

Журнал физической химии, 2020, T. 94, № 5, стр. 698-705
Влияние заместителей в гидролизованных цефалоспоринах на внутримолекулярную связь O–H···N
Е. О. Левина a, b, *, М. Г. Хренова a, c, В. Г. Цирельсон a, d
a Российская академия наук, ФИЦ Биотехнологии
Москва, Россия
b МФТИ (ГУ)
Долгопрудный, Россия
c Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
d Российский химико-технологический университет имени Д.И. Менделеева
Москва, Россия
* E-mail: levina.eo@phystech.edu
Поступила в редакцию 18.07.2019
После доработки 18.07.2019
Принята к публикации 17.09.2019
Аннотация
Изучены модельные молекулярные системы, имитирующие переходное состояние лимитирующей стадии реакции гидролиза цефалоспориновых антибиотиков в активном центре L1 металло-β-лактамазы. На примере фторзамещенных соединений цефалоспоринового ряда показано, что природа заместителей в тиазиновом и β-лактамном кольцах существенно влияет на силу внутримолекулярной водородной связи O–H···N, определяющей каталитические параметры в реальных биологических системах. Усиление или ослабление связи O–H···N во фторзамещенных соединениях установлено при помощи квантово-топологического анализа электронной плотности, дополненного анализом других дескрипторов, характеризующих химическое связывание. Полученные данные подтверждены анализом сдвига частот валентных колебаний –OH фрагмента карбоксильной группы, формирующего связь O–H···N, по отношению к нефторированному соединению. Отсутствие установленной монотонной зависимости силы водородной связи от степени донорности/акцепторности заместителя, свидетельствует о том, что имеющиеся многоцелевые дескрипторы не обеспечивают в полной мере понимание механизмов связывания в активном центре L1 металло-β-лактамазы.
Современные методы молекулярного моделирования позволяют проводить рациональный дизайн соединений с заданной реакционной способностью, например, для ферментативных реакций [1]. В частности, важно их применение для создания биологически активных молекул, которые действуют на определенные белковые мишени, ингибируют их, и при этом медленно разрушаются под действием других ферментов [2]. К таким задачам относится создание антибиотиков, не гидролизующихся или медленно гидролизующихся β-лактамазами. Для определения реакционной способности соединения требуется изучение механизма реакции. Комбинированный метод квантовой механики / молекулярной механики (КМ/ММ) [3] позволяет решить эту задачу на основании данных о полной энергии системы и вычисления констант скоростей элементарных стадий ферментативных реакций. К сожалению, погрешность метода КМ/ММ [4], связанная с ошибками расчета квантово-механической, молекулярно-механической подсистем и их взаимодействий, не позволяет достоверно ранжировать соединения со схожей реакционной способностью. Для преодоления этого ограничения мы предлагаем использовать комбинацию методов КМ/ММ и квантово-топологического анализа электронной плотности (QTAIM – “quantum theory of atoms in molecules” [5]), дополненных анализом современных дескрипторов химического связывания [6, 7].
QTAIM изучает химическое связывание в атомно-молекулярных системах в терминах критических точек (КТ) электронной плотности (ЭП) и их характеристик [5]. Существует четыре типа КТ, отвечающих элементам молекулярной структуры: ядерные – локальные максимумы ЭП, клеточные – локальные минимумы ЭП, циклические – образованные замкнутыми циклами атомов и имеющие два положительных значения гессиана ЭП, и критические точки связи – седловые точки с одним положительным собственным значением гессиана ЭП вдоль межъядерного вектора. Через каждую КТ-связь проходит связевый путь, образованный двумя градиентными линиями, начинающимися в данной седловой точке и заканчивающиеся на ближайших локальных максимумах (ядрах). Связевый путь отвечает мостику ЭП между двумя соседними ядрами, максимальному по отношению ко всем малым боковым смещениям. Наличие КТ-связи (связевого пути) между двумя атомами, в рамках QTAIM постулируется как признак наличия связывания между этими атомами. Значения дескрипторов ЭП в критической точке связи (электронная плотность и ее лапласиан, плотности кинетической и потенциальной энергии электронов, эллиптичности связи и др.) позволяют охарактеризовать каждое межатомное взаимодействие и выявить его особенности [8–13].
Данная работа инициирована исследованиями реакционной способности цефалоспориновых соединений в активном центре L1 металло-β-лактамазы (L1 MβL). В работе [14] предложен механизм реакции гидролиза окрашенного субстрата цефалоспоринового ряда нитроцефина L1 MβL. Расчеты методом КМ/ММ в варианте PBE0-D3/6-31G**/AMBER показали, что лимитирующей является стадия переноса протона, сопровождающаяся образованием временной внутримолекулярной водородной связи O–H···N (рис. 1). Дальнейшие теоретические исследования реакционной способности набора из 10 цефалоспоринов [15, 16] проведены с применением комбинированного подхода КМ/ММ и QTAIM [17]. Анализ свойств критических точек связи в сопоставлении с каталитическими свойствами этих соединений показал, что каталитическая константа непосредственно связана с силой внутримолекулярной водородной связи O–H···N. Установлено, что более реакционноспособные соединения обладают более слабой Н-связью, а менее реакционноспособные соединения – сильной [17].
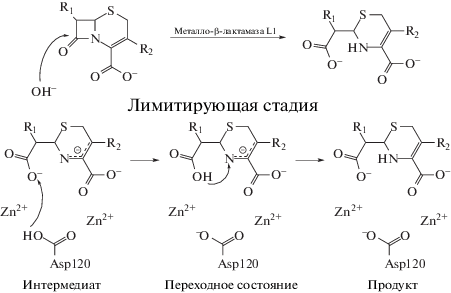
Однако нерешенным остался вопрос о том, как, без дорогостоящих КМ/ММ расчетов, определить какие заместители будут ускорять реакцию, а какие – замедлять. Если обратиться к экспериментальным данным по гидролизу цефалоспориновых соединений в активном центре L1 MβL [15, 16], то можно найти пары субстратов, у которых один из заместителей одинаков, а другой различается. Например, для пар цефокситин и цефуроксим, цефалоспорин C и цефотаксим заместители в тиазиновом кольце совпадают (табл. 1). При этом в первой паре это приводит к изменению константы скорости в 80 раз, а во второй паре – практически не влияет на гидролитическую активность.
Таблица 1.
Структуры переходных состояний антибиотиков цефалоспоринового ряда, рассмотренных в данной работе
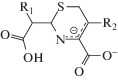 |
R1 | R2 | kcat, с–1 |
|---|---|---|---|
| Цефокситин |  |
 |
1.1 |
| Цефуроксим | 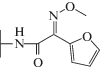 |
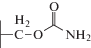 |
80 |
| Цефалоспорин C | 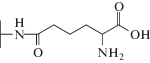 |
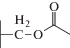 |
62 |
| Цефотаксим | 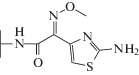 |
 |
66 |
Цель данной работы – выбрать небольшие модельные системы, сохраняющие важные свойства ферментативной системы: водородную связь O‒H···N и координационную связь N···Zn2+, для определения влияния заместителей на силу водородной связи O–H···N. Один из факторов, который может определять силу водородной связи – донорная/акцепторная природа заместителей. Для проверки этой гипотезы изучен набор цефалоспориновых соединений, в которых проводилось последовательное фторирование заместителей и оценивалась сила водородной связи по частоте соответствующего валентного колебания.
МЕТОДИЧЕСКАЯ ЧАСТЬ
Моделирование малых молекулярных комплексов
В качестве малых моделей выбраны молекулярные кластеры, состоящие из цефалоспориновых соединений с катионом Zn2+, координированным двумя молекулами ДМСО (рис. 2). Данные комплексы структурно воспроизводят 3D геометрию переходного состояния цефалоспориновых антибиотиков и соответствуют локальным минимумам на поверхности потенциальной энергии. Рассмотренный набор цефалоспориновых соединений включает в себя базовое соединение и его фторированные аналоги (рис. 3). Проводилось фторирование атома СL и метильной группы при атоме CR (рис. 2).
Рис. 2.
Структура комплекса модельного соединения (1), координированного одним катионом цинка. В системах (2)–(8) проводились замены атомов водорода на атомы фтора у атома CL и метильной группы при атоме СR. Внутримолекулярная водородная связь O–H···N отмечена черной пунктирной линией, координационные связи иона Zn2+ отмечены серыми пунктирными линиями. Атомы водорода, заменяемые на атомы фтора выделены пунктирными окружностями.
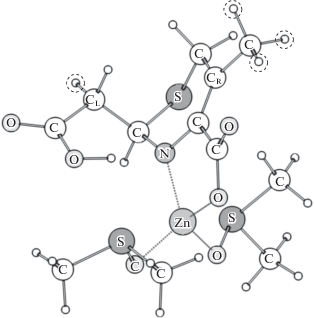
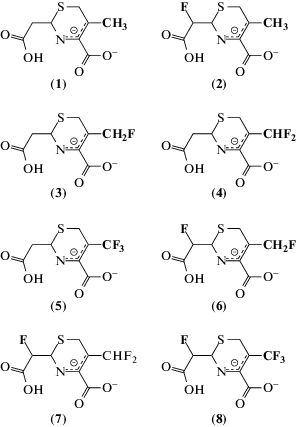
Рис. 3.
Структуры модельных соединений с цефалоспориновым остовом и различными вариантами фторирования атома углерода CL (2, 6, 7, 8) и метильной группы при CR (3, 4, 5, 6, 7, 8).
Все расчеты проводились в программном пакете Firefly [18] методом функционала плотности в варианте PBE0-D3/6-31G** [19–21], аналогичном описанию КМ подсистемы в КМ/ММ расчетах [17]. Локализация минимума ППЭ подтверждалась анализом частот нормальных колебаний соответствующих систем.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Вычисленные геометрические и топологические характеристики рассмотренных изолированных комплексов представлены в табл. 2. Вне зависимости от природы заместителей, связь O–H···N характеризуется схожими расстояниями H···N, O···N и углами O–H···N. В сравнении с аналогичной водородной связью в комплексах цефалоспоринов (RH···N ≈ 1.9–2.0 Å), в модельных соединениях наблюдается ее заметное укорочение (усиление). Для явного выявления особенностей влияния заместителей на связь O–H···N в модельных комплексах проведен анализ частот нормальных колебаний. Полоса валентных колебаний –OH фрагмента карбоксильной группы, формирующего водородную связь O–H···N, лежит в области 3000–3200 см–1. Это отвечает положению характеристической полосы валентных колебаний –OH фрагмента карбоксильной группы, вовлеченной в водородную связь (широкая полоса в области 2500–3300 см–1) [22].
Таблица 2.
Геометрические и топологические характеристики водородной связи O–H···N в комплексах модельных соединений 1–8, координированных одним катионом цинка (см. рис. 3). ρb – электронная плотность в критической точке связи H···N, ∇2ρb – лапласиан электронной плотности
| Ком-плекс | RH···N, Å | RO···N, Å | Угол (O–H···N), град. | ρb, а.е. | ∇2ρb, а.е. |
|---|---|---|---|---|---|
| 1 | 1.715 | 2.660 | 155.5 | 0.055 | 0.109 |
| 2 | 1.699 | 2.642 | 154.2 | 0.058 | 0.111 |
| 3 | 1.691 | 2.637 | 155.2 | 0.058 | 0.114 |
| 4 | 1.717 | 2.652 | 153.9 | 0.054 | 0.113 |
| 5 | 1.724 | 2.656 | 153.6 | 0.053 | 0.111 |
| 6 | 1.680 | 2.624 | 154.0 | 0.059 | 0.114 |
| 7 | 1.683 | 2.631 | 153.4 | 0.058 | 0.114 |
| 8 | 1.715 | 2.642 | 152.2 | 0.054 | 0.112 |
Сдвиг частоты валентных колебаний группы X–H характеризует силу взаимодействия X–H···Y [23], поэтому нами проанализированы положения частот OH валентных колебаний, ${{\bar {\nu }}_{{{\text{O}} - {\text{H}}}}}$, для фторированных соединений (2)–(8), относительно исходного комплекса (1). В случае значительной степени фторирования метильной группы при атоме углерода CR (рис. 3 – соединения 4, 5, 8) наблюдается сдвиг в более высокочастотную область. Это свидетельствует о тенденции к ослаблению связи O–H···N в случае значительного повышения акцепторных свойств заместителя при атоме углерода CR. Фторирование СL приводит к обратному эффекту, т.е. усилению O–H···N связи, выражающемуся в сдвиге ${{\bar {\nu }}_{{{\text{O}} - {\text{H}}}}}$ в область более низких частот (табл. 3). Следует отдельно отметить неоднозначный эффект слабого фторирования заместителя при атоме CR, который проявляет себя в усилении связи O–H···N.
Таблица 3.
Частота валентного колебания O–H, ${{\bar {\nu }}_{{{\text{O}} - {\text{H}}}}}$, в комплексах соединений 1–8 и сдвиг этой частоты относительно значения для комплекса с соединением 1. OH-группа участвует в формировании водородной связи O–H···N
| Соеди-нение | Заместителиа) | RH···N, Å | ${{\bar {\nu }}_{{{\text{O}} - {\text{H}}}}}$, см–1 | $\Delta {{\bar {\nu }}_{{{\text{O}} - {\text{H}}}}}$, см–1 |
|---|---|---|---|---|
| 1 | – | 1.715 | 3097 | – |
| 2 | СLFH | 1.699 | 3037 | –61 |
| 3 | CR–CH2F | 1.691 | 3060 | –37 |
| 4 | CR–CHF2 | 1.717 | 3150 | 53 |
| 5 | CR–CF3 | 1.724 | 3172 | 75 |
| 6 | СLFH, CR–CH2F | 1.680 | 3007 | –90 |
| 7 | СLFH, CR–CHF2 | 1.683 | 3058 | –39 |
| 8 | СLFH, CR–CF3 | 1.715 | 3130 | 33 |
а) нумерация атомов представлена на рис. 3.
Поскольку водородная связь O–H···N в комплексах цефалоспорин – L1 MβL наблюдается в переходном состоянии, смещение частоты валентного колебания ${{\bar {\nu }}_{{{\text{O}} - {\text{H}}}}}$ не может рассматриваться как индикатор влияния заместителей в цефалоспориновом остове на силу связи O–H···N. Для преодоления этого ограничения нами были проанализированы зависимости соответствующих частот от различных характеристик водородной связи (рис. 4 и 5). Обычно изменение частоты валентных колебаний группы X–H сопряжено с изменением расстояния X···Y [24], однако, в случае систем с цефалоспориновым остовом более показательным является расстояние H···Y (рис. 4а). Необходимо отметить, что мы использовали линейную аппроксимацию зависимостей частоты от различных характеристик связи O‒H···N [25, 26]. Это обусловлено относительно небольшим изменением данных характеристик в рассмотренных комплексах. В случае значительных различий в силе связей, не исключена и нелинейная (например) экспоненциальная зависимость частоты валентных колебаний OH от длины водородной связи [24].
Рис. 4.
Зависимости значений расстояний H···N и O···N от частоты валентного колебания ${{\bar {\nu }}_{{{\text{O}} - {\text{H}}}}}$ для водородной связи O–H···N в модельных комплексах. Линейная аппроксимация представлена черным цветом, темно-серым и светло-серым обозначены границы доверительного интервала и интервала предсказания (p = 95%).
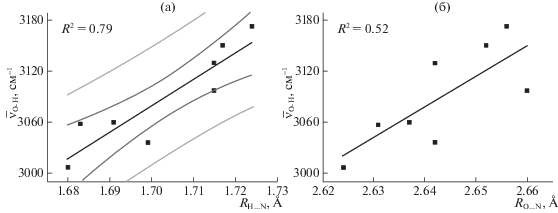
Рис. 5.
Зависимости (а–в) значений электронной плотности (ρb), плотностей потенциальной и кинетической энергий электронов (νb и gb) в критической точке связи H···N от валентной частоты ${{\bar {\nu }}_{{{\text{O}} - {\text{H}}}}}$ для водородной связи O–H···N в модельных комплексах. Линии линейной аппроксимации представлены черным цветом, темно-серым и светло-серым обозначены границы доверительных интервалов и интервалов предсказания (p = 95%).
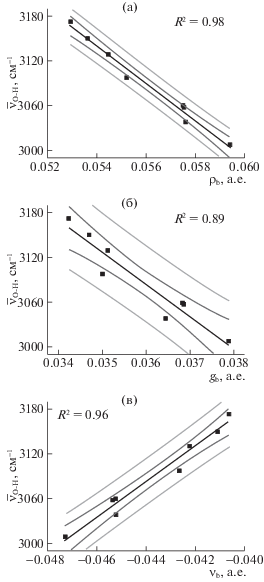
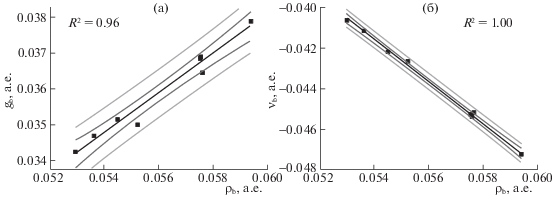
Значительную корреляцию с изменением частоты валентного колебания ${{\bar {\nu }}_{{{\text{O}} - {\text{H}}}}}$ показали значения ЭП (ρb), а также плотности потенциальной и кинетической энергий электронов (νb и gb) в КТ-связи H···N (рис. 5). Наличие корреляции ${{\bar {\nu }}_{{{\text{O}} - {\text{H}}}}}$ со значениями ρb согласуется с данными о зависимости энергий водородных связей различной силы от соответствующих значений ρb [27]. Корреляция плотностей потенциальной и кинетической энергий с валентными частотами ${{\bar {\nu }}_{{{\text{O}} - {\text{H}}}}}$ согласуется с моделью из работы [28]. Целесообразность использования значений ρb, νb и gb (даже при малом их изменении) как индикатора влияния заместителей на силу связи O–H···N в комплексах соединений цефалоспоринового типа дополнительно подтверждается наличием хорошей корреляцией этих дескрипторов друг с другом (рис. 6). При этом использование корреляции ρb – νb в КТ-связи O–H···N является предпочтительным, поскольку показывает лучшие статистические параметры и демонстрирует согласие с данными из частот валентных колебаний OH-группы.
Рис. 6.
Зависимости значений плотностей кинетической gb (а) и потенциальной νb энергий (б) электронов от ρb для водородной связи O–H···N в модельных комплексах. Линии линейной аппроксимации представлены черным цветом, темно-серым и светло-серым обозначены границы доверительных интервалов и интервалов предсказания (p = 95%).
Изменение частоты валентного колебания ${{\bar {\nu }}_{{{\text{O}} - {\text{H}}}}}$ демонстрирует еще один значимый эффект. При введении акцепторного заместителя у атома СL частота снижается вне зависимости от природы заместителя у атома CR (см. рис. 7). То есть при наличии одинаковых заместителей у атома CR в цефалоспориновом остове усиление связи O‒H···N может быть ассоциировано с увеличением акцепторных свойств заместителя при СL. Данный эффект позволяет интерпретировать изменения в значениях дескрипторов в КТ-связи O–H···N для цефалоспоринов с одинаковыми заместителями при атоме CR (табл. 4). Так, цефокситин и цефалоспорин C демонстрируют бóльшие значения ρb и |νb|, чем цефуроксим и цефотаксим, соответственно [17]. Это свидетельствует о некотором усилении связи O–H···N для первой пары антибиотиков по сравнению со второй. Учитывая одинаковую природу заместителей у цефокситина и цефуроксима, цефалоспорина C и цефотаксима при атоме CR можно предполагать больший акцепторный эффект заместителей при атоме углерода СL.
Рис. 7.
Изменение частоты валентных колебаний ${{\bar {\nu }}_{{{\text{O}} - {\text{H}}}}}$ в фрагменте O–H···N в парах модельных комплексов c катионом цинка и молекулами ДМСО, отличающихся заместителями при атоме CL.
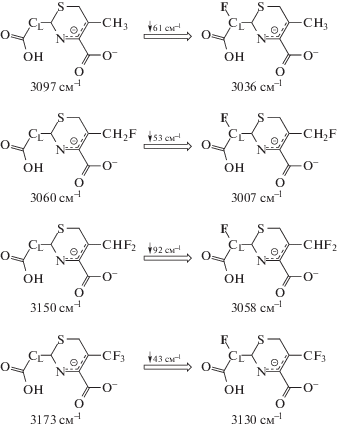
Таблица 4.
Геометрические и топологические характеристики водородной связи O–H···N в переходных состояниях рассмотренных антибиотиков цефалоспоринового ряда (табл. 1)
| Переходное состояние цефалоспорина | RH···N, Å | RO···N, Å | Угол (O–H···N), град. | ρb, а.е. | ∇2ρb, а.е. | gb, а.е. | νb, а.е. |
|---|---|---|---|---|---|---|---|
| Цефокситин | 1.913 | 2.618 | 125.2 | 0.037 | 0.093 | 0.026 | –0.029 |
| Цефуроксим | 2.071 | 2.674 | 117.6 | 0.028 | 0.083 | 0.021 | –0.022 |
| Цефалоспорин C | 1.993 | 2.693 | 125.6 | 0.032 | 0.081 | 0.022 | –0.024 |
| Цефокситин | 2.068 | 2.699 | 120.1 | 0.028 | 0.079 | 0.021 | –0.022 |
Для более детального анализа природы заместителей в цефалоспоринах требуется явное выявление влияния окружения на связь O–H···N. Для этого необходим поиск других дескрипторов электронной плотности, что явится предметом дальнейших исследований.
Таким образом, по результатам анализа модельных систем, схожих по своему геометрическому строению с переходным состоянием лимитирующей стадии установлено, что природа заместителей у углеродных атомов CR и СL существенно влияет на силу водородной связи O‒H···N, определяющей каталитические параметры цефалоспориновых соединений в активном центре L1 металло-β-лактамазы. Частоты валентных колебаний –OH находятся в широком коридоре рассчитанных значений, 3000–3200 см–1. Фторирование по атому CL приводит к ослаблению водородной связи по отношению к нефторированному аналогу. Последовательное фторирование метильной группы у атома CR не приводит к монотонному изменению частоты валентного колебания OH. Установление монотонной зависимости между силой водородной связи O–H···N и электронодонорными/акцепторными свойствами заместителей требует выборки, более широкой, чем использованные 8 модельных молекулярных кластеров и расширение круга дескрипторов для более полного понимания механизмов связывания в активном центре L1 металло-β-лактамазы. Это позволит усилить выводы данной работы, расширить их на биологически активные цефалоспориновые соединения и провести поиски других возможных дескрипторов заместителей, определяющих силу внутримолекулярной водородной связи O–H···N.
Исследование выполнено при финансовой поддержке РНФ в рамках научного проекта № 18-74-10056. Работа выполнена с использованием оборудования Центра коллективного пользования сверхвысокопроизводительными вычислительными ресурсами МГУ имени М.В. Ломоносова.
Список литературы
Sliwoski G., Kothiwale S., Meiler J., Lowe E.W. // Pharmacol. Rev. 2014. V. 66. P. 334. https://doi.org/10.1124/pr.112.007336
Mandal S., Moudgil M., Mandal S.K. // Eur. J. Pharmacol. 2009. V. 625. P. 90. https://doi.org/10.1016/j.ejphar.2009.06.065
Warshel A., Levitt M. // J. Mol. Biol. 1976. V. 103. № 2. P. 227. https://doi.org/10.1016/0022-2836(76)90311-9
Mardirossian N., Head-Gordon M. // Mol. Phys. 2017. V. 115. № 19. P. 2315. https://doi.org/10.1080/00268976.2017.1333644
Bader R.F.W. Atoms in molecules. Quantum Theory. Oxford: Clarendon press, 1994. P. 432.
Khrenova M.G., Tsirelson V.G. // Mendeleev Commun. 2019. V. 29. №. 5. P. 492. https://doi.org/10.1016/j.mencom.2019.09.004
Хренова М.Г., Томилко А.В., Цирельсон В.Г. // Вестн. МГУ. Сер. 2. Химия. 2019. Т. 60. № 3. С. 141.
Bader R.F.W., Essén H. // J. Chem. Phys. 1984. V. 80. P. 1943. https://doi.org/10.1063/1.446956
González L. et al. // J. Chem. Phys. 1998. V. 109. № 7. P. 2685. https://doi.org/10.1063/1.476868
Grabowski S.J., Robinson T.L., Leszczynski J. // Chem. Phys. Lett. 2004. V. 386. № 1–3. P. 44. https://doi.org/10.1016/j.cplett.2004.01.013
Vener M.V., Levina E.O., Astakhov A.A. et al. // Chem. Phys. Lett. 2015. V. 638. P. 233. https://doi.org/10.1016/j.cplett.2015.08.053
Espinosa E., Alkorta I., Elguero J. et al. // J. Chem. Phys. 2002. V. 117. № 12. P. 5529. https://doi.org/10.1063/1.1501133
Cremer D., Kraka E., Slee T.S. et al. // J. Am. Chem. Soc. 1983. V. 105. № 15. P. 5069. https://doi.org/10.1021/ja00353a036
Khrenova M.G., Nemukhin A.V. // J. Phys. Chem. B. 2018. V. 122. № 4. P. 1378. https://doi.org/10.1021/acs.jpcb.7b10188
Crowder M.W., Walsh T.R., Banovic L. et al. // Antimicrob. Agents Chemother. 1998. V. 42. № 4. P. 921. https://doi.org/10.1128/AAC.42.4.921
Felici A., Amicosante G. // Antimicrob. Agents Chemother. 1995. V. 39. № 1. P. 192. https://doi.org/10.1128/AAC.39.1.192
Khrenova M.G., Krivitskaya A.V., Tsirelson V.G. // New J. Chem. 2019. V. 43. № 19. P. 7329.https://doi.org/10.1039/C9NJ00254E
Granovsky A.A., Firefly version 8, www http://classic.chem.msu.su/gran/firefly/index.html
Adamo C., Barone V. // J. Chem. Phys. 1999. V. 110. № 13. P. 6158. https://doi.org/10.1063/1.478522
Grimme S., Antony J., Ehrlich S. et al. // J. Chem. Phys. 2010. V. 132. № 15. P. 154104. https://doi.org/10.1063/1.3382344
Lu T., Chen F. // J. Comput. Chem. 2012. V. 33. № 5. P. 580. https://doi.org/10.1002/jcc.22885
Socrates G. Infrared and Raman characteristic group frequencies: tables and charts. John Wiley & Sons, 2004.
Rozenberg M., Loewenschuss A., Marcus Y. // Phys. Chem. Chem. Phys. 2000. V. 2. № 12. P. 2699. https://doi.org/10.1039/B002216K
Steiner T. // Angew. Chem. Int. Ed. 2002. V. 41. № 1. P. 48. https://doi.org/10.1002/1521-3773(20020104)41:1<48::AID-ANIE48>3.0.CO;2-U
Rozenberg M. // RSC Adv. 2014. V. 4. № 51. P. 26928. https://doi.org/10.1039/C4RA03889D
Afonin A.V., Vashchenko A.V., Sigalov M.V. // Org. Biomol. Chem. 2016. V. 14. № 47. P. 11199. https://doi.org/10.1039/C6OB01604A
Parthasarathi R., Subramanian V., Sathyamurthy N. // J. Phys. Chem. A. 2006. V. 110. № 10. P. 3349. https://doi.org/10.1021/jp060571z
Mata I., Alkorta I., Espinosa E. et al. // Chem. Phys. Lett. 2011. V. 507. № 1–3. P. 185. https://doi.org/10.1016/j.cplett.2011.03.055
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии