

Журнал физической химии, 2020, T. 94, № 6, стр. 910-919
Особенности газохроматографического разделения таутомеров этилацетоацетата
И. Г. Зенкевич a, *, В. М. Лукина a
a Санкт-Петербургский государственный университет, Институт химии
Санкт-Петербург, Россия
* E-mail: izenkevich@yandex.ru
Поступила в редакцию 09.10.2019
После доработки 09.10.2019
Принята к публикации 15.10.2019
Аннотация
Рассмотрены особенности газохроматографического разделения кето- и енольной таутомерных форм этилацетоацетата на капиллярной колонке со стандартной неполярной полидиметилсилоксановой неподвижной фазой BPX-1. Подтверждено, что хроматограммы смеси таутомеров имеют специфический профиль, а именно – “плато” между пиками таутомеров, что соответствует обратимому превращению “кето $ \rightleftarrows $ енол в процессе разделения. Показано, что енольная и кето-формы этилацетоацетата имеют разные коэффициенты температурной зависимости индексов удерживания (0.19 ± 0.03 и 0.02 ± 0.02 соответственно). Установлено отсутствие зависимости относительных площадей пиков таутомеров от природы растворителя (полярный этиловый спирт и неполярный гексан) при разных температурах, т.е. такие отношения преимущественно отражают положение равновесия “кето $ \rightleftarrows $ енол” в паровой фазе испарителя хроматографа. Сделан вывод, что следствием этого оказываются близкие значения термодинамических параметров (стандартные энтальпия и энтропия) таутомерного равновесия, определяемые при дозировании проб в различных растворителях. Обсуждены возможные искажения результатов определений за счет эффектов дискриминации состава проб, дозируемых в капиллярные колонки с делением потока газа-носителя. Примесь в длительно хранившемся образце этилацетоацетата идентифицирована как этил-2-гидрокси-3-оксобутаноат – продукт окисления растворенным кислородом атмосферного воздуха.
Прототропная таутомерия (1,3-сдвиг атома водорода) вызывает особый интерес в химии, так как усложняет характеристику физико-химических и спектральных свойств органических соединений вследствие их зависимости от условий определения (температура, растворитель, рН растворов и т.д.). Типичным примером структур, для которых типична прототропная таутомерия, являются β-дикарбонильные соединения:

Существование подобного таутомерного равновесия определяет сложности и неоднозначности результатов хроматографического разделения способных к таутомерии аналитов [1]. Чаще всего в химической практике встречаются такие представители β-дикетонов как 2,4-пентандион (ацетилацетон, I) и этил-3-оксобутаноат (этилацетоацетат, ацетоуксусный эфир, II):
По литературным данным содержание енольной формы в (I) при нормальных условиях (н.у.) составляет 80–96% [2], а в (II) – от 6.8–8.0 [3] до 9.4–10.5% [4]. Основной метод определения соотношения кето- и енольных форм в растворах при н.у. – спектроскопия ЯМР 1Н, причем оценки их количеств по разным сигналам спектров могут несколько различаться между собой [4]. Содержание в растворах менее полярных енольных форм β-дикарбонильных соединений увеличивается при переходе от полярных к неполярным растворителям [5], а также в паровой фазе.
Все отмеченные выше закономерности установлены для жидких индивидуальных веществ или их растворов. Газохроматографическое же разделение подразумевает перевод компонентов проб в паровую фазу в испарителе хроматографа и перемещение их зон по колонке при повышенных температурах, что изменяет соотношение таутомеров. Однако во всех оригинальных работах [6–13], использованных в качестве источников информации в базе данных NIST [14], указаны газохроматографические индексы удерживания (RI) этилацетоацетата (II) на стандартных неполярных и полярных фазах без уточнения их отнесения к какому-либо из таутомеров. То же относится к масс-спектру ионизации электронами (ИЭ) эфира (II) [14]. Подобная ситуация наблюдается и для ацетилацетона (I). В то же время для изомерного (II) метилового эфира 2-оксопентановой кислоты в базе [14] приведены два разных масс-спектра кето- и енольной форм, но значение RI приписано только первой из них. Между тем, еще в конце 1980-х гг. появились сообщения о газохроматографическом разделении кето- и енольных форм 1,3-дикетонов [15], а несколько позже и β-кетоэфиров [16]; подтверждены различия их масс-спектров ИЭ. Данные работ [15, 16] позволяют обобщить закономерности последовательностей газохроматографического элюирования таутомеров β-дикарбонильных соединений, по крайней мере, на стандартных неполярных фазах. Если при атоме углерода, расположенном между карбонильными группами, нет заместителей, то енольная форма имеет меньшие параметры удерживания по сравнению с кето-таутомером. При наличии заместителей порядок элюирования заменяется обратным.
Измерение соотношения интенсивностей сигналов таутомеров эфира (II) в спектрах ЯМР 1Н при разных температурах эквивалентно определению констант равновесия “кето $ \rightleftarrows $ енол”, на основании которых можно вычислять термодинамические параметры активации (ΔН# и ΔS#) этого процесса [3–5, 17, 18]. Газохроматографические данные для этих целей не использовали, так как оставалось неясным, к какой фазе (системе) относить полученные данные. Есть сообщения об использования таких данных для оценки энтальпий испарения таутомеров [3].
Задачи настоящей работы:
– выявление особенностей газохроматографического разделения двух таутомерных форм этилацетоацетата (II), в том числе температурной зависимости соотношений площадей пиков S(енол) – S(кето) в растворителях разной полярности;
– уточнение значений газохроматографических индексов удерживания обоих таутомеров этилацетоацетата и характеристик их температурной зависимости;
– сопоставление термодинамических параметров активации процесса установления таутомерного равновесия “кето $ \rightleftarrows $ енол” для растворов этилацетоацетата в разных растворителях с целью уточнения природы этого процесса в условиях газохроматографического разделения;
- идентификация примеси ранее не охарактеризованного соединения, обнаруженной в хранившемся в течение длительного времени образце этилацетоацетата.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Используемые реактивы и приготовление образцов. Использовали этилацетоацетат (“х.ч.”, “Реахим”, Москва), н-алканы С6–С12 с четным числом атомов углерода (“х.ч.” или “х.ч. для хроматографии”, “Реахим”, Москва); растворители н‑гексан (“х.ч.”, Вектон, Санкт-Петербург) и этиловый спирт (ООО “Гатчинский спиртовый завод”, Ленинградская обл.). Образцы готовили растворением 150 мкл этилацетоацетата в 1.35 мл этилового спирта или н-гексана с добавкой по 50 мкл н-октана и н-декана (реперные углеводороды).
Условия газохроматографического и хромато-масс-спектрометрического анализа. Газохроматографический анализ растворов этилацетоацетата в этиловом спирте и гексане проводили на газовом хроматографе Хроматэк-Кристалл 5000.2 с пламенно-ионизационным детектором и WCOT колонкой из плавленого кварца со стандартной неполярной полидиметилсилоксановой фазой BPX-1 длиной 10 м, внутренним диаметром 0.53 мм и толщиной пленки неподвижной фазы 2.65 мкм. Температура колонки 70°С при варьировании температуры испарителя от 100 до 220°С с шагом 20 K, или изотермические режимы от 60 до 100°С с шагом 10 K при постоянной температуре испарителя 160°С. Температура детектора 200°С, газ-носитель азот, объемная скорость 5.2 мл/мин (линейная скорость 43.9 см/с), деление потока 1 : 3, объем дозируемых проб 0.5 мкл (микрошприц “МШ-1”). Число параллельных определений для одних и тех же образцов в одинаковых условиях 2–4. Для определения изотермических индексов удерживания (Ковача) в образцы добавляли по 50 мкл н-алканов С8 и С10.
Хромато-масс-спектрометрический анализ проводили на хромато-масс-спектрометре Shimadzu QP-2010 SE с ИЭ (энергия ионизации 70 эВ) с колонкой Optima 1 длиной 25 м, внутренним диаметром 0.32 мм и толщиной пленки неподвижной фазы 0.35 мкм. Режим анализа: программирование температуры от 50 до 250°С со скоростью 5 K/мин, температура испарителя 180°С, температура детектора 200°С, газ-носитель гелий, расход 1.82 мл/мин (линейная скорость 53.6 см/с), деление потока 1:10, объем дозируемых проб 0.5 мкл. Температуры интерфейса и источника ионов 200°С. Время перекрывания потока газа-носителя из хроматографической колонки в источник ионов (“отсечка растворителя”) 1.6 мин. Для определения линейно-логарифмических индексов удерживания в образцы добавляли смесь реперных н-алканов С6–С12 с четным числом атомов углерода.
Обработка результатов. Компоненты реакционных смесей характеризовали стандартными масс-спектрами и индексами удерживания с их последующим усреднением. В качестве оценки “мертвого” времени для растворов в н-гексане принимали время выхода пика первой из примесей в растворителе, для растворов в этаноле – время выхода растворителя. Таутомеры характеризовали отношениями площадей пиков кето- и енольной форм. В случае автоматической регистрации площадей в области “плато” между пиками таутомеров эти значения прибавляли к площади первого пика при условии tR < [tR(1) + + tR(2)]/2 и второго при невыполнении этого условия. Вычисление индексов удерживания и статистическую обработку данных проводили с использованием ПО Excel (Microsoft Office, 2010), Origin (версия 4.2) и программы QBasic.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Особенности хроматографических профилей таутомеров этилацетоацетата. Фрагмент хроматограммы раствора этилацетоацетата (II) в этиловом спирте в режиме программирования температуры приведен на рис. 1. По соотношению интенсивностей пиков и в соответствии с литературными данными о порядке элюирования таутомеров [15] менее интенсивный пик с меньшим временем удерживания должен быть отнесен к енольной форме эфира. Интенсивность сигналов молекулярных ионов енольной формы закономерно выше благодаря наличию системы сопряжения. Масс-спектры ИЭ таутомеров в целом сходны, но в них наблюдаются некоторые заметные отличия, например, в интенсивностях пиков с m/z = 85 и 69:
Рис. 1.
Фрагмент хроматограммы раствора этилацетоацетата в этиловом спирте в условиях программирования температуры; компонент “Х” – примесь в образце, С10 – реперный н-алкан С10Н22, t – время удерживания.

№ 1 (енольная форма), m/z ≥ 39 (Iотн, %): 130(17)М, 115(5), 102(12), 88(16) [M – CH2CO], 87(16), 86(3), 85(31) [M – C2H5O], 84(15), 70(3), 69(24), 61(4), 60(8), 58(4), 56(2), 55(2), 45(5), 44(3), 43(100) [CH3CO], 42(12), 41(3), 39(3).
№ 2 (кето-таутомер), m/z ≥ 39 (Iотн, %): 130(6)М, 115(2), 102(6), 88(20) [M – CH2CO], 87(4), 85(13) [M – C2H5O], 84(2), 70(3), 69(4), 61(5), 60(11), 58(2), 45(4), 44(3), 43(100) [CH3CO], 42(12), 41(2).
Единственный приведенный в базе [14] масс-спектр этилацетоацетата приблизительно соответствует суперпозиции двух приведенных спектров. В частности, интенсивность сигнала с m/z = = 69 в нем составляет 10%.
Обращает на себя внимание специфический контур хроматографического сигнала в области между пиками таутомеров, уровень которого не достигает базовой линии и образует некоторое “плато” (рис. 1). Регистрация масс-спектров в разных точках подобных “плато” на примерах других соединений, обратимо или необратимо превращающихся друг в друга в процессе разделения в хроматографической колонке [19, 20], показывает, что они также представляют собой суперпозиции масс-спектров индивидуальных таутомеров в разных соотношениях. Такой вид хроматограмм однозначно свидетельствует о взаимопревращении “кето $ \rightleftarrows $ енол” в процессе перемещения зон аналитов по колонке. Аналогичные профили хроматографических сигналов наблюдались для четырех кетоэфиров [15] и для диацетила на дополнительно окисленной полярной неподвижной фазе RTx-1701 [21].
Газохроматографический анализ растворов (II) в изотермических условиях при разных температурах колонки (от 60 до 100°С) и постоянной температуре испарителя позволяет охарактеризовать температурную зависимость индексов удерживания каждого из таутомеров и обнаруженной в образце (II) примеси, а также установить характер зависимости констант равновесия (K), равных отношениям площадей пиков таутомеров K = Sотн = S(енол)/S(кето) от температуры колонки.
На рис. 2 сопоставлены фрагменты хроматограмм таутомеров (II) в изотермических условиях при температурах колонки 60°С (а) и 100°С (б), иллюстрирующие заметную зависимость степени их разделения от температуры. При 60°С “плато” между пиками имеет минимальную величину, но при повышении температуры его высота увеличивается. В результате при 100°С уже можно сделать вывод о неполном разделении таутомеров, что объясняется двумя причинами. Прежде всего, енольная и кето-формы этилацетоацетата характеризуются разными коэффициентами (β) температурной зависимости индексов удерживания RI(T):
где RI(T) – значение индекса удерживания при температуре Т, RI(T0) – значение при температуре Т0 = 0°С (условно), β = dRI/dT – коэффициент температурной зависимости индексов удерживания. Значения RI(T0) и β вычисляют методом наименьших квадратов. В результате обработки данных в интервале температур 60–100°С получаем результаты (в сравнении со значениями RIпрогр в условиях программирования температуры), приведенные в табл. 1.Рис. 2.
Фрагменты хроматограмм раствора этилацетоацетата в этиловом спирте в изотермических условиях; температура колонки 60 (а) и 100°С (б); С8 – реперный н-алкан С8Н18.

Таблица 1.
Значения RI(T0) и β для енольной и кето-форм этилацетоацетата
| Компонент | RIпрогр | RI(T0) | β |
|---|---|---|---|
| Енольная форма (II) | 889 ± 2 | 875 ± 2 | 0.19 ± 0.03 |
| Кето-форма (II) | 920 ± 1 | 919 ± 1 | 0.02 ± 0.02 |
| Примесь | 957 ± 2 | 964 ± 1 | –0.10 ± 0.01 |
Из значения β = 0.02 ± 0.02 ед. индекса/K следует, что значение RI кето-формы этилацетоацетата практически не зависит от температуры. В то же время положительное значение β = 0.19 ± 0.03 для енольной формы означает, что при повышении температуры хроматографической колонки “расстояние” между пиками таутомеров в шкале индексов удерживания уменьшается. Причина бóльшего значения коэффициента β енольной формы (II) – наличие в ее молекуле сопряженной системы >C=C–C=O. В связи с этим следует отметить аномальное отрицательное значение коэффициента β для примеси (–0.10 ± 0.01). Столь необычная для большинства органических соединений величина, во-первых, исключает присутствие в молекуле систем сопряжения и, во-вторых, типична для соединений с функциональными группами, содержащими активные атомы водорода. Скорость установления равновесия “кето $ \rightleftarrows $ енол” при повышении температуры возрастает, что увеличивает высоту “плато” между пиками таутомеров. Дальнейшее ее повышение теоретически должно приводить к слиянию пиков таутомеров (аналогично температуре коалесценции в спектроскопии ЯМР [18]).
Анализ растворов (II) при разных температурах газохроматографической колонки подтверждает существование температурной зависимости относительных площадей пиков. Например, значения Sотн = S(енол)/S(кето) < 1 уменьшаются при повышении температуры, что иллюстрируют данные табл. 2. Однако линеаризовать зависимость Sотн от температуры хроматографической колонки уравнением Антуана:
где коэффициенты a и b вычисляют методом наименьших квадратов, не удается.Таблица 2.
Зависимость средних значений отношений площадей пиков таутомеров этилацетоацетата (раствор в этиловом спирте) от температуры хроматографической колонки при постоянной температуре испарителя (160°С)
| Т, °С | S(енол)/S(кето) | S(кето)/S(енол) | S(енол)/S(С8) | S(кето)/S(С8) |
|---|---|---|---|---|
| 60 | – | 4.1 | 0.27 | 1.08 |
| 70 | 0.25 | 4.1 | 0.27 | 1.11 |
| 80 | 0.26 | 3.9 | 0.28 | 1.10 |
| 90 | 0.24 | 4.1 | 0.26 | 1.10 |
| 100 | 0.23 | 4.4 | 0.26 | 1.08 |
Причина этого – неодинаковые времена нахождения аналитов в нагретой хроматографической колонке, зависимость которых от температуры характеризуется уравнением [22]:
В результате линеаризация зависимости Sотн = = f(T) возможна только при объединении соотношений (2) и (3) в одно, содержащее такую “экзотическую” функцию как двойной логарифм lnln Sотн [21, 23], что не входило в задачи настоящей работы. Вместо этого предпочтительнее рассмотреть проще интерпретируемую зависимость Sотн от температуры испарителя при постоянной температуре хроматографической колонки.
Зависимость относительных площадей пиков таутомеров этилацетоацетата от температуры испарителя. В табл. 3 приведены средние значения относительных площадей Sотн = S(енол)/S(кето) и обратные им величины при различных температурах испарителя (от 100 до 220°С) для растворов этилацетоацетата в этиловом спирте, а в табл. 4 – аналогичные данные для растворов в гексане. Температуру колонки при этом сохраняли постоянной (70°С). Таблицы 3 и 4 дополнительно содержат отношения площадей пиков енольной и кето-форм этилацетоацетата к площади пика инертного компонента смеси – н-алкана С8Н18 (см. обсуждение далее). Согласно литературным данным, содержание енольной формы в неполярном растворителе (гексан) выше, чем в полярном (этанол) (39 и 7.2% соответственно [5]). Однако величины Sотн таутомеров для обоих растворителей при всех температурах испарителя отличаются незначительно. Например, значения Sотн = = S(кето)/S(енол) при увеличении температуры от 100 до 200°С для растворов (II) в этаноле увеличиваются в 1.9, а для растворов в гексане – в 2.2 раза Этот результат позволяет утверждать, что температурные вариации положения таутомерного равновесия “кето $ \rightleftarrows $ енол”, устанавливаемые по газохроматографическим данным, относятся не к конденсированной, а преимущественно к паровой фазе, в которой влияние природы растворителя на соотношение таутомеров минимально. Кроме того, наблюдаемые различия могут быть обусловлены не влиянием природы растворителей на положение таутомерного равновесия, а проявлениями эффектов дискриминации состава анализируемых проб (см. далее).
Таблица 3.
Зависимость средних значений отношений площадей пиков таутомеров этилацетоацетата (раствор в этиловом спирте) от температуры испарителя при постоянной температуре хроматографической колонки (70°С)
| Т, °С | S(енол)/S(кето) | S(кето)/S(енол) | S(енол)/S(С8) | S(кето)/S(С8) | [S(енол) + S(кето)]/S(C8) |
|---|---|---|---|---|---|
| 100 | 0.42 | 2.36 | 0.20 | 0.46 | 0.66 |
| 120 | 0.36 | 2.83 | 0.19 | 0.54 | 0.73 |
| 140 | 0.30 | 3.36 | 0.17 | 0.58 | 0.75 |
| 160 | 0.27 | 3.76 | 0.17 | 0.64 | 0.81 |
| 180 | 0.25 | 4.05 | 0.15 | 0.65 | 0.80 |
| 200 | 0.22 | 4.54 | 0.14 | 0.67 | 0.81 |
| 220 | 0.20 | 5.09 | 0.14 | 0.70 | 0.84 |
Таблица 4.
Зависимость cредних значений отношений площадей пиков таутомеров этилацетоацетата (раствор в гексане) от температуры испарителя при постоянной температуре хроматографической колонки (70°С)
| Т, °С | S(енол)/S(кето) | S(кето)/S(енол) |
|---|---|---|
| 100 | 0.45 | 2.21 |
| 120 | 0.36 | 2.72 |
| 140 | 0.32 | 3.14 |
| 160 | 0.28 | 3.57 |
| 180 | 0.24 | 4.21 |
| 200 | 0.21 | 4.77 |
Вычисление параметров линейной регрессии ln Sотн от обратной температуры (в виде 1000/Т с последующим пересчетом) методом наименьших квадратов по уравнению:
(4)
$\ln [S({\text{енол)/S(кето}})] = --\Delta {{H}^{\# }}{\text{/}}RT + \Delta {{S}^{\# }}{\text{/}}R$На рис. 3 и 4 приведены зависимости (4) для растворов этилацетоацетата в этиловом спирте и гексане. Параметры уравнений линейной регрессии указаны в подписях к этим рисункам. Вычисленные по этим данным значения ΔН# и ΔS# в случае этанола составляют –9.3 ± 0.3 кДж/моль и –32.3 ± 0.7 Дж/(K моль), а в случае гексана –10.9 ± ± 0.3 кДж/моль и –35.8 ± 0.8 Дж/(K моль). Отрицательные знаки величин ΔН# и ΔS# и порядок их абсолютных величин, в целом, согласуются с определенными в работе [21], в которой, правда, они были приписаны не равновесию в паровой фазе, а взаимодействию таутомеров с полярной неподвижной фазой хроматографической колонки.
Рис. 3.
Зависимость ln Sотн от температуры испарителя (1/Т) для раствора этилацетоацетата в этиловом спирте. Параметры линейной регрессии: a = 1.12 ± ± 0.03, b = –3.89 ± 0.08, R = 0.998, S0 = 0.02.

Рис. 4.
Зависимость ln Sотн от температуры испарителя (1/Т) для раствора этилацетоацетата в гексане. Параметры линейной регрессии: a = 1.31 ± 0.04, b = = ‒4.31 ± 0.10, R = 0.998, S0 = 0.02.

Незначительная величина |ΔН#| ∼ 10 ± 1 кДж/моль объясняет легкость взаимных превращений таутомеров при относительно невысоких температурах газохроматографического разделения, проявляющихся в специфических профилях хроматограмм.
Влияние эффектов дискриминации состава проб. Любые газохроматографические определения, предполагающие дозирование проб в капиллярные колонки с делением потока при разных температурах испарителя или в разных растворителях, должны учитывать возможное искажение результатов за счет так называемых эффектов дискриминации состава проб [23–26]. Одно из наиболее типичных их проявлений – вариации абсолютных и относительных площадей пиков в зависимости от температуры испарителя хроматографа и природы растворителя. В рассматриваемом в настоящей работе случае характеристика зависимости Sотн = f(T) для таутомеров как раз и предполагает вариации температуры испарителя, а сравнение параметров этой зависимости для полярного этанола и неполярного гексана – влияние второго фактора.
Для обсуждения эффектов дискриминации состава проб при их дозировании в капиллярные колонки с делением потока целесообразно вернуться к рассмотрению данных табл. 2. Помимо относительных площадей пиков таутомеров (II) (раствор в этиловом спирте) при разных температурах хроматографической колонки в ней дополнительно представлены отношения площадей пиков каждого из таутомеров к площадям пиков инертного компонента – углеводорода н-С8Н18: S(енол)/S(С8) и S(кето)/S(С8), равные 0.27 ± 0.01 и 1.10 ± 0.01 соответственно. Это подтверждает отсутствие какой-либо зависимости указанных отношений от температуры колонки (нет эффектов дискриминации), так что вариации S(енол)/S(кето) и S(кето)/S(енол) обусловлены исключительно зависимостью Sотн = f(T) для таутомеров.
В табл. 3 помимо значений S(енол)/S(С8) и S(кето)/S(С8) приведены аналогичные отношения S(енол)/S(C8) и S(кето)/S(C8), демонстрирующие выраженные температурные зависимости (убывающая для первого отношения и возрастающая для второго). Графическая иллюстрация этих зависимостей представлена на рис. 5. Особый интерес представляют не температурные зависимости каждого из этих отношений по отдельности, а температурная зависимость их суммы. В табл. 3 дополнительно приведены значения [S(енол) + S(кето)]/S(C8), а на графике для наглядности отображена кривая, соответствующая полиномиальной аппроксимации температурной зависимости полусуммы [S(енол)/S(C8) + S(кето)/S(C8)]/2). Кето- и енольные формы этилацетоацетата изомерны, что предопределяет близкую чувствительность к ним пламенно-ионизационного детектора, но данные табл. 3 и рис. 5 иллюстрируют заметное увеличение отношений [S(енол) + S(кето)]/S(C8) при увеличении температуры от 100 до 220°С (в 1.27 раза). Это и может быть объяснено проявлением эффектов дискриминации [23–26], так как при увеличении температуры испарителя увеличиваются относительные площади пиков более высококипящих компонентов (для этилацетоацетата Ткип =180.8°С, для н-октана – 125.7°С).
Рис. 5.
Графическое представление зависимостей относительных площадей пиков: 1 – S(енол)/S(С8), 2 – S(кето)/S(С8), 3 – S(С10)/S(С8) (для сравнения) от температуры испарителя. Сплошная линия – результат полиномиальной аппроксимации возрастающей зависимости средних значений [S(енол)/S(C8) + + S(кето)/S(C8)]/2 от температуры (по данным для раствора этилацетоацетата в этиловом спирте).
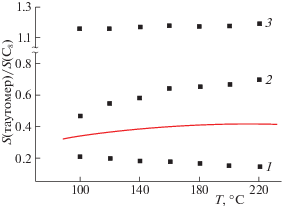
Дополнительно на рис. 5 приведен набор точек, соответствующих относительным площадям пиков соединений одной химической природы – реперным углеводородам н-С10Н22 и н-С8Н18, из чего можно сделать вывод об отсутствии явно выраженной температурной зависимости для этой пары компонентов. Аналогичная температурная зависимость отношений [S(енол) + S(кето)]/S(C8) наблюдается и для растворов эфира (II) в гексане, но для сокращения объема обсуждения результатов соответствующие данные не включены в табл. 4.
Таким образом, эффекты дискриминации, безусловно, влияют на характеристики температурной зависимости относительных площадей пиков таутомеров и, следовательно, на надежность определения термодинамических параметров таутомерного равновесия “кето $ \rightleftarrows $ енол”. В рассматриваемом случае можно отметить, что отношения S(кето)/S(C8) для растворов (II) в этаноле (табл. 3) при увеличении температуры увеличиваются в 2.2 раза, тогда как оценка с учетом эффектов дискриминации соответствует увеличению всего приблизительно в 1.3 раза. Более точный учет их влияния требует дальнейшего совершенствования методов обработки экспериментальных данных и специального рассмотрения.
Идентификация неизвестной примеси в образце этилацетоацетата. Кроме двух таутомеров с RI = 880 и 909 (значения для режима программирования температуры), в хранившемся в течение длительного времени образце этилацетоацетата была обнаружена примесь с RI = 957 со следующим масс-спектром ИЭ:
m/z ≥ 39 (Iотн ≥ 2%): 146(1), 119(1), 75(17), 74(74), 73(24), 59(8), 57(17), 56(17), 47(8), 46(7), 45(100), 44(18), 43(40), 42(6), 41(11).
Характерная особенность этого масс-спектра – присутствие двух интенсивных сигналов с m/z = = 45 и 74, относящихся к гомологическим группам у = 3 и 4. Такое сочетание массовых чисел редко встречается в масс-спектрах органических соединений; библиотечный поиск по этим двум пикам с использованием базы данных NIST [14] привел всего к 10 альтернативным ответам с факторами совпадения менее 0.5, так что ни один из них не может быть принят к дальнейшему рассмотрению. Идентификация по номерам гомологических групп главных сигналов и масс-спектрам ионных серий [27] также не дала положительных результатов. Безрезультатными оказались попытки предположить природу этого компонента исходя из схемы синтеза этилацетоацетата и, в том числе, гипотезы о возможном образовании этилового эфира енольной формы [28]. Следует заметить, что любые производные енольной формы (II) должны быть исключены на основании отрицательного значения коэффициента температурной зависимости индексов удерживания (β = –0.10 ± 0.01). Таким образом, пик с RI = 957 принадлежит неидентифицированному компоненту “Х”.
Тем не менее, установление его структуры возможно в результате совместной интерпретации масс-спектра ИЭ и индекса удерживания на стандартной неполярной неподвижной фазе. Слабый сигнал масс-спектра с максимальным значением m/z = 146, скорее всего, принадлежит молекулярным ионам. Такое отнесение подтверждается наличием в масс-спектре еще одного слабого сигнала с m/z = 119, соответствующего осколочным ионам [M – C2H3]+, типичным для этиловых эфиров карбоновых кислот (разрыв связей с “двойной” перегруппировкой водорода [29]). Если так, то образование соединения с М = 146 из этилацетоацетата С6Н10О3 с М = 130 представляет собой его окисление, что согласуется с длительным хранением образца в контакте с атмосферным воздухом. Окисление протекает растворенным в эфире (II) кислородом воздуха по свободнорадикальному механизму.
Образующиеся при этом гидропероксиды и полимерные пероксиды нестабильны при нагревании и, следовательно, недоступны для газохроматографического анализа, так что наблюдаемый компонент “Х”, наиболее вероятно, имеет структуру этил-2-гидрокси-3-оксобутаноата С6Н10О4:

Это соединение упоминается в литературе, о чем свидетельствует его CAS № 15863-59-9, однако ни каких-либо его физико-химических характеристик, ни спектральных данных для него найти не удалось. На основании масс-спектра можно только предположить, что интенсивный сигнал масс-спектра с m/z = 74 соответствует ионам, образующимся в результате разрыва связи C–C с миграцией атома водорода, например, М+ (m/z = = 146) → [M – CH3COCHO]+ ([M – 72] = 74). Следовательно, основным источником информации при доказательстве структуры этого соединения должно быть совпадение экспериментального значения его газохроматографического индекса удерживания (957 в режиме программирования температуры) с теоретически вычисленной исходя из структуры величиной.
Из всех известных способов предсказания газохроматографических индексов удерживания наиболее информативным в данном случае представляется алгоритм, основанный на гипотетической “сборке” требуемой молекулярной структуры из структур более простых аналогов с последующими арифметическими операциями (суммирование и вычитание) с известными значениями RI таких аналогов [30–32]. Фактически, это – один из вариантов аддитивных схем. В данном случае решение задачи осложняется наличием внутримолекулярной водородной связи, вследствие чего сначала необходимо оценить ее вклад в аддитивную оценку RI. Для этого сравним, например, оценку RI 2-гидрокси-3-пентанона, содержащего аналогичную водородную связь в молекуле, с экспериментальным значением RI этого соединения. В качестве справочных значений RI использованы данные базы [14]:
Следовательно, не учитываемый такой аддитивной схемой инкремент внутримолекулярной водородной связи равен 862 – (787 ± 10) = 75 ± 10 ед. индекса. Далее проведем аналогичную сборку структуры целевого этил-2-гидрокси-3-оксобутаноата уже с учетом полученного инкремента:

Окончательно получаем: 1027 – 75 = 952.
Оценкой стандартного отклонения полученного результата является квадратный корень из суммы квадратов стандартных отклонений всех используемых при расчетах данных, т.е. (42 + 52 + + 102 + 22 + 22)1/2 ≈ 12. Следовательно, вычисленное значение RI = 952 ± 12 совпадает с экспериментальной величиной (957), что в данном случае можно рассматривать как решающее доказательство правильности предполагаемой структуры компонента “X” – этил-2-гидрокси-3-оксобутаноат.
Таким образом, рассмотрение особенностей газохроматографического разделения таутомеров этилацетоацетата (II) позволило сделать следующие выводы.
- Отсутствие зависимости относительных площадей пиков таутомеров от природы растворителя при разных температурах показывает, что результаты газохроматографического анализа преимущественно отражают положение равновесия “кето $ \rightleftarrows $ енол” в паровой фазе испарителя хроматографа.
- Следствием этого оказываются близкие значения термодинамических параметров (стандартных энтальпии и энтропии активации) таутомерного равновесия, определяемые с использованием растворов этилацетоацетата в различных растворителях.
- Температурные вариации относительных площадей пиков таутомеров заметно превышают их температурные вариации за счет эффектов дискриминации состава проб, дозируемых в капиллярные колонки с делением потока. Тем не менее, можно полагать, что именно эффекты дискриминации вносят некоторые погрешности в результаты определений.
- Следствием малых абсолютных значений ΔН# оказывается легкость обратимых взаимных превращений таутомеров в процессе газохроматографического разделения. При температурах колонок 100°С и выше их разделение, видимо, невозможно.
- Установлено, что кето- и енольный таутомеры этилацетоацетата характеризуются значительными различиями температурных коэффициентов газохроматографических индексов удерживания на стандартных неполярных фазах.
- Использование модифицированного варианта аддитивных схем оценки газохроматографических индексов удерживания позволило идентифицировать примесь в образце этилацетоацетата как продукт его окисления растворенным кислородом атмосферного воздуха, а именно этил-2-гидрокси-3-оксобутаноат.
Список литературы
Minkin V.I., Olekhnovich L.P., Zhdanov Yu.A. Molecular design of tautomeric compounds. Kluwer Publ.: Dordrecht-Boston-Tokio, 1958. 312 p. https://doi.org/10.1007/978-94-009-1429-2
Spencer J.N., Holmboe E.S., Kirshembaum M.R., Firth D.W., Pinto P.B. // Can. J. Chem. 1982. V. 68. P. 1178.
Umnahanant P., Chickos J.S. // J. Chem. Eng. Data, 2005. V. 50. № 5. P. 1720. https://doi.org/10.1021/je050179z
Antic D. Measuring the Equilibrium Constant of a Keto-Enol Tautomerism Using Benchtop NMR. ThermoScientific Application Note, AN52327. 2017. 3 p.
Rogers M.T., Burdett J.L. // Can. J. Chem. 1965. V. 43. P. 1516.
Tiess D. // Wiess Z. Willhelm-Pieck-Univ. Rostock Math. Naturwiss. Reiche. 1984. V. 33. P. 6.
Peppard T.L. // J. Agric. Food Chem. 1992. V. 40. № 2. P. 257. https://doi.org/10.1021/jf00014a018
Tudor E. // J. Chromatogr. A. 1997. V. 779. P. 287. https://doi.org/10.1016/S0021-9673(97)00453-6
Tudor E., Moldovan D., Zarna N. // Rev. Roum. Chim. 1999. V. 44. № 2. P. 665.
Jordan M.J., Goodner K.L., Shau P.E. // J. Agric. Food Chem. 2002. V. 50. № 6. P. 1523. https://doi.org/10.1021/jf011077p
Adamova M., Orinak A., Halas L. // J. Chromatogr. A. 2005. V. 1087. P. 131. https://doi.org/10.1016/j.chroma.2005.01.003
Pino J.A., Mesa J., Munos Y., Marti M.P., Marbot R. // J. Agric. Food Chem. 2005. V. 53. № 6. P. 2213. https://doi.org/10.1021/jf0402633
Bianchi F., Careri M., Mangia A., Musci M. // J. Sep. Sci. 2007. V. 39. № 4. P. 563.
The NIST 17 Mass Spectral Library (NIST17/2017/EPA/NIH). Software/Data Version (NIST17); NIST Standard Reference Database, Number 69, June 2017. National Institute of Standards and Technology, Gaithersburg, MD 20899: http://webbook.nist.gov (дата обращения: октябрь 2019 г.).
Masur M., Grutzmascher H.-F., Munster H., Budzikieicz H. // Org. Mass Spectrom. 1987. V. 22. P. 493.
Allegretti P.E., Schiavoni M.M., Di Loreto H.E., Furlong J.J.P., Della Vedova C.O. // J. Mol. Struct. 2001. V. 560. P. 327.
Ruggiero S.J., Luaces V.-M. // J. Chem. Educ. 1988. V. 65. № 7. P. 629. https://doi.org/10.1021/ed065p629
Krishman V. // Inventions. 2019. V. 4. 15 p. https://doi.org/10.3390/inventions4010013
Kornilova T.A., Ukolov A.I., Kostikov R.R., Zenkevich I.G. // Rapid Commun. Mass Spectrom. 2013. V. 27. № 3. P. 461. https://doi.org/10.1002/rcm.6457
Зенкевич И.Г., Подольский Н.Е. // Аналитика и контроль. 2017. Т. 21. № 2. С. 125. https://doi.org/10.15825/analitika.2017.21.2.002
Skrdla P.J., Antomucci V., Lindemann C. // J. Chromatogr. Sci. 2001. V. 39. P. 431.
Руководство по газовой хроматографии. Под ред. Э. Лейбница и Х.Г. Штруппе. М.: Мир, 1988. В 2 т.
Grob K., Neukom H.P. // J. Chromatogr. A. 1982. V. 236. P. 297. https://doi.org/10.1016/S0021-9673(00)84878-5
Зенкевич И.Г., Олисов Д.А. // Лаборатория и производство. 2018. № 2. С. 92.
Зенкевич И.Г., Лелеев Е. // Аналитика и контроль. 2019. Т. 23. № 1. С. 110. https://doi.org/10.15826/analitika.2019.23.1.012
Зенкевич И.Г., Олисов Д.А. // Журн. аналит. химии. 2019. Т. 74. № 7. С. S40. https://doi.org/10.1134/S1061934819070190
Зенкевич И.Г., Иоффе Б.В. Интерпретация масс-спектров органических соединений. Л.: Химия, 1986, 176 с.
Зенкевич И.Г., Лукина В.М. // Аналитика и контроль. 2019. Т. 23. № 3. С. 410. https://doi.org/10.15826/analitika.2019.23.3.009
Hamming M.C., Foster N.G. Interpretation of Mass Spectra of Organic Compounds. New York: Academic Press, 1979. 694 p.
Zenkevich I.G., Moeder M., Koeller G., Schrader S. // J. Chromatogr. A. 2004. V. 1025. P. 227. https://doi.org/10.1016/j.chroma.2003.10.106
Зенкевич И.Г., Уколов А.И. // Журн. общ. химии. 2011. Т. 81. № 9. С. 1479. https://doi.org/10.1134/1070363211090143
Зенкевич И.Г., Уколов А.И. // Масс-спектрометрия. 2011. Т. 8. № 4. С. 264. https://doi.org/10.1134/S1061934812130114
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии