

Журнал физической химии, 2021, T. 95, № 4, стр. 568-573
Порядок связи как мера электрохимической стабильности замещенных сульфоланов
Э. М. Хамитов a, *, Е. В. Кузьмина a, Е. В. Карасева a, В. С. Колосницын a
a Российская академия наук, Уфимский федеральный исследовательский центр, Уфимский институт химии
Уфа, Россия
* E-mail: khamitovem@gmail.com
Поступила в редакцию 29.04.2020
После доработки 29.04.2020
Принята к публикации 12.05.2020
Аннотация
С целью оценки влияния заместителей на прочность связей в молекулах замещенных сульфоланов и их электрохимическую стабильность выполнено квантово-химическое исследование 35 структур: сульфолана, 4 моно- и 10 дифторозамещенных структур, по четыре структуры со следующими углеводородными радикалами в качестве заместителей: метил, этил, пропил, винил, аллил. Рассчитаны порядки связей для этих структур тремя методами: Wiberg (метод Вайберга), Mayer (метод Майера), Fuzzy (метод определения порядков связей для “нечетких” атомов).
Разработка литиевых и литий-ионных аккумуляторов, обладающих высокой удельной энергией и длительным сроком эксплуатации, – актуальная задача электрохимической энергетики [1]. Высокая плотность энергии в аккумуляторах может быть достигнута применением активных материалов с низким эквивалентным весом и высоким электродным потенциалом. Применение активных материалов с высокими электродными потенциалами требует использования электролитов с широким окном электрохимической устойчивости [2]. Сульфолан – перспективный растворитель для электролитов литиевых и литий-ионных аккумуляторов, поскольку он обладает высокими диэлектрической проницаемостью, температурой кипения и температурой вспышки. Электролитные системы на основе сульфолана имеют высокие окислительные потенциалы (>5.5 В) и представляют большой интерес для применения в высоковольтных литиевых и литий-ионных аккумуляторах [3–6].
Окислительная и восстановительная стабильность апротонных органических растворителей может быть улучшена путем введения в их молекулы заместителей различной природы. Из патента [7] следует, что в качестве заместителя в сульфолане чаще всего используют фтор, реже – углеводородные радикалы. Поскольку синтез новых растворителей с различными заместителями и исследования их электрохимической стабильности представляет собой сложную и трудоемкую задачу, целесообразно использование методов вычислительной химии для предварительной оценки электрохимической стабильности (окислительной и восстановительной устойчивости) растворителей.
Окисление молекулы растворителя происходит путем удаления электрона с высшей занятой молекулярной орбитали (ВЗМО), поэтому положение энергетического уровня ВЗМО обычно указывает на степень сложности удаления электрона и, следовательно, на высоту потенциала окисления. Эта простая корреляция может быть использована для поиска молекул с высокой окислительной стабильностью. Один из способов изменить уровень ВЗМО – введение функциональных групп в молекулы растворителей. Так, например, в работе [8] проведены квантово-химические расчеты производных сульфонов, где в качестве функциональных групп использовали фтор-, циано-, сложно эфирную и карбонатную группы. Наиболее интересны результаты расчетов авторов, показывающие, что две сильные электроноакцепторные группы, такие как ‒F и ‒CN, расположенные на концах линейной молекулы сульфона, могут значительно понизить энергию ВЗМО, тем самым затруднить отрыв электрона и привести к повышению окислительного потенциала, относительно незамещенного сульфона.
Наиболее используемыми численными характеристиками стабильности молекулы служат энергии граничных молекулярных орбиталей, а также вертикальный и адиабатический потенциалы ионизации. Эти численные характеристики могут быть получены в результате квантово-химических расчетов. Поскольку вертикальный потенциал ионизации рассчитывают без оптимизации геометрических параметров иона, т.е. без учета релаксации ионизированной частицы, его значение не дает точной оценки стабильности молекул. Более точную оценку стабильности молекул растворителей дает адиабатический потенциал ионизации, который рассчитывается с учетом релаксации иона. Большое различие между вертикальным и адиабатическим потенциалами ионизации, которое может достигать 1.8 В, указывает на важность учета релаксации частицы в процессе окисления. Существенное влияние эффекта релаксации на результаты вычислительной оценки потенциала ионизации было обнаружено при вычислении потенциалов восстановления [9].
Основная проблема при изучении свойств растворителей и реакций электрохимического окисления и восстановления вычислительными методами – размер модельной системы [10], т.е. количество частиц, которое необходимо учесть для адекватного описания системы. Увеличение размера модельной системы, с одной стороны, приводит к большей точности результатов расчетов, с другой стороны, существенно увеличивает время расчетов. Поэтому актуален поиск вычислительных методов оценки свойств растворителей, которые бы позволяли достаточно точно и быстро оценивать влияние заместителей на свойства молекул растворителей.
Известно, что порядок [11] и длина [12] связей коррелируют с их прочностью: чем выше порядок, тем короче связь и тем больше ее прочность. Соответственно данные о порядке связей можно использовать для оценки изменения прочности и длины связей, в особенности в схожих по строению молекулах. Такой подход ранее не был использован для оценки электрохимической устойчивости растворителей. Данный подход привлекателен благодаря незначительным вычислительным затратам. Для его реализации необходимо провести оптимизацию геометрических параметров молекулы растворителя квантово-химическим методом с последующим анализом порядков всех связей.
Существует несколько методов расчета порядков связей. Каждый из методов имеет свои ограничения и области применимости для описания того или иного типа молекул. Для небольших молекул с полярными связями хорошо подходят метод Вайберга (Wiberg bond order) [13] и метод “нечетких” атомов (bond order for “fuzzy” atoms) [14], так как они мало зависимы от базисного набора, в отличие от довольно распространенного метода Майера (Mayer bond order) [15], и при этом могут описывать полярные связи. Данные методы анализа реализованы в программе Multiwfn [16].
В настоящей работе мы предприняли попытку использовать величину порядков связей в молекулах замещенных сульфоланов как меру их прочности и попытались найти оптимальный заместитель для повышения электрохимической устойчивости сульфолана.
МЕТОДИКА РАСЧЕТОВ
Квантово-химические расчеты проводили с использованием программного пакета Gaussian09 [17]. Поиск равновесных геометрических параметров и расчет энергетических характеристик сульфолана и всех замещенных продуктов выполняли методом TPSS/cc-pVTZ [18, 19]. Все рассчитанные структуры являются стационарными точками на поверхности потенциальной энергии (ППЭ), что доказано решением колебательной задачи. Для визуализации полученных данных использовали программы VMD [20]. Анализ волновых функций и расчет порядков связи методами Мейера, Вайберга и “ Fuzzy” проводили в программном комплексе MultiWFN [17].
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Нами были рассчитаны порядки связей как для моно-, так и для дизамещенных молекул сульфолана. Положения заместителей в молекуле сульфолана показаны на рис. 1. Слева изображены варианты монозамещенных производных, всего четыре структуры. Ввиду симметрии второго порядка в молекуле сульфолана, четырех вариантов достаточно для получения всех возможных комбинаций замещения в молекуле. Справа на рис. 1 изображены пары атомов и все возможные комбинации дизамещения в молекуле сульфолана, всего было рассмотрено 10 вариантов.
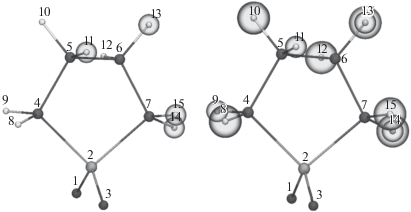
Влияние электроноакцепторных заместителей на порядки связей на примере моно- и дифторпроизводных сульфолана
В качестве электроноакцепторного заместителя был выбран фтор. На рис. 2 показаны результаты квантово-химических расчетов и анализа порядка связи O1–S2 в монофторпроизводных сульфолана. Слева представлены порядки связи, полученные тремя методами (Майера, нечетких атомов и Вайберга), а справа – их значения относительно незамещенного сульфолана.
Рис. 2.
Результаты расчетов порядка связи O1–S2 в ряду моно- и дифторпроизводных сульфолана. Справа показаны положения заместителей при наибольшем и наименьшем значении порядка связи O1–S2.
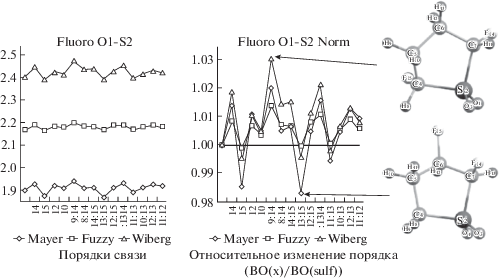
При замещении первого атома водорода на атом фтора наибольшее значение порядка связи O1–S2 соответствует положению 14, наименьшее – 15. При замещении двух атомов водорода диапазон порядка связи увеличивается. Максимальное значение порядка связи наблюдается в положениях 9; 14, а наименьшее в 13; 15. Аналогичным образом были проанализированы остальные связи и было установлено, что все три метода оценки порядка связей дают аналогичные профили, поэтому далее будет упоминаться только метод “Fuzzy” или метод оценки порядка связи для “нечетких” атомов.
В табл. 1 приведены относительные изменения порядков связей в процентах. Установлено, что все варианты ввода фтора в молекулу сульфолана имеют отрицательные суммарные значения. Для связи O3–S2 в ряду монофторпроизводных максимальное значение порядка связи соответствует положению атома фтора в 15 позиции, минимальное – в 14. При добавлении второго атома фтора диапазон значений порядка связи увеличивается. Определены соответствующие экстремумы: минимум – при 9; 14, максимум – при 13; 15.
Таблица 1.
Изменение порядков связей (%) во фторзамещенных сульфоланах относительно незамещенной молекулы
| Связь | – | 14 | 15 | 12 | 10 | 9; 14 | 8; 14 | 14; 15 |
|---|---|---|---|---|---|---|---|---|
| O1–S2 | 0.00 | 0.83 | –0.13 | 0.66 | 0.36 | 1.39 | 0.70 | 0.66 |
| O3–S2 | 0.00 | –0.45 | 0.75 | –0.30 | 0.26 | –0.32 | 0.70 | 0.47 |
| C4–S2 | 0.00 | 2.13 | –0.16 | 0.95 | 0.13 | –9.85 | –11.74 | 1.29 |
| C7–S2 | 0.00 | –11.39 | –10.83 | –1.11 | –0.56 | –11.72 | –11.74 | –21.74 |
| C4–C5 | 0.00 | –1.08 | –0.37 | –1.48 | –0.73 | –4.93 | –4.60 | –0.83 |
| C5–C6 | 0.00 | –0.41 | –1.04 | –5.02 | –5.14 | –1.36 | –0.50 | –0.72 |
| C6–C7 | 0.00 | –4.78 | –4.24 | –5.34 | –6.12 | –4.88 | –4.60 | –8.26 |
| Сумма | 0.00 | –15.14 | –16.01 | –11.64 | –11.79 | –31.67 | –31.78 | –29.13 |
| Связь | 13; 15 | 12; 15 | 13; 14 | 11; 13 | 10; 13 | 12; 13 | 11; 12 | диап. |
| O1–S2 | –0.05 | 0.80 | 1.07 | 0.03 | 0.56 | 0.89 | 0.56 | 1.52 |
| O3–S2 | 1.06 | 0.40 | 0.00 | 0.97 | 0.56 | 0.07 | 0.57 | 1.51 |
| C4–S2 | –0.17 | 0.39 | 1.60 | –1.55 | –0.09 | 0.69 | –1.36 | 13.87 |
| C7–S2 | –10.66 | –12.67 | –11.87 | –0.13 | –0.09 | –1.39 | –1.36 | 21.74 |
| C4–C5 | –1.34 | –1.47 | –1.66 | –5.48 | –6.54 | –1.88 | –5.02 | 6.54 |
| C5–C6 | –5.94 | –4.85 | –5.30 | –10.41 | –10.37 | –9.79 | –12.19 | 12.19 |
| C6–C7 | –10.90 | –11.30 | –10.85 | –7.17 | –6.54 | –10.94 | –5.03 | 11.30 |
| Сумма | –28.00 | –28.69 | –27.02 | –23.74 | –22.50 | –22.34 | –23.84 |
При оценке порядка связи C4–S2 (табл. 1) найдены следующие экстремумы: максимум в положении 14 и минимум в положении 15 для моно- фторпроизводных; максимум в положении 14; 15 и заметно выделяющийся минимум в положении 8; 14 для дифторпроизводных. Аналогичная ситуация наблюдается и для связи C7–S2 – найден выраженный минимум в положении 14; 15 для дифторзамещенных. Следует отметить, что ни один вариант замещения атомов водорода на атомы фтора в молекуле сульфолана не дает повышения порядка связи.
При оценке порядка связи С4–С5 определены следующие экстремумы: минимум в положении 12 для монофторзамещенных, минимум в положении 10;13 для дифторзамещенных. Для связи C5–C6 (табл. 1) найдены следующие экстремумы: минимум в положении 10 для моно- и в положении 11; 12 для дизамещенных. В случае связи C6–C7 наблюдается аналогичная картина: найдены минимумы в положениях 10 и 12; 15 для моно- и дизамещенных соответственно. Повышения значений порядка всех связей С–С в молекуле сульфолана не наблюдается. Так, судя по суммам изменений порядков связей (табл. 1, нижняя строка) введение фтора не делает молекулу растворителя прочнее, а из величин диапазона изменения порядков связей (табл. 1, правый столбец) следует, что прочность связи увеличивается в ряду C–S < C–C < O=S.
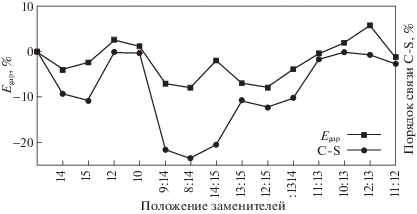
Так как разность энергий граничных орбиталей используется для оценки величины электрохимического окна, была проведена проверка адекватности подхода оценки электрохимической стабильности с помощью порядков связей. Сопоставлены относительные величины порядков связей и разности энергий граничных орбиталей (Egap), абсолютные значения которых были рассчитаны в данной работе. Наибольшая корреляция с различием энергий граничных орбиталей наблюдается у суммы порядков связей C–S. На рис. 3 приведена диаграмма, где представлена соответствующая зависимость. Коэффициент корреляции составляет 0.79. Таким образом, порядок связи можно использовать для проведения оценки электрохимической стабильности.
Влияние электронодонорных заместителей на порядки связей на примере моноалкил- и моноалкенил производных сульфолана
В качестве электронодонорных выбраны пять заместителей: метил, этил, пропил, винил и аллил. На данном этапе работы рассмотрены только монозамещенные структуры сульфолана. В табл. 2 приведены относительные порядки всех связей для всех вариантов монозамещенного сульфолана относительно незамещенной формы.
Таблица 2.
Изменение порядков связей (%) в алкил- и алкенилзамещенных сульфоланах относительно незамещенной молекулы
| Связь | – | Meтил | Этил | Пропил | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 8 | 11 | 13 | 15 | 8 | 11 | 13 | 15 | 8 | 11 | ||
| O1–S2 | 0.00 | –0.15 | –0.01 | –0.01 | –0.04 | –0.18 | –0.03 | –0.01 | –0.01 | –0.19 | –0.06 |
| O3–S2 | 0.00 | –0.48 | –0.18 | –0.03 | –0.59 | –0.46 | –0.23 | –0.09 | –0.88 | –0.47 | –0.17 |
| C4–S2 | 0.00 | –5.83 | 0.06 | –0.05 | 0.29 | –6.08 | 0.08 | 0.26 | 0.71 | –6.06 | –0.06 |
| C7–S2 | 0.00 | 0.24 | –0.10 | 0.16 | –6.24 | 0.35 | 0.08 | 0.23 | –6.19 | 0.31 | 0.14 |
| C4–C5 | 0.00 | –3.31 | –4.12 | –0.05 | –0.11 | –3.59 | –4.51 | –0.10 | –0.15 | –3.61 | –4.58 |
| C5–C6 | 0.00 | 0.11 | –4.20 | –3.95 | –0.19 | 0.11 | –4.29 | –4.36 | –0.42 | 0.12 | –4.27 |
| C6–C7 | 0.00 | –0.18 | –0.32 | –4.24 | –3.93 | –0.18 | –0.40 | –4.38 | –4.20 | –0.16 | –0.40 |
| Сумма | 0.00 | –9.59 | –8.86 | –8.17 | –10.81 | –10.03 | –9.29 | –8.44 | –11.14 | –10.07 | –9.40 |
| Связь | Пропил | Винил | Аллил | Диап. | |||||||
| 13 | 15 | 8 | 11 | 13 | 15 | 8 | 11 | 13 | 15 | ||
| O1–S2 | –0.07 | –0.05 | –0.02 | –0.21 | –0.05 | –0.60 | –0.35 | 0.03 | –0.12 | –0.02 | 0.63 |
| O3–S2 | –0.04 | –0.88 | –0.25 | –0.04 | 0.09 | –0.22 | –0.60 | –0.19 | 0.05 | –0.68 | 0.97 |
| C4–S2 | 0.11 | 0.59 | –8.81 | –0.33 | –0.09 | 0.03 | –6.19 | 0.08 | –0.15 | 0.37 | 9.53 |
| C7–S2 | 0.38 | –6.26 | –0.05 | 0.61 | 0.11 | –5.12 | 1.34 | 0.00 | 0.38 | –6.37 | 7.72 |
| C4–C5 | –0.07 | –0.10 | –4.37 | –4.38 | –0.13 | –0.56 | –3.64 | –4.33 | –0.13 | –0.16 | 4.53 |
| C5–C6 | –4.34 | –0.36 | 0.01 | –6.28 | –4.32 | –0.63 | 0.02 | –4.59 | –4.30 | –0.18 | 6.39 |
| C6–C7 | –4.42 | –4.15 | –0.09 | –0.66 | –6.02 | –6.48 | –0.58 | –0.41 | –4.58 | –4.35 | 6.39 |
| Сумма | –8.45 | –11.22 | –13.58 | –11.29 | –10.41 | –13.58 | –10.00 | –9.41 | –8.85 | –11.38 | |
Практически все положения метилового заместителя в молекуле сульфолана приводят к понижению порядков связей. Наибольшее влияние введение заместителя оказывает на связи C–S. Для моноэтилпроизводных сульфолана общая тенденция аналогична монометилпроизводным, наибольший эффект понижения порядка связи наблюдается для связей C4–S2 и C7–S2. Для пропилпроизводных закономерность остается аналогичной предыдущим двум случаям. Для винилпроизводных сульфолана наименее чувствительными к замене атомов оказались связи O–S, а связи C–S и С–С ослабляются в равной мере. Заметного увеличения порядка связей не обнаружено. В ряду аллилпроизводных сульфолана закономерности схожи с наблюдаемыми для алкилпроизводных: наименьшее изменение порядков связей наблюдается для связей O–S, а наибольшее − для связей C–S и С–С.
Так же, как и в случае введения электроноакцепторного заместителя, электронодонорные заместители не оказывают положительного эффекта (табл. 2, нижняя строка) на стабильность молекулы растворителя. Величины диапазонов изменения порядков связей в случае введения электронодонорных заместителей меньше по сравнению с электроноакцепторными заместителями, но ряд относительной прочности связей остается таким же, она уменьшается в ряду O=S > C–C > C–S (табл. 2, правый столбец).
Проведенные расчеты порядков связей показали, что самой слабой в молекуле сульфолана является связь C–S. Введение различных заместителей не позволяет существенно увеличить ее прочность, в самом лучшем случае – при β-дизамещении водорода на фтор порядок связи C–S увеличивается всего на 2.5%, при этом суммарный эффект остается отрицательным. Замещение водорода фтором в молекуле сульфолана понижает порядок ближайших к фтору связей, но повышает порядок отдаленных связей. Наибольшее понижение порядка связи C–S на 21% наблюдается в случае двойного альфа-замещения, а наибольшее повышение на 2.5% – в случае бета-замещения.
Для связи O–S порядок повышается на 2–3%, если фтор не находится в той же плоскости над кольцом, и понижается на 2.5%, если находится. Для связи C–C происходит только понижение порядков ближайших связей как электронодонорными группами, так и электроноакцепторными максимум на 11–12% при двойном замещении на атом фтора. Углеводородные заместители понижают порядки связей на 4–6% при альфа-замещении и практически не влияют при бета-замещении.
Авторы выражают благодарность за помощь в проведении расчетов Ясько Андрею Сергеевичу.
Работа выполнена в рамках государственного задания по теме № AAAA-A20-120012090022-1 (В.С. Колосницын) и при финансовой поддержке РНФ (проект №17-73-20115) “Экспериментальные и теоретические исследования механизмов необратимых процессов в литий-серных аккумуляторах” (Э.М. Хамитов, Е.В. Кузьмина, Е.В. Карасева).
Список литературы
Chen S., Wen K., Fan J. et al. // J. Mater. Chem. A. 2018. V. 6. I.25. P. 11631. https://doi.org/10.1039/C8TA03358G
Жоу Ж.-Ф., Куи К.-Л., Жан Х.-М. и др. // Электрохимия. 2017. Т. 4, С.399. https://doi.org/10.7868/S0424857017040144
Shao N., Sun X.-G., Dai S. et al. // J. Phys. Chem. B. 2011. V. 115. I. 42. P. 12120. https://doi.org/10.1021/jp204401t
Wu F., Zhou H., Bai Y. et al. // ACS Appl. Mater. Interfaces. 2015. V. 7. I. 27. P. 15098. https://doi.org/10.1021/acsami.5b04477
Kolosnitsyn V.S., Sheina L.V., Mochalov S.E. // Russ. J. Electrochem. 2008. V. 44. I. 5. P. 575. https://doi.org/10.1134/S102319350805011X
Sheina L.V., Kuz’mina E.V., Karaseva E.V. // Russ. J. Appl. Chem. 2018. V. 91. I. 9. P. 1427. https://doi.org/10.1134/S1070427218090045
An Y-H., Yang D.K., Lee C-H. et al. // Non-Aqueous Electrolyte Solution for Lithium Secondary Battery and Lithium Secondary Battery Comprising Same. 2012. EP2755272.
Shao N., Sun X.-G., Dai S. et al. // J. Phys. Chem. B. 2012. V. 116. I. 10. P. 3235. https://doi.org/10.1021/jp211619y
Borodin O., Olguin M., Spear C.E. et al. // Nanotechnology. 2015. V. 26. I. 35. P. 354003. https://doi.org/10.1088/0957-4484/26/35/354003
Borodin O., Ren X., Vatamanu J. // Acc. Chem. Res. 2017. V. 50. I. 12. P. 2886. https://doi.org/10.1021/acs.accounts.7b00486
Lu T., Chen F. // J. Phys. Chem. A. 2013. V. 117. I. 14. P. 3100. https://doi.org/10.1021/jp4010345
Jenkins H.O. // J. Am. Chem. Soc. 1955. V. 77. V. 11. P. 3168. https://doi.org/10.1021/ja01616a097
Wiberg K.B. // Tetrahedron. 1968. V. 24. I. 3. P. 1083. https://doi.org/10.1016/0040-4020(68)88057-3
Mayer I., Salvador P. // Chem. Phys. Lett. 2004. V. 383. I. 3–4. P. 368. https://doi.org/10.1016/j.cplett.2003.11.048
Mayer I. // Chem. Phys. Lett. 1983. V. 97. I. 3. P. 270. https://doi.org/10.1016/0009-2614(83)80005-0
Lu T., Chen F. // J. Comp. Chem. 2012. V. 33. I. 5. P. 580. https://doi.org/10.1002/jcc.22885
Gaussian 09, Revision C.01, M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J.A. Montgomery, Jr., J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J.M. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R.L. Martin, K. Morokuma, V. G. Zakrzewski, G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, O. Farkas, J.B. Foresman, J.V. Ortiz, J. Cioslowski, and D.J. Fox, Gaussian, Inc., Wallingford CT, 2010.
Tao J., Perdew J.P., Staroverov V.N. // Physical Review Letters. 2003. V. 91. I. 14. https://doi.org/10.1103/PhysRevLett.91.146401
Woon D.E., Dunning T.H. // J. Chem. Phys. 1993. V. 98. I. 2. P. 1358. https://doi.org/10.1063/1.464303
Humphrey W., Dalke A., Schulten K. // J. Mol. Graphics. 1996. V. 14. I. 1. P. 33. https://doi.org/10.1016/0263-7855(96)00018-5
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии