∼7 Å, а при более высоких значениях n (8–12) до ∼11–12 Å. В системах с катионом лития диапазон изменения расстояния между ионами оказывается немного больше, чем в системах с аммонием. Это можно объяснить тем, что в первую координационную сферу Li+ входят четыре молекулы ДМСО, тогда как к аммонию координируются пять таких молекул, хотя последняя – на большем расстоянии (∼0.3–0.4 Å), и слабо связанных молекул пластификатора для формирования прослойки между ионами остается меньше.
Данная работа выполнена на ВЦ ИПХФ РАН при финансовой поддержке Российского фонда фундаментальных исследований (код проекта № 17-79-30054). Моделирование комплексов ДМСО с катионом выполнено по теме Государственного задания № АААА-А19-119061890019-5.
Список литературы
Bruce P.G., Scrosati B., Tarascon J.-M. // Angew. Chem. Int. Edit. 2008. V. 47. P. 2930.
Tarascon J.-M., Armand M. // Nature. 2011. V. 414. P. 359.
Скундин А.М., Ефимов О.Н., Ярмоленко О.В. // Успехи химии. 2002. Т. 71. С. 378.
Xu K. // Chem. Rev. 2004. V. 104. P. 4303.
Баскакова Ю.В., Ярмоленко О.В., Ефимов О.Н. // Успехи химии. 2012. Т. 81. С. 367.
Yaroslavtsev A.B., Dobrovolsky Yu.A., Shaglaeva N.S., et al. // Russ. Chem. Rev. 2012. V. 81. P. 191.
Kim J.G., Son B., Mukherjee S. et al. // J. Power Sources. 2015. V. 282. P. 299.
Doyle M., Lewittes M.E., Roelofs S.A. // J. Membr. Sci. 2001. V. 184. P. 257.
Laoire C.O., Mukerjee S., Abraham K.M. // J. Phys. Chem. C. 2010. V. 114. P. 9178.
Escalante-Garcia I.L., Wainright J.S., Thompson L.T., Savinell R.F. // J. Electrochem. Soc. 2015. V. 162. P. A363.
Mozhzukhina N., Longinotti M.P., Corti H.R., Calvo E.J. // Electrochim. Acta. 2015. V. 154. P. 456.
Liang S., Darling R.M., Gallagher K.G. et al. // J. Electrochem. Soc. 2016. V. 163. P. A5253.
Sanginov E.A., Kayumov R.R., Shmygleva L.V. et al. // Solid State Ionics. 2017. V. 300. P. 26.
Voropaeva D.Yu., Novikova S.A., Kulova T.L., Yaroslavtsev A.B. // Ionics. 2018. V. 24. P. 1685.
Voropaeva D.Yu., Novikova S.A., Kulova T.L., Yaroslavtsev A.B. // Solid State Ionics. 2018. V. 324. P. 28.
Cheddie D. Hydrogen Energy: Challenges and Perspectives. Ammonia as a Hydrogen Source for Fuel Cells: A Review. Edited by Dragica Minic, Belgrade, BoD–Books on Demand, 2012. ISBN 978-953-51-0812-2.
Suzuki S., Muroyama H., Matsui T., Eguchi K. // J. Power Sources. 2012. V. 208. P. 257.
Rees N.V., Compton R.G. // Energy Environ. Sci. 2011. V. 4. P. 1255.
Hejze T., Besenhard J.O., Kordesch K. et al. // J. Power Sources. 2008. V. 176. P. 490.
Зюбина Т.С., Зюбин А.С., Добровольский Ю.А., Волохов В.М. // Журн. неорган. химии. 2016. Т. 61. С. 1606.https://doi.org/10.7868/S0044457X16120230
Зюбина Т.С., Зюбин А.С., Добровольский Ю.А., Волохов В.М. // Журн. неорган. химии. 2017. Т. 62. С. 1061. https://doi.org/10.7868/S0044457X17080074
Zyubina T.S., Prokhorov A.I., Zyubin A.S. et al. // Solid State Ionics. 2018. V. 325. P. 214.
Kayumov R.R., Shmygleva L.V., Radaeva A.P. et al. // J. Electrochem. Soc. 2019. V. 166(7). P. F3216. https://doi.org/10.1149/2.0261907jes
Becke A.D. // J. Chem. Phys. 1993. V. 98. P. 5648.
Johnson B.J., Gill P.M.W., Pople J.A. // J. Chem. Phys. 1993. V. 98. P. 5612.
Frisch M.J., Trucks G.W., Schlegel H.B. et al. Gaussian 09, Revision B.01. Gaussian, Inc., Wallingford CT, 2010.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии