

Журнал физической химии, 2020, T. 94, № 6, стр. 862-865
Изменение кристалличности пшеничной соломы в процессе обработки озоном
Е. М. Бенько a, *, Д. Г. Чухчин b, А. В. Малков c, И. В. Выдрина b, Е. В. Новожилов b, В. В. Лунин a
a Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
b Северный (Арктический) федеральный университет имени М.В. Ломоносова
Архангельск, Россия
c ФГБУН ФИЦКИА РАН
Архангельск, Россия
* E-mail: benko_elena@mail.ru
Поступила в редакцию 11.06.2019
После доработки 11.06.2019
Принята к публикации 09.07.2019
Аннотация
Новый способ дифрактометрического определения степени кристалличности веществ, основанный на оценке скорости изменения функции I = f(2Θ) по всему диапазону углов рассеяния дифрактограммы исследуемого образца, использован для исследования изменения кристалличности пшеничной соломы в процессе обработки озоном. Показано, что степень кристалличности образца обратно пропорциональна содержанию в нем лигнина. Корреляций между кристалличностью лигноцеллюлозного субстрата и его реакционной способностью в процессах ферментативного гидролиза в сахара не обнаружено.
Делигнификация растительного сырья с помощью озона рассматривается в качестве перспективного метода его предобработки в процессах ферментативного гидролиза в сахара [1–4]. Озонирование позволяет достичь высокой степени конверсии биомассы с минимальным негативным воздействием на окружающую среду. Реакции ферментативного гидролиза целлюлозосодержащих материалов протекают в гетерогенной среде, поэтому скорость гидролиза существенно зависит от структурных свойств субстрата [1]. Установлено, что аморфные структуры целлюлозы разрушаются ферментами быстрее, чем кристаллические [5]. В работе [6] показано, что кристалличность хвойной и лиственной сульфатной целлюлозы при ферментной обработке проходит через максимум, поскольку на начальной стадии процесса ферменты гидролизуют аморфные участки микрофибрилл целлюлозы и гемицеллюлозы, на более поздних стадиях – кристаллические участки целлюлозы. Любые способы обработки, которые способствуют увеличению содержания аморфной целлюлозы в лигноцеллюлозном материале, ускоряют процесс ферментации.
Для целлюлозных субстратов, не содержащих лигнин, отмечена четкая отрицательная линейная зависимость между индексом кристалличности и скоростью ферментативного гидролиза субстрата [7, 8]. Однако в случае лигнинсодержащих материалов такой явной корреляции не прослеживается. Изменение кристалличности целлюлозы может не иметь заметного влияния на фоне такого более значимого фактора как доступность целлюлозы для ферментов, что определяется величиной удельной поверхности, размерами пор, экранирующим действием лигнина и гемицеллюлоз [1].
Литературные сведения об изменении кристалличности лигноцеллюлозных материалов после озонирования неоднозначны. Согласно работам [9–11], кристалличность образцов древесины после обработки озоном немного возрастает вследствие удаления аморфного лигнина. Напротив, в [12] при длительном озонировании лиственной древесины наблюдали падение индекса кристалличности. Этими же авторами проведено исследование структурных превращений чистой хлопковой целлюлозы при озонировании [13]. Установлено, что в начале озонирования в кристаллитах целлюлозы происходит удаление деформированных частей макромолекул, и наблюдается некоторое уменьшение индекса кристалличности. Затем идет кристаллизация аморфной части, и кристалличность возрастает. При длительной обработке озоном межплоскостное расстояние приобретает свое равновесное значение, степень кристалличности и размер кристаллита стабилизируются на уровне 0.78 и 6.6 нм соответственно.
Цель настоящей работы – изучение изменений кристаллической структуры пшеничной соломы в процессе ее обработки озоном, а также выяснение влияния кристалличности субстрата на его реакционную способность в реакциях ферментативного гидролиза в сахара. В работе на примере соломы апробирован новый метод расчета степени кристалличности веществ по данным рентгеновской дифрактометрии, изложенный в работе [14].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Озонировали образцы пшеничной соломы (Tritcum L.) с размером частиц 0.6–1.0 мм и влажностью (абсолютной) 100%. Образцы предоставлены и охарактеризованы Алтайским государственным университетом (Барнаул). Содержание основных компонентов: лигнин – 20%, целлюлоза – 40%, гемицеллюлозы – 24%. Методика озонирования в проточной системе в реакторе с неподвижным слоем и способ расчет расхода озона в реакции описаны в нашей работе [15]. Содержание остаточного лигнина определяли по стандартной методике с помощью двухстадийного гидролиза серной кислотой [16]. Характеристики образцов пшеничной соломы (время озонирования, количество поглощенного озона, полное содержание остаточного лигнина) приведены в табл. 1.
Таблица 1.
Характеристика образцов соломы
| № | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Время озонирования, мин | 0 | 15 | 30 | 50 | 90 | 240 |
| Расход озона, ммоль/г | 0 | 0.8 | 1.8 | 2.2 | 3.4 | 7.0 |
| Содержание общего лигнина, % | 19.1 | 15.5 | 10. 8 | 9.3 | 7.3 | 6.6 |
Для рентгеновских исследований образцы соломы после озонирования промывали водой и подвергали лиофильной сушке. Затем образцы размалывали в планетарной шаровой мельнице Retsch PM100 в течение 2 мин при частоте вращения 400 об./мин. Материал гарнитуры карбид вольфрама. Таблетки толщиной около 1 мм готовили в цилиндрической пресс-форме диаметром 25 мм на прессе Retsch PP 25 (усилие 10 тс). Для записи дифрактограмм использовали рентгеновский дифрактометр XRD-7000S (Shimadzu, Япония), оборудованный приставкой для вращения образца и системой поликапиллярной оптики. Оптическая схема гониометра θ–θ, детектор сцинтилляционный, с монохроматором с длиной волны 1.5406 Å (линия CuKα1). Рентгеновские дифрактограммы образцов записывали с использованием неотражающего держателя со скоростью вращения 30 об./мин. Параметры работы рентгеновской трубки: ускоряющее напряжение 50 кВ, ток 30 мА, материал мишени – Cu. Диапазон сканирования по углу 2θ: 10°–70°, скорость сканирования 0.5 град/мин, шаг 0.02°.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
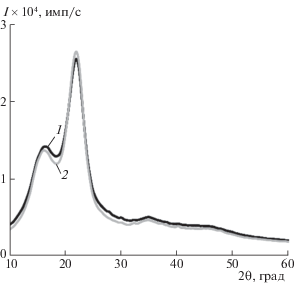
На рис. 1 представлены дифрактограммы образцов соломы до и после обработки озоном. Дифрактограммы содержат два пика с максимумами при 2θ = 22.5° и 16°, характерными для кристаллической решетки целлюлозы [17]. Для определения степени кристалличности образцов использовали новый метод, разработанный авторами [14], основанный на оценке скорости изменения функции I = f(2θ) по всему диапазону углов рассеяния дифрактограммы исследуемого образца. Величину индекса кристалличности IC рассчитывали как отношение интеграла модуля первой производной функции I = f(2θ) к интегралу площади под кривой дифрактограммы:
Рис. 1.
Дифрактограммы образцов исходной (1) и озонированной (2) соломы. Расход озона 3.4 ммоль/г (2).
Для нахождения значения степени кристалличности полученную величину IC соотносят с данными для эталонного кристаллического тела, в качестве которого выбрано золото:
где XAu – значение индекса кристалличности образца, град–1; ICAu – значение индекса кристалличности стандарта (Аu), град–1.В отличие от других методов [18–21], расчет ведется по всему диапазону углов сканирования. Корректность метода подтверждена тем, что на трех субстанциях, содержащих известные доли аморфной и кристаллической составляющих (аморфный углерод–графит; кварцевое стекло–горный хрусталь; лигнин–целлюлоза) установлена линейная зависимость (R2 > 0.99) между заданным соотношением кристаллической и аморфной частей и вычисленным показателем кристалличности [14].
На рис. 2 показаны зависимости степени кристалличности образцов озонированной соломы от расхода озона и от содержания лигнина. Из приведенных данных следует, что по мере увеличения количества поглощенного озона происходит увеличение степени кристалличности, что может быть обусловлено удалением из лигноуглеводного композита аморфных компонентов – лигнина и, частично, гемицеллюлоз.
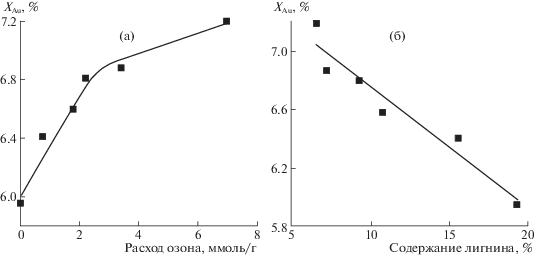
В работе [10] при делигнификации озоном древесины (осина) также отмечено увеличение доли кристаллической фазы. Расчет индекса кристалличности (IC) проводился по методу Сегала [18]. Показано, что при расходе озона 4 ммоль/г значение IC для древесины возрастает с 73.2 до 80.5%, а при совместном использовании озона с пероксидом водорода – до 85.0% [10]. Согласно [22], величина IC для образцов исходной и интенсивно обработанной озоном (8 ммоль/г) осиновой древесины составляет соответственно 38 и 44%. Увеличение индекса кристалличности образцов биомассы за счет удаления аморфных составляющих наблюдали также в результате обработки с помощью различных других методов (паровой взрыв [23], высокотемпературная обработка СО2, SO2 [24]).
В работах [15, 25] нами показано, что при ферментативном гидролизе обработанной озоном соломы выход сахаров возрастает пропорционально количеству поглощенного озона и удаленного лигнина (в интервале расхода озона от 0 до 2–3 ммоль/г). Согласно данным рис. 2, кристалличность образца при этом возрастает. Таким образом, на реакционную способность делигнифицированного субстрата в большей степени оказывает влияние не индекс кристалличности целлюлозы, а увеличение доступности целлюлозы и снижение потерь ферментов за счет их адсорбции на лигнине.
Работа выполнена при поддержке Российского фонда фундаментальных исследований (код проекта № 16-08-00876) с использованием оборудования, приобретенного при поддержке Программы развития Московского университета, а также ЦКП НО “Арктика” (САФУ) при финансовой поддержке Минобрнауки РФ.
Список литературы
Sun Y., Cheng J. //Bioresour. Technol. 2002. V. 83. P. 1.
Travaini R., Martín-Juárez J., Lorenzo-Hernando A., Bolado-Rodríguez S. // Bioresour. Technol. 2016. V. 199. P. 2.
Wu J.S., Upreti J., Ein-Mozaffari F. // Int. J. Hydrogen Energy. 2013. V. 38. P. 10270.
Bhattarai S., Bottenus D., Ivory C.F. et al. // Bioresour. Technol. 2015. V. 196 P. 78.
Chang V.S., Holtzapple M.T. // Appl. Biochem. Biotechnol. 2000. V. 84. P. 5.
Novozhilov E.V., Sinel’nikov I.G., Aksenov A.S. et al. // Catalysis in Industry. 2016. V. 8. P. 95.
Fan L.T., Lee Y., Beardmore D.H. // Biotechnol. Bioeng. 1980. V. 22. P. 177.
Hall M., Bansal P., Lee J.H. et al. // FEBS Journal. 2010. V. 277. P. 1571.
Yu Z., Jameel H., Chang H., Park S. // Bioresour. Technology. 2011. V. 102. P. 9083.
Mamleeva N.A., Abrosimova G.E., Kharlanov A.N., Lunin V.V. // Russ. J. Phys. Chem A. 2013. V. 87. P. 1102.
Liu M., Chen X., Tian Xi. // Intern. J. Polymer Sci. 2018. Article ID 3452586. 7 pages https://doi.org/10.1155/2018/3452586
Демин В.А. Окислительная деструкция лигнина и целлюлозы пероксиреагентами: Автореф. дис. … докт. хим. наук. Уфа: Ин-т органической химии РАН, 1997. 44 с
Секушин Н.А., Кочева Л.С., Демин В.А. // Химия растительного сырья. 1999. P. 59.
Чухчин Д.Г., Малков А.В., Тышкунова И.В. и др. // Кристаллография. 2016. V. 61. P. 1.
Ben’ko E.M., Chukhchin D.G., Lunin V.V. // Russ. J. Phys. Chem. A. 2017. V. 91. P. 2092.
Aldaeus F., Sjöholm E. COST Action FP0901 Round Robins of Lignin Samples. Part 1: Lignin Content, Version 3 (2010-12-14), Innventia Report No.: IR 108. 2011.
Иоелович М.Я., Веверис Г.П. // Химия древесины. 1987. V. 5. P. 72.
Segal L., Creely J.J., Martin A.E., Conrad C.M. // Text. Res. J. 1959. V. 29. P. 786.
Vonk C.G. // Appl. Cryst. 1973. V. 6. P. 148.
Terinte N., Ibbett R., Schuster K. // Lenzinger Berichte. 2011. V. 89. P. 118.
Rietveld H. // J. Appl. Cryst. 1969. V. 2. P. 65.
Ben’ko E.M., Manisova O.R., Murav’eva G.P., Lunin V.V. // Russ. J. Phys. Chem. A. 2013. V. 87. P. 1097.
Asada C., Sasaki C., Hirano T., Nakamura Y. // Bioresour. Technol. 2015. V. 182. P. 245.
Corrales R.C.N.R., Mendes F.M.T., Perrone C.C. et al. // Biotechnol. Biofuels. 2012. V. 5. P. 1.
Ben’ko E.M., Chukhchin D.G., Mamleeva N.A. et al. // Russ. J. Phys. Chem. A. 2019. V. 93 (in press).
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии