

Журнал физической химии, 2020, T. 94, № 5, стр. 680-685
Учет конформационной подвижности в квантово-химических расчетах потенциалов окисления нитроксильных радикалов
a Российская академия наук, Институт проблем химической физики
142432 Московская обл., Черноголовка, Россия
b Московский государственный университет им. М.В. Ломоносова
119991 Москва, Россия
* E-mail: vbl@icp.ac.ru
Поступила в редакцию 17.07.2019
После доработки 17.07.2019
Принята к публикации 17.09.2019
Аннотация
Проведены расчеты редокс-потенциалов в воде для пары R2N+=O/R2N–${{{\text{O}}}^{ \bullet }}$ циклических нитроксильных радикалов на основе теории функционала плотности и модели поляризуемого диэлектрика. Оценки потенциалов окисления нитроксилов учитывали свободные энергии конформационных вкладов. Для определения последних изучены три расчетные схемы, включающие как использование термодинамического цикла для переноса электрона в газе и воде, так и прямые квантово-химические вычисления равновесных свободных энергий Гиббса реактантов в воде. Проведено сравнение корреляционных зависимостей между рассчитанными и экспериментальными значениями редокс-потенциалов для рассмотренных подходов. Наименьшие погрешности расчета получены для методов, использующих оптимизированные в воде структуры молекул.
Стабильные свободные нитроксильные радикалы являются важным классом химических соединений с многообразной реакционной способностью и широким прикладным использованием [1–5]. Многие химические и биологические функции нитроксилов связаны с их участием в окислительно-восстановительных процессах. В частности, биологическая активность нитроксилов проявляется за счет ингибирования паталогических процессов перекисного окисления биомолекул [2, 4, 6]. Важной реакцией в этом ряду является окисление нитроксильных радикалов (NR) R2N–${{{\text{O}}}^{ \bullet }}$ до оксоаммониевого катиона (OC) R2N+=O перекисными радикалами в водном окружении [6, 7]. Легкость окисления нитроксилов определяет их антиокислительную способность. Нитроксилы также участвуют в электрокаталитическом окислении спиртов и других химических соединений [1, 8]. В связи с перечисленными примерами, количественная оценка и предсказание окислительной способности нитроксилов в воде имеют большое значение.
Современные методы вычислительной химии предоставляют широкие возможности в изучении реакционной способности и физико-химических свойств молекул. Квантово-химические расчеты редокс-свойств нитроксилов, а также определяющих их структурных параметров и количественных соотношений структура–активность, представлены в ряде публикаций [9–19]. В указанных работах изучено влияние уровня вычислительных методов на точность расчета электрохимических потенциалов, прослежено влияние заместителей, установлена роль конформационных свойств и электростатических эффектов растворителя. В частности, было обнаружено различие доступных конформаций циклических нитроксилов в газовой фазе и воде, что вносит систематическую погрешность в расчеты редокс потенциала при использовании подхода, основанного на рассмотрении термодинамического цикла [19].
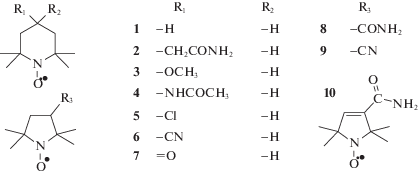
В настоящей работе, проведены расчеты редокс потенциалов $E_{{{\text{OC/NR}}}}^{ \circ }$ пары OC/NR (потенциалов окисления) в воде для десяти циклических нитроксилов (рис. 1) на основе теории функционала плотности (DFT) и модели поляризуемой непрерывной среды (PCM) растворителя. Целью работы явилось изучение и сравнение схем расчета свободных энергий и $E_{{{\text{OC/NR}}}}^{ \circ }$ для конформационно-подвижных нитроксилов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Методы вычислений
Редокс-потенциал $E_{{{\text{OC/NR}}}}^{ \circ }$ одноэлектронного окисления нитроксильного радикала в оксоаммониевый катион в воде для полуреакции R2N–${{{\text{O}}}^{ \bullet }}$ → R2N+=O + e–(g) был рассчитан (для стандартных условий 1 M идеального раствора) согласно уравнению
(1)
$E_{{{\text{OC/NR}}}}^{ \circ } = {{\Delta {{G}_{{{\text{ox,sol}}}}}} \mathord{\left/ {\vphantom {{\Delta {{G}_{{{\text{ox,sol}}}}}} F}} \right. \kern-0em} F} - E_{{{\text{abs}}}}^{ \circ }({\text{SHE}}).$Вклад Gox,sol представляет свободную энергию Гиббса реакции в растворе и равен разности равновесных свободных энергий реактантов в растворе, F – константа Фарадея, $E_{{{\text{abs}}}}^{ \circ }$(SHE) – абсолютный потенциал стандартного водородного электрода, равный −4.44 В [13, 20]
(2)
$\Delta {{G}_{{{\text{ox,sol}}}}} = {{G}_{{{\text{sol}}}}}({{X}^{ + }}) - {{G}_{{{\text{sol}}}}}({{X}^{ \bullet }}{\text{)}}.$Свободные энергии Гиббса Gsol(X) реактантов X были получены из квантово-химических расчетов по программе Gaussian-09 [21]. Схемы расчета свободных энергий Gox,sol обсуждаются далее. В частности, значения Gsol(X) определяли на основе модели термодинамического цикла (ТЦ) (рис. 2), что включало расчеты вклада идеального газа Gg(X) и свободных энергий сольватации ΔGs(X). При расчете Gsol(X) учитывали все термически доступные конформации реактантов Xi в пределах ≤6 kBT, где kB – константа Больцмана, T – абсолютная температура. Свободные энергии конформеров Xi в газе рассчитывали как сумму полных электронных энергий Etot, энергий нулевых колебаний ZPVEg и термической поправки Gtherm,g. Значения ΔGs были рассчитаны в рамках модели PCM [21, 22] согласно уравнению
(3)
$\begin{gathered} \Delta {{G}_{{\text{s}}}}({{X}_{i}}) \equiv \Delta {{G}_{{\text{s}}}}({{{\mathbf{R}}}_{s}}({{X}_{i}}){\text{/}}{{{\mathbf{R}}}_{{\text{g}}}}({{X}_{i}})) = \\ = \;E_{{{\text{tot}}}}^{{{\text{PCM}}}}({\mathbf{U}}\, = \,{{{\mathbf{U}}}_{{\text{s}}}}{\text{/}}2,\;{{{\mathbf{R}}}_{{\text{s}}}}({{X}_{i}})) - {{E}_{{{\text{tot}}}}}({\mathbf{U}}\, = \,0,\;{{{\mathbf{R}}}_{{\text{g}}}}({{X}_{i}})). \\ \end{gathered} $Рис. 2.
Термодинамический цикл окисления нитроксилов. Фигурные скобки указывают, что рассматривается один конформер из возможных наборов конформаций в газе и в растворе.
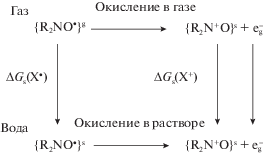
Матрицы Rg(Xi) и Rs(Xi) описывают равновесные молекулярные структуры в газе и в воде, U – вектор кулоновского потенциала растворителя на центрах атомов растворенной молекулы.
Трехмерные структуры молекул в газе и в воде были рассчитаны с полной оптимизацией геометрии с использованием функционала M06–2X [23] в базисе 6–31+G(d, p), включающем диффузные и поляризационные функции. Электронная структура свободных радикалов рассчитывалась в рамках неограниченного метода Хартри–Фока. Значения Etot рассчитаны методом M06–2X в расширенном базисе 6–311+G(2df, 2p) [23, 24]. Значения ΔGs были получены методом PCM (SMD) [21, 22] с использованием функционалов M06-2X и PBE [25]. Вклады Gtherm и ZPVE рассчитывали использованием вычисленных на уровне M06-2X/6-31+G(d,p) частот гармонических колебаний равновесных структур. Значения свободных энергий определяли для T = 298.15. Дополнительные настройки программы Gaussian-09 при расчете электронной и молекулярной структур соответствовали описанным ранее [19].
Свободные радикалы и катионы циклических нитроксильных радикалов способны находиться в нескольких конформациях при комнатных температурах (рис. 3) [17–19]. Так, пиперидиновый цикл может принимать как форму кресла, так и скрученную конформацию. Боковые группы R дают наборы вращательных и стереоизомеров (рис. 3). Энергии активации конформационных переходов циклических структур лежат в пределах 11−17 кДж моль–1 [18], и большинство переходов между изученными конформерами нитроксилов допустимы при комнатной температуре. Рассчитанные конформационные свойства нитроксилов, в целом, хорошо согласуются с известными данными для нитроксилов и замещенных пиперидинов [26–30]. Учет множественных конформаций увеличивает точность квантово-химических расчетов $E_{{{\text{OC/NR}}}}^{ \circ }$ нитроксилов по сравнению с одно-конформационным подходом [17–19].
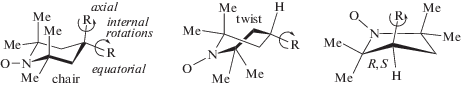
Расчеты показали, что относительные энергии конформаций циклических нитроксилов могут иметь различающийся порядок чередования в газовой фазе и в воде [18, 19]. Далее, некоторые вращательные конформеры боковых групп нитроксилов, как например для ОС 2, могут представлять локальный минимум в воде, но отсутствовать в газе [19]. Последнее наблюдение показывает, что наборы термически разрешенных конформаций реактантов в газе {Xi, i = 1, …, ng}g и в растворе {Xi, i = = 1, …, ns}s (см. рис. 2) могут иметь различные размеры. При этом, число разрешенных конформеров также может различаться для NR и OC. В следствие этого, не удается установить соответствие между конформерами в газе и воде, и свободная энергия сольватации дополнительного конформера в воде остается неопределенной. Эта неопределенность вносит систематическую погрешность в расчет потенциала окисления в рамках модели ТЦ [19]. Рассмотрим далее возможные подходы для разрешения подобных противоречий при расчете $E_{{{\text{OC/NR}}}}^{ \circ }$ конформационно-подвижных нитроксилов.
Значения Gsol(X) определяли в предыдущих работах [13, 18, 19] по формуле для свободной энергии набора конформационных состояний Xi в растворе [31]
(4)
${{G}_{{{\text{sol}}}}}(X) = - {{k}_{{\text{B}}}}T\ln \sum\limits_{i = 1}^n {{{e}^{{ - {{G}_{{{\text{sol}}}}}({{X}_{i}}){\text{/}}{{k}_{{\text{B}}}}T}}}} .$(5)
${{G}_{{{\text{sol}}}}}({{{\text{X}}}_{i}}) = {{G}_{{\text{g}}}}({{{\text{X}}}_{i}}) + \Delta {{G}_{{\text{s}}}}({{{\text{X}}}_{i}}).$В первом предложенном способе, используется вычисление свободных энергий наборов конформаций NR и OC в газе и воде (метод В). Вследствие равенства нулю суммы свободных энергий по отрезкам замкнутого ТЦ (рис. 2) , выражение для Gox,sol можно представить как
(6)
$\begin{gathered} \Delta {{G}_{{{\text{ox,sol}}}}}({\text{X}}) = \Delta {{G}_{{\text{g}}}}({{{\text{X}}}^{ \bullet }} \to {{{\text{X}}}^{{\text{ + }}}}) \\ + \;\Delta {{G}_{{\text{s}}}}({{{\text{X}}}^{{\text{ + }}}}) - \Delta {{G}_{{\text{s}}}}({{{\text{X}}}^{\centerdot }}). \\ \end{gathered} $(7)
$\Delta {{G}_{{\text{g}}}}({{{\text{X}}}^{ \bullet }} \to {{{\text{X}}}^{{\text{ + }}}}) = {{G}_{{\text{g}}}}({{{\text{X}}}^{{\text{ + }}}}) - {{G}_{{\text{g}}}}({{{\text{X}}}^{ \bullet }}),$(8)
${{G}_{{\text{g}}}}({\text{X}}) = - {{k}_{{\text{B}}}}T\ln \sum\limits_{i = 1}^{{{n}^{{\text{g}}}}} {{{e}^{{ - {{G}_{{\text{g}}}}({{{\text{X}}}_{i}})/{{k}_{{\text{B}}}}T}}}} .$(9)
$\begin{gathered} \Delta {{G}_{{\text{s}}}}({\text{X}}) = {{k}_{{\text{B}}}}T\ln \sum\limits_{i = 1}^{{{n}^{{\text{g}}}}} {{{e}^{{ - E_{{{\text{tot}}}}^{{\text{g}}}({{{\text{X}}}_{i}})/{{k}_{{\text{B}}}}T}}}} - \\ - \;{{k}_{{\text{B}}}}T\ln \sum\limits_{j = 1}^{{{n}^{{\text{s}}}}} {{{e}^{{ - E_{{{\text{tot}}}}^{{{\text{PCM}}}}({{{\text{X}}}_{j}})/{{k}_{{\text{B}}}}T}}}} , \\ \end{gathered} $Второй способ включает прямой расчет свободных энергий Gsol(X) наборов конформационных состояний молекул в воде по формуле (4) (метод С). Величину Gox,sol оценивают далее по формуле (2). Свободные энергии конформационных состояний Xi в этом случае вычисляются как суммы
(10)
$\begin{gathered} {{G}_{{{\text{sol}}}}}({{{\text{X}}}_{i}}) = E_{{{\text{tot}}}}^{{{\text{PCM}}}}({\mathbf{U}} = {{{\mathbf{U}}}_{{\text{s}}}}{\text{/}}2,\;{{{\mathbf{R}}}_{{\text{s}}}}({{{\text{X}}}_{i}})) + \\ + \;ZPV{{E}_{{\text{s}}}}({{{\text{X}}}_{i}}) + {{G}_{{{\text{therm,s}}}}}({{{\text{X}}}_{i}}). \\ \end{gathered} $Значения $E_{{{\text{OC/NR}}}}^{ \circ }$ циклических нитроксилов, полученные методами А−С для различных уровней расчета свободных энергий, приведены в таблице 1 и на рис. 4. При сопоставлении подходов А и В (таблица 1), учет релаксации геометрии реактантов в воде (Rs ≠ Rg) и переход от метода PBE/6-31G(d) к методу M06-2X/6-31+G(d,p) с расширенным базисом дают улучшение корреляции между экспериментальными и расчетными данными. Значения $E_{{{\text{OC/NR}}}}^{ \circ }$ нитроксила 2, для которого нет полного соответствия между конформерами в газе и воде, демонстрирует наибольшие отклонения от расчетных прямых. В целом, результаты расчетов методом А по формулам (4), (5) несколько лучше коррелируют с экспериментом, чем методом В с использованием формул (6)–(9).
Таблица 1.
Экспериментальные [32] и рассчитанные методами А и B значения редокс-потенциалов $E_{{{\text{OC/NR}}}}^{ \circ }$ нитроксилов 1–10
| NR/OC | $E_{{{\text{OC/NR}}}}^{ \circ }$ (В) | |||
|---|---|---|---|---|
| эксп. [32] | Rs = ${\mathbf{R}}_{{\text{g}}}^{{\text{a}}}$ А [19]; В |
Rs ≠ ${\mathbf{R}}_{{\text{g}}}^{{\text{a}}}$ А [19]; В |
Rs ≠ ${\mathbf{R}}_{{\text{g}}}^{{\text{b}}}$ А [19]; В |
|
| 1 | 0.734 | 0.594; 0.593 | 0.555; 0.553 | 0.593; 0.591 |
| 2 | 0.784 | 0.715; 0.712 | 0.679; 0.690 | 0.670; 0.679 |
| 3 | 0.818 | 0.660; 0.687 | 0.648; 0.676 | 0.658; 686 |
| 4 | 0.843 | 0.731; 0.748 | 0.684; 0.701 | 0.693; 0.706 |
| 5 | 0.865 | 0.702; 0.691 | 0.692; 0.681 | 0.713; 0.710 |
| 6 | 0.890 | 0.735; 0.731 | 0.724; 0.722 | 0.736; 0.748 |
| 7 | 0.920 | 0.747; 0.741 | 0.737; 0.730 | 0.739; 0.729 |
| 8 | 0.877 | 0.748; 0.800 | 0.727; 0.739 | 0.722; 0.783 |
| 9 | 0.970 | 0.800; 0.800 | 0.789; 0.789 | 0.811; 0.817 |
| 10 | 0.972 | 0.848; 0.847 | 0.839; 0.838 | 0.839; 0.835 |
| Rc | – | 0.908; 0.858 | 0.948; 0.919 | 0.973; 0.937 |
| ac | – | 0.830; 0.802 | 0.960; 0.904 | 0.912; 0.882 |
| SD(В) c | – | 0.028; 0.035 | 0.023; 0.028 | 0.016; 0.024 |
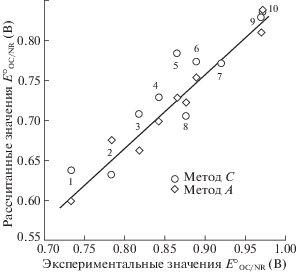
Результаты расчетов методами А и С с учетом релаксации геометрии реактантов в воде (Rs ≠ Rg) приведены на рис. 4. Соответствующие значения $E_{{{\text{OC/NR}}}}^{ \circ }$ показали более хорошие статистические корреляции с экспериментом (метод А: R = 0.982, a = 0.913, SD = 0.013 В; метод С: R = 0.932, a = = 0.861, SD = 0.024 В), чем значения потенциалов окисления из табл. 1. В представленных на рис. 4 данных, значения Etot в газе и ΔGs были рассчитаны на одинаковом уровне. Вследствие этого, различие значений $E_{{{\text{OC/NR}}}}^{ \circ }$, полученных методами А и С, определяется лишь вкладами ZPVE и Gtherm в газовой фазе и воде.
Полученные данные показывают, что подход С, использующий гармонические частоты с учетом влияния воды, дает больший статистический разброс точек, чем подход А, использующий гармонические частоты из расчетов для изолированных молекул. Исключение составляет нитроксил 2, который вносит систематическую погрешность в расчет методом А и более, чем другие молекулы, отклоняется от соответствующей корреляционной прямой на рис. 4. Использование гармонических частот в воде (метод С) дает значительное снижение потенциала окисления для соединения 2 по сравнению с использованием частот из расчетов в газовой фазе (методы А и В). Также, метод С предсказывает значительное сближение потенциалов окисления соединений 9 и 10, как это имеет место в эксперименте. Однако, для молекул 4, 5, 6, 8 использование колебательных частот в растворе увеличивает отклонение от корреляционной прямой (рис. 4). Неоднозначное влияние растворителя на значения $E_{{{\text{OC/NR}}}}^{ \circ }$ в этих случаях может быть обусловлено погрешностями континуальной модели в оценке частот колебаний в растворе. Более достоверные оценки влияния воды возможно могут быть получены из рассмотрения частот в кластерах сольватированных молекул с молекулами воды. Так, правильную оценку величины равновесного изотопного эффекта 16О/18О для депротонирования фосфатов удается получить лишь при учете специфической сольватации атомов исследуемых молекул и анионов [33].
В заключение, в настоящей работе проведено сравнение трех подходов по оценке вкладов свободных энергий в редокс потенциалы $E_{{{\text{OC/NR}}}}^{ \circ }$ конформационно-подвижных циклических нитроксильных радикалов. Использованный ранее метод [13, 18, 19], основанный на рассмотрении ТЦ и оценке свободных энергий Гиббса для наборов конформаций в растворе, дает наименьший статистический разброс значений потенциала окисления, но включает систематическую погрешность, связанную с невозможностью непротиворечивой оценки свободной энергии сольватации отдельных конформаций. Предложенные методы вычисления свободных энергий наборов конформационных состояний, полученных как из рассмотрения ТЦ, с использованием модифицированной процедуры усреднения по состояниям (В), так и из прямого расчета свободных энергий в воде (С), устраняют отмеченную систематическую погрешность, но дают несколько больший статистический разброс значений $E_{{{\text{OC/NR}}}}^{ \circ }$. Учет релаксации структуры реактантов в воде повышает точность расчетов. Подход, основанный на прямом расчете равновесных свободных энергий в воде, может быть использован в качестве замены методов, включающих рассмотрение термодинамического цикла.
Работа поддержана гос. темой Министерства Науки № 0089-2019-0014 ИПХФ РАН. Автор выражает признательность за помощь в проведении расчетов сотрудникам вычислительного центра ИПХФ РАН.
Список литературы
Golubev V.A., Kozlov Yu.N., Petrov A.N. et al., in Bioactive Spin Labels / Ed. R.I. Zhdanov. Berlin: Springer, 1992. P. 119.
Soule B.P., Hyodo F., Matsumoto K. et al. // Free Radical Biol. Med. 2007. V. 42. P. 1632.
Nishide H., Oyaizu K. // Science. 2008. V. 319. P. 737.
Wilcox C.S. // Pharmacol. Ther. 2010. V. 126. P. 119.
Tebben L., Studer A. // Angew. Chem. Int. Ed. 2011. V. 50. P. 5034.
Mitchell J.B., Samuni A., Krishna M.C. et al. // Biochemistry. 1990. V. 29. P. 2802.
Griesser M., Shah R., Van Kessel A.T. et al. // J. Am. Chem. Soc. 2018. V. 140. P. 3798.
Nutting J.E., Rafiee M., Stahl S.S. // Chem. Rev. 2018. V. 118. P. 4834.
Hodgson J.L., Namazian M., Bottle S.E. et al. // J. Phys. Chem. A. 2007. V. 111. P. 13595.
Blinco J.P., Hodgson J.L., Morrow B.J. et al. // J. Org. Chem. 2008. V. 73. P. 6763.
Yamasaki T., Mito F., Ito Y. et al. // Ibid. 2011. V. 76. P. 435.
Billone P.S., Johnson P.A., Lin S. et al. // Ibid. 2011. V. 76. P. 631.
Marenich A.V., Ho J., Coote M.L. et al. // Phys. Chem. Chem. Phys. 2014. V. 16. P. 15068.
Hickey D.P., Schiedler D.A., Matanovic I. et al. // J. Am. Chem. Soc. 2015. V. 137. P. 16179.
Mendkovich A.S., Luzhkov V.B., Syroeshkin M.A. et al. // Russ. Chem. Bulletin, Int. Ed. 2017. V. 66. P. 683.
Levitskiy O.A., Magdesieva T.V. // Mendeleev Commun. 2018. V. 28. P. 187.
Zhang K., Noble B.B., Mater A.C. et al. // Phys. Chem. Chem. Phys. 2018. V. 20. P. 2606.
Krapivin V.B., Mendkovich A.S., Sen’ V.D. et al. // Mendeleev Commun. 2019. V. 29. P. 77.
Krapivin V.B., Sen’ V.D., Luzhkov V.B. // Chem. Phys. 2019. V. 522. P. 214.
Trasatti S. // Pure Appl. Chem. 1986. V. 58. P. 955.
Frisch M.J., Trucks G.W., Schlegel H.B. et al. / Gaussian 09, Revision B.01. Gaussian, Inc., Wallingford CT, 2010.
Marenich V., Cramer C.J., Truhlar D.G. et al. // J. Phys. Chem. B. 2009. V. 113. P. 6378.
Zhao Y., Truhlar D.G. // Theor. Chem. Acta. 2008. V. 120. P. 215.
Lynch B.J., Zhao Y., Truhlar D.G. // J. Phys. Chem. A. 2003. V. 107. P. 1384.
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. P. 3865.
Shibaeva R.N. // J. Struct. Chem. 1975. V. 16. P. 318. https://doi.org/10.1007/BF00747164
Yonekuta Y., Oyaizu K., Nishide H. // Chem. Lett. 2007. V. 36. P. 866.
Sen’ V.D., Shilov G.V., Golubev V.A. // Russ. J. Org. Chem. 2014. V. 50. P. 1124. https://doi.org/10.1134/S1070428014080090
Martins F.A., Silla J.M., Freitas M.P. // Carbohydrate Res. 2017. V. 451. P. 29.
Korneichuk A.Ya., Senyavin V.M., Kuramshina G.M. // Russ. J. Phys. Chem. A. 2017. V. 91. P. 351. https://doi.org/10.1134/S0036024417020170
Флори П. Статистическая механика молекул. М.: Мир, 1971. 440 с.
Sen’ V.D., Tikhonov I.V., Borodin L.I. et al. // J. Phys. Org. Chem. 2015. V. 28. P. 17.
Kolmodin K., Luzhkov V.B., Åqvist J. // J. Am. Chem. Soc. 2002. V. 124. P. 10130.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии