

Журнал физической химии, 2020, T. 94, № 5, стр. 686-691
Теоретические колебательные спектры реакционных интермедиатов в активном центре гуанозинтрифосфат-связывающих белков
Б. Л. Григоренко a, b, А. В. Немухин a, b, *
a Московский государственный университет имени М.В. Ломоносова,
Химический факультет
Москва, Россия
b Российская академия наук, Институт биохимической физики имени Н.М. Эмануэля
Москва, Россия
* E-mail: anem@lcc.chem.msu.ru
Поступила в редакцию 18.07.2019
После доработки 18.07.2019
Принята к публикации 17.09.2019
Аннотация
Показано, что результаты расчетов структур и энергий интермедиатов реакции ферментативного гидролиза гуанозинтрифосфата, выполненных методами квантовой механики/молекулярной механики (КМ/ММ), позволяют предложить механизм химических превращений реакционных частиц в активном центре, предполагающий амид-имидную таутомерию боковой цепи глутамина. Для экспериментальной проверки данного механизма методами ИК-спектроскопии предсказаны положения полос колебаний и соответствующие сдвиги полос при изотопном замещении 14N → 15N в боковой цепи реакционного глутамина.
Изучение интермедиатов химических реакций, катализируемых ферментами, необходимо для выяснения деталей реакционных механизмов, которыми можно управлять при оптимизации биотехнологических процессов. Прогресс в теоретических подходах к исследованию ферментативного катализа в существенной степени связан с развитием метода квантовой механики/молекулярной механики (КМ/ММ) [1], позволяющего проводить расчеты строения и свойств реакционных частиц в активных центрах ферментов и предоставлять данные для интерпретации или предсказания экспериментальных результатов [2, 3].
Цель настоящей работы – моделирование колебательных спектров интермедиатов одной из важнейших биохимических реакций – реакции гидролиза гуанозинтрифосфата (GTP), катализируемой ферментом Ras, представляющим обширный класс гуанозинтрифосфат-связывающих белков (ГТФаз), в комплексе с ускорителем GAP [4]. Значение исследований фермента человека Ras трудно переоценить – мутации в данном белке ассоциируются примерно с 30% онкологических пациентов; но, несмотря на десятилетние усилия, терапевтических решений пока не найдено. Результатом реакции Ras · GAP · GTP + H2O → → Ras · GAP · GDP + Pi является отщепление γ‑фосфатной группы от молекулы GTP с образованием гуанозиндифосфата (GDP) и остатка фосфорной кислоты, традиционно называемого неорганическим фосфатом (Pi). Мы рассматриваем молекулярные системы, в которых и реагенты (GTP + H2O), и продукты (GDP + Pi) находятся в связанном состоянии с белковыми макромолекулами (Ras · GAP). В этом смысле и фермент-субстратный комплекс Ras · GAP · GTP · H2O, т.е. геометрическая конфигурация системы в точке минимума энергетической поверхности в начале реакции, и конфигурации локальных минимумов энергетической поверхности вдоль координаты реакции рассматриваются как реакционные интермедиаты. Моделированию реакционного пути гидролиза гуанозинтрифосфата комплексом Ras · GAP посвящено достаточное количество публикаций (см. один из последних обзоров [5]), однако дискуссии по механизму химических преобразований в активном центре фермента пока еще не привели к полностью согласованной позиции. Согласно результатам расчетов методом КМ/ММ, наиболее предпочтителен механизм, при котором активную роль играет критический аминокислотный остаток глутамина Gln61 в активном центре Ras, причем в процессе образования неорганического фосфата боковая цепь Gln61 участвует в таутомерных превращениях между амидной и имидной формами [6–8]. Этот механизм согласуется с экспериментальными данными по кинетике ферментативной реакции и с косвенными данными рентгеноструктурного анализа для продуктов реакции. Другие обсуждаемые варианты [5] механизма реакции гидролиза гуанозинтрифосфата (GTP), катализируемой ферментом Ras в комплексе с ускорителем GAP, основаны либо на т.н. субстрат-ассистирующем катализе, либо на гипотезе об участии двух молекул воды в химических преобразованиях. В первом случае предполагается, что молекула субстрата (GTP), а не какая-либо молекулярная группа фермента, выступает в роли обобщенного основания. Однако расчеты показывают [6], что на этом пути должен быть высокий энергетический барьер, не согласующийся с наблюдаемой константой скорости реакции. Во втором случае разрыв связи фосфор–кислород в GTP и перераспределение протонов с участием двух молекул воды могут проходить с разумными энергетическими барьерами [6], но анализ доступных кристаллических структур комплексов ферментов с аналогами субстрата исключает присутствие двух молекул воды в активном центре. И субстрат-ассистирующий, и “двухводный” механизмы не подразумевают участие в реакции гидролиза боковой цепи Gln61 фермента Ras.
Проверить предлагаемый в теоретических работах механизм реакции принципиально возможно методами колебательной спектроскопии, если удастся идентифицировать ИК-спектры интермедиатов [9]. Для гидролиза фосфатов подобное сопоставление теоретических и экспериментальных полос в области колебаний фосфорно-кислородных групп дает полезную информацию [10], но не позволяет подтвердить механизм, предполагающий таутомерные превращения боковой цепи Gln61.
Расчеты, представленные в данной работе, были инициированы исследованиями другого класса белков, в которых также осуществляются молекулярные процессы с участием глутамина. Именно, фотохимический цикл флавинсодержащих бактериальных фоторецепторных белков включает стадии таутомерных превращений боковой цепи глутамина, расположенного около хромофора. Для прямой проверки этого молекулярного механизма, предсказанного теоретически [11], в работе [12] измерялись время-разрешенные ИК-спектры в области волновых чисел 1620–1750 см–1 для природных белков и белков с изотопной меткой 15N для атомов азота в боковых цепях глутамина. Дополнительные расчеты [12], показывают, что полосы амидной формы глутамина (которая присутствует в темном состоянии белка) практически не сдвигаются при изотопном замещении 14N → 15N, в то время как характерная полоса, относящаяся к колебанию C=N в имидной форме глутамина (присутствующего в фотоактивированном белке), должна заметно смещаться в сторону меньших частот. Сопоставление рассчитанных и измеренных разностных спектров позволило авторам работы [12] заключить, что стадии таутомерных превращений глутамина действительно реализуются в экспериментах.
В настоящей работе мы предсказываем колебательные спектры для двух реакционных интермедиатов в реакции гидролиза гуанозинтрифосфата комплексом Ras · GAP для природного белка Ras и белка с изотопной меткой 15N в боковой цепи Gln61. Сопоставление этих данных с возможными экспериментальными исследованиями позволит подтвердить или опровергнуть механизм элементарных превращений в активном центре гуанозинтрифосфат-связывающих белков при гидролизе субстрата.
МОДЕЛИ И МЕТОДЫ
Построение молекулярных моделей для фермент-субстратного комплекса Ras · GAP · GTP · H2O, обозначаемого далее ES, и интермедиатов Int выполнено в соответствии с протоколом, детально описанным в работах [6, 7]. Исходный набор атомных координат взят из кристаллографической структуры PDBID 1WQ1 [13] банка данных белковых структур, содержащих аналог GTP в активном центре белкового комплекса Ras · GAP. Методами молекулярного моделирования были восстановлены фосфатные группы субстрата и добавлены атомы водорода в предположении, что боковые цепи Arg и Lys положительно заряжены и Glu и Asp – отрицательно заряжены. Белковая система была помещена в оболочки молекул воды и предварительно уравновешена методами классической молекулярной динамики. Далее равновесные геометрические параметры фермент-субстратного комплекса были оптимизированы методом КМ/ММ с отнесением к квантовой подсистеме фосфатных групп GTP, нуклеофильной молекулы воды, боковых цепей Lys16, Ser17, Thr35, Gln61 белка Ras, Mg2+ и двух молекул воды из координационной сферы магния, а также боковой цепи Arg789 от GAP. На этом этапе КМ-подсистема включала более 90 атомов, ММ-подсистема – более 5000 атомов. Энергии и силы в КМ-подсистеме рассчитывались методом функционала электронной плотности в варианте Кона–Шэма с гибридным функционалом PBE0 [14] с дисперсионной поправкой D3 [15] и базисом cc-pVDZ. ММ-подсистема описывалась в рамках классического силового поля AMBER [16]. Расчеты методом КМ/ММ выполнялись с использованием программы NWChem [17].
От найденной точки минимума на энергетической поверхности, отвечающей структуре ES, выполнялось сканирование потенциальной поверхности вдоль координат реакций на отдельных элементарных стадиях. Для целей настоящей работы достаточно рассмотреть структуры двух критических точек на энергетической поверхности – ES с амидной формой Gln61 и Int(iGln) с имидной формой Gln61. Обе структуры были оптимизированы методом КМ/ММ с двумя вариантами выбора квантовой подсистемы – в первом случае так, как описано выше, во втором случае – с уменьшенной квантовой частью, к которой были отнесены фосфатные группы GTP, нуклеофильная молекула воды и боковые цепи Thr35, Gln61 белка Ras. В этом варианте удобнее проводить колебательный анализ в гармоническом приближении при полном расчете матрицы Гесса в соответствующих точках минимумов. Коррекция вычисленных частот колебаний проводилась с использованием масштабирующего множителя 0.939 [8]. Интенсивности ИК-колебаний получены с помощью производных дипольного момента для соответствующих мод. Моделирование изотопного замещения проведено при изменении атомной массы ядра азота. Для описания формы линии применялось лоренцево уширение 10 см–1 на полувысоте полосы.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
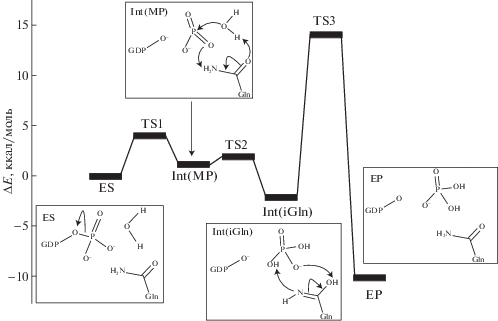
Основные качественные особенности механизма гидролиза GTP белковым комплексом Ras · GAP описаны ранее [6, 7] и воспроизведены в данной работе. Как показано на рис. 1, на первой стадии в результате сближения нуклеофильной молекулы воды с γ-фосфатной группой GTP происходит разрыв связи PG–O3B с отщеплением метафосфата (MP); в результате образуется интермедиат Int(MP). На следующей стадии происходит перераспределение протонов в подсистеме, включающей метафосфат, нуклеофильную молекулу воды и боковую цепь Gln61, и система переходит в следующий интермедиат Int(iGln) уже со сформированным неорганическим фосфатом. В названии Int(iGln) подчеркивается, что боковая цепь Gln61 находится в имидной форме (iGln). Реакция завершается восстановлением основной таутомерной амидной формы Gln61 [6, 7].
Рис. 1.
Рассчитанный методом КМ/ММ энергетический профиль реакции гидролиза гуанозинтрифосфата, катализируемой ферментом Ras в комплексе с ускорителем GAP. На вставках показаны формулы интермедиатов и направления химических превращений между интермедиатами.
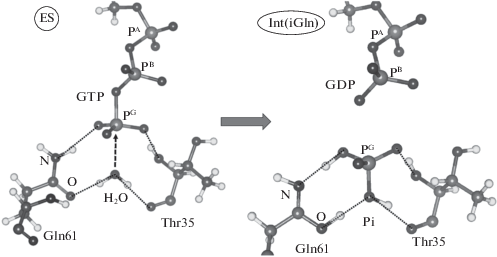
На рис. 2 показаны фрагменты структур фермент-субстратного комплекса ES и интермедиата Int(iGln), оптимизированных методом КМ/ММ. В структуре ES боковая цепь Gln61 находится в основной амидной таутомерной форме; нуклеофильная молекула воды ориентирована водородными связями с карбоксильными группами Gln61 и Thr35. В результате нуклеофильной атаки сначала отщепляется метафосфат PO$_{3}^{ - }$, затем происходит перенос протонов с образованием неорганического фосфата Pi и имидной таутомерной формы Gln61. Основные участники в структуре интермедиата Int(iGln) показаны справа на рис. 2.
Рис. 2.
Фрагменты структур фермент-субстратного комплекса ES (слева) и интермедиата Int(iGln), оптимизированных в расчетах методом КМ/ММ.
Для целей настоящей работы важно отметить, что интермедиат Int(iGln) соответствует точке локального минимума на поверхности энергии, расположенной ниже уровня ES на 2 ккал/моль и отделенной барьерами высотой 4 ккал/моль (TS2) и 16 ккал/моль (TS3) от соседних точек минимальной энергии (рис. 1). Поэтому в ходе реакции ES → EP будет происходить накопление и медленное расходование интермедиата Int(iGln), что принципиально позволяет индентифицировать его спектральные полосы экспериментальными методами.
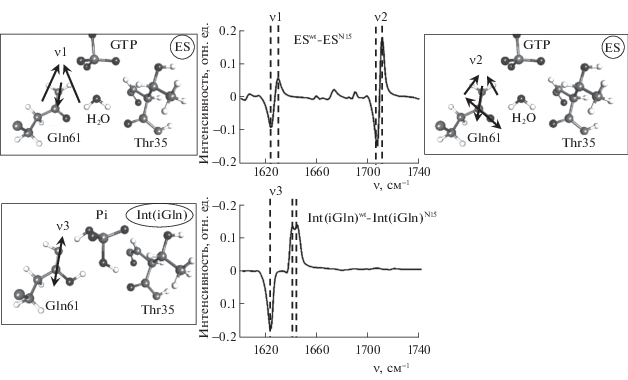
Рассчитанные структуры ES и Int(iGln) характеризуются полностью действительными частотами гармонических колебаний. В интересующую с точки зрения потенциального экспериментального исследования данной реакции область волновых чисел 1620–1750 см–1 попадают три моды с частотами ν1, ν2, ν3, формы колебаний которых для структур ES и Int(iGln) изображены на вставках слева и справа на рис. 3. В центре рис. 3 показаны рассчитанные разностные спектры для природных белков (с изотопом 14N в боковой цепи Gln61) и для белков с помеченным изотопом 15N азотом в боковой цепи Gln61. Видно, что полосы ν1, ν2, которые относятся к связанным колебаниям групп C=O и NH2 амидной формы Gln61 в структуре ES малочувствительны к изотопному замещению, в то время как полоса ν3, соотносимая с колебанием C=N имидной формы Gln61, смещается на величину до 20 см–1 при изотопном замещении.
Рис. 3.
В центре – предсказанные по результатам моделирования методом КМ/ММ разностные колебательные спектры для интермедиатов ES и Int(iGln) реакции гидролиза гуанозинтрифосфата природной ГТФазой Ras и белком с изотопной меткой 15N в боковой цепи Gln61. Вставки слева и справа иллюстрируют на молекулярных моделях формы колебаний, соответствующие полосам ν1, ν2, ν3.
Предсказанный эффект смещения полос должен быть идентифицирован в экспериментах, если реакция протекает по механизму с участием глутамина. Для альтернативных вариантов механизма реакции предлагаемое изотопное замещение не должно проявляться.
Таким образом, моделирование колебательных спектров для двух характерных структур на пути реакции гидролиза гуанозинтрифосфата ГТФазой Ras, а именно, для фермент-субстратного комплекса ES и интермедиата Int(iGln), в состав которого входит имидная таутомерная форма боковой цепи Gln61, показывает, что изотопное замещение 14N → 15N в глутамине позволит четко различить композиции реакционных частиц. В области волновых чисел 1620–1750 см–1, которая сканировалась в экспериментальной работе [12] при исследовании молекулярных процессов с участием глутамина в флавинсодержащих фоторецепторных белках, разностные спектры (рис. 3) показывают, что сдвиг полосы ν3, относимой к колебанию C=N имидной формы глутамина в интермедиате Int(iGln), составляет примерно 20 см–1. Это должно уверенно регистрироваться в инструментальных исследованиях реакций ферментативного катализа методами время-разрешенной ИК-спектроскопии.
Исследование выполнено при финансовой поддержке Российского научного фонда (проект № 19-73-20032). Расчеты проведены с использованием оборудования Центра коллективного пользования сверх-высокопроизводительными вычислительными ресурсами МГУ имени М.В. Ломоносова и межведомственного суперкомпьютерного центра РАН.
Список литературы
Warshel A., Levitt M. // J. Mol. Biol. 1976. V. 103. P. 227. https://doi.org/10.1016/0022-2836(76)90311-9
Nemukhin A.V., Grigorenko B.L., Lushchekina S.V. et al. // Russ. Chem. Rev. 2012. V. 81. №. 11. P. 1011. https://doi.org/10.1070/RC2012v081n11ABEH004311
Kots E.D., Khrenova M.G., Nemukhin A.V. et al. // Russ. Chem. Rev. 2019. V. 88. № 1. P. 1. https://doi.org/10.1070/RCR4842
Mishra A.K., Lambright D.G. // Biopolymers. 2016. V. 105. P. 431. https://doi.org/10.1002/bip.22833
Carvalho A.T.P., Szeler K., Vavitsas K. et al. //Arch. Biochem. Biophys. 2015. V. 582. P. 80. https://doi.org/10.1016/j.abb.2015.02.027
Grigorenko B.L., Kots E.D., Nemukhin A.V. // Org. Biomol. Chem. 2019. V. 17. P. 4879. https://doi.org/10.1039/C9OB00463G
Khrenova M.G., Grigorenko B.L., Kolomeisky A.B., Nemukhin A.V. // J. Phys. Chem. B. 2015. V. 119. P. 12838. https://doi.org/10.1021/acs/jpcb.5b07238
Grigorenko B.L., Khrenova M.G., Nemukhin A.V. // Phys. Chem. Chem. Phys. 2018. V. 20. P. 23827. https://doi.org/10.1039/c8cp04817g
Kötting C., Gerwert K. // Biol. Chem. 2015. V. 396. P. 131. https://doi.org/10.1515/hsz-2014-0219
Khrenova M.G., Grigorenko B.L., Nemukhin A.V. // Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2016. V. 166. P. 68. https://doi.org/10.1016/j.saa.2016.04.056
Domratcheva T., Grigorenko B.L., Schlichting I., Nemukhin A.V. // Biophys. J. 2008. V. 94. P. 3872. https://doi.org/10.1529/biophysj.107.124172
Domratcheva T., Hartmann E., Schlichting I., Kottke T. // Sci. Rep. 2016. V. 6. P. 22669. https://doi.org/10.1038/srep22669
Scheffzek A.K., Ahmadian M.R., Kabsch W. et al. // Science. 1997. V. 277. P. 333. https://doi.org/10.1126/science.277.5324.333
Adamo C., Barone V. // J. Chem. Phys. 1999. V. 110. № 13. P. 6158. https://doi.org/10.1063/1.478522
Grimme S., Antony J., Ehrlich S. et al. // J. Chem. Phys. 2010. V. 132. № 15. P. 154104. https://doi.org/10.1063/1.3382344
Cornell W.D., Cieplak P., Bayly C.I. et al. // J. Am. Chem. Soc. 1995. V. 117. № 19. P. 5179. https://doi.org/10.1021/ja00124a002
Valiev M., Bylaska E.J., Govind N. et al. // Comput. Phys. Commun. 2010. V. 181. № 9. P. 1477. https://doi.org/10.1016/j.cpc.2010.04.018
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии