

Журнал физической химии, 2021, T. 95, № 6, стр. 825-837
Формирование и модифицирование металлооксидных подложек для контролируемых процессов адсорбции и превращения молекул на их поверхности
a Северо-Осетинский государственный университет им. К.Л. Хетагурова
Владикавказ, Россия
* E-mail: t_magkoev@mail.ru
Поступила в редакцию 01.08.2020
После доработки 01.08.2020
Принята к публикации 22.11.2020
Аннотация
Приведен обзор результатов изучения влияния свойств оксидов металлов и металлооксидных систем, образующихся при нанесении наноразмерных кластеров металлов на поверхности оксидов, на характер адсорбции и взаимодействия молекул (CO, CO2, NO, O2, H2, H2O) на их поверхности. Акцент сделан на исследования, проводимые в условиях сверхвысокого вакуума с достаточно высоким уровнем контроля чистоты и свойств in-situ формируемых систем. Проведен сравнительный анализ оксидов простых, переходных и редкоземельных металлов (Al2O3, MgO, TiO2, FeO, MoO3, CeO2), поскольку свойства нанесенного металла (в данном случае Au, Pt, Pd, Rh) и характер реализуемых адсорбционно-реакционных процессов в значительной мере определяется типом и состоянием оксидного адсорбента. Эффективность таких систем как катализаторов обусловлена сочетанием ряда параметров – свойствами нанесенного нанокластера металла, структурным, стехиометрическим, электронным состоянием оксида, характером взаимодействия на границе раздела металл/оксид. Показано, что наличие такого широкого спектра параметров позволяет осуществлять их подбор так, чтобы контролируемым образом настраивать активность и селективность катализатора.
За последние 35–40 лет наблюдается повышенный интерес к изучению свойств поверхности оксидов металлов и систем, образованных нанесением атомов, кластеров и тонких пленок металлов на поверхности оксидов металлов (металлооксидных систем). Это обусловлено тем, что поверхности таких систем обладают рядом уникальных электронных, адсорбционных и реакционных свойств, обусловленных многокомпонентностью их строения (наличие катионов и анионов в той или иной пропорции в зависимости от стехиометрии, их зарядового состояния), свойствами нанесенного наноразмерного кластера металла, которые могут существенно отличаться от свойств соответствующего массивного металла, особенностями межфазной границы раздела металл/оксид. В связи с этим оксидные и металлооксидные системы привлекают значительный интерес как подложки для изучения процессов адсорбции и превращения атомов и молекул на их поверхности – как модельные системы для дальнейшего развития фундаментальных представлений физики и химии поверхности, с одной стороны, и как прототипы гетерогенных катализаторов – с другой. Как следствие, результатом интенсивных исследований поверхностных свойств систем рассматриваемого типа стало появление ряда обзоров, опубликованных к началу 2000-х годов [1–6]. По мере развития данного направления акцент исследователей стал смещаться от изучения свойств оксидных и металлооксидных систем, формируемых тем или иным способом, к изучению способов их модифицирования с целью придания им определенных свойств, в частности, в плане реализации управляемых процессов адсорбции и превращения атомов и молекул на их поверхности. Данный аспект, однако, остается практически не освещенным в литературе. Лишь в последние годы стали появляться обзорные работы, касающиеся отдельных систем, например, на основе СеО2, FeO, TiO2 [7–10]. В связи с этим в настоящей работе приведен обзор результатов этих и других исследований по данной теме, опубликованных к настоящему времени. Основное внимание уделено системам, формируемым в условиях сверхвысокого вакуума, для уменьшения неконтролируемого влияния чужеродных частиц и процессов на свойства исследуемых систем. Это позволяет в наибольшей степени установить фундаментальные физико-химические закономерности их формирования и свойства. Преимущественно рассмотрены оксиды TiO2, Al2O3, CeO2, FeO, MgO, MoO3, ZrO2, формируемые контролируемым образом в виде тонких пленок на поверхности металлических подложек. Это позволяет управлять структурой, стехиометрией, морфологией оксидных пленок, что имеет существенное влияние на процессы адсорбции и превращения молекул на их поверхности. Рассмотрены металлооксидные системы, образующиеся при нанесении атомов, кластеров и тонких пленок металлов, преимущественно благородных – Pt, Au, Pd, Rh – на поверхности указанных оксидов. Обобщены результаты исследования процессов их формирования, адсорбции, взаимодействия и взаимного превращения на их поверхности молекул СО, NO, O2, H2, CO2, H2O. Основной акцент сделан на выявление взаимосвязи между состоянием поверхности оксидной и металлооксидной системы – атомной структуры оксида, его стехиометрии, природы и плотности дефектов, электронного состояния, размера и морфологии нанесенных частиц металлов – и характером адсорбции и взаимного превращения молекул на их поверхности. Рассмотрены различные способы формирования оксидных и металлооксидных систем и способов их модифицирования, в частности, за счет ионной и электронной бомбардировки, светового воздействия, термической обработки. Приведены методы анализа, используемые при исследовании систем и процессов рассматриваемого типа. На основе анализа существующих результатов изучения взаимосвязи характера адсорбции и превращении молекул от состояния поверхности оксидных и металлооксидных подложек обозначены перспективные пути дальнейших исследований в этом направлении.
МЕТОДИЧЕСКИЕ ОСОБЕННОСТИ ИССЛЕДОВАНИЯ АДСОРБЦИОННО-РЕАКЦИОННЫХ СВОЙСТВ МЕТАЛЛООКСИДНЫХ СИСТЕМ В УСЛОВИЯХ СОХРАНЕНИЯ АТОМНОЙ ЧИСТОТЫ
Поскольку адсорбционно-реакционные процессы атомов и молекул происходят на поверхности материала, подавляющее большинство методов, используемых в исследованиях данного типа, – методы анализа поверхности, реализующиеся, как правило, в условиях сверхвысокого вакуума [11]. Большинство этих методов основано на использовании пучков заряженных частиц и фотонов или на нагреве поверхности. Это накладывает определенные методические ограничения на материалы, обладающие низкой электро- и теплопроводностью, например, оксиды металлов. Для преодоления этих ограничений оксиды металлов формируют в виде тонких пленок на проводящих металлических подложках [2, 5, 6]. Обоснование такого широко используемого подхода – то, что такие пленки, толщиной в пределах единиц нанометров, обладают свойствами, близкими к свойствам соответствующих массивных оксидов [2, 6]. Для изучения процессов адсорбции и превращения молекул на поверхности оксидных и металлооксидных систем используются практически все известные к настоящему времени методы анализа поверхности. Методы, преимущественно используемые для исследования свойств самих подложек:
– электронная оже-спектроскопия (ЭОС), рентгеновская фотоэлектронная спектроскопия (РФЭС) для изучения элементного и химического состояния;
– ультрафиолетовая фотоэлектронная спектроскопия (УФЭС) и спектроскопия характеристических потерь энергии электронов (СХПЭЭ) для изучения электронного состояния валентной зоны;
– сканирующая туннельная микроскопия и спектроскопия (СТМ, СТС), атомно-силовая микроскопия (АСМ), дифракция медленных электронов (ДМЭ), спектроскопия обратного рассеяния ионов низкой энергии (СОРИНЭ) для изучения атомной структуры, морфологии поверхности, локальной плотности состояний.
Все приведенные выше методы основаны на использовании высокоэнергетических пучков заряженных частиц и фотонов, в связи с чем применение их к таким “деликатным” системам как молекулы газов (CO, NO, O2, H2 и др.) может приводить к неконтролируемому изменению их свойств или разрушению. Для таких систем применяются “мягкие” и в то же время эффективные методы анализа такие как инфракрасная фурье-спектроскопия (ИКС), спектроскопия комбинационного рассеяния (СКР), спектроскопия энергетических потерь высокого разрешения (СЭПВР). Эти методы основаны на использовании световых сигналов или электронных лучей низкой энергии и интенсивности и, являясь неразрушающими, позволяют получать информацию о состоянии адсорбированных молекул и характере их взаимодействия с высокой степенью точности и разрешающей способности. Кроме того, в сочетании с методами АСМ возможно, наряду с этим, достичь высокой пространственной локальности анализа (TERS). Для повышения качества исследований часто используются фотоэлектронные исследования с использованием синхротронного излучения, а также резонансные ядерные реакции, в частности, 15N + 1H – 12C + + 4He + γ, как эффективный метод изучения адсорбции водорода и реакций с его участием [12]. Широко используются теоретические методы для изучения процессов роста оксидов и металлооксидных систем, механизмов дефектообразования природы и плотности дефектов, реконструкции поверхности, адсорбционно-реакционных свойств поверхности и границ раздела. Наиболее распространен метод функционала плотности (DFT + U) с использованием, в частности, Венского расчетного пакета из первых принципов (VASP). Тенденция последнего времени в исследованиях данного типа – объединение исследовательских групп из разных организаций и стран, обладающих взаимодополняющими друг друга методами; результаты, приведенные в литературе, как правило, получены с использованием большинства из них.
КОНТРОЛИРУЕМОЕ ФОРМИРОВАНИЕ И АДСОРБЦИОННО-РЕАКЦИОННЫЕ СВОЙСТВА ПЛЕНОК ОКСИДОВ МЕТАЛЛОВ
В последнее время изучению процессов формирования и свойств тонких пленок оксидов (1–2 нм) на поверхности металлических подложек уделяется большое внимание [2, 5, 6, 13]. При этом, несмотря на то, что такие пленки имеют свойства, близкие к свойствам соответствующих массивных оксидов, они в то же время обладают новыми уникальными особенностями, проявляемыми, в частности, в процессах адсорбции и взаимодействия молекул на их поверхности [14, 15]. Это обусловлено, с одной стороны, формированием особых структурных и электронных особенностей пленок пониженной размерности, с другой – формированием границы раздела металл/оксид. При этом свойства образующихся пленок и способы их контролируемого роста и модификации зависят от типа самого оксида. Один из наиболее исследованных в этом плане оксидов – оксид титана. Например, пленки различной стехиометрии, полученные реактивным испарением в атмосфере кислорода на поверхности Pt(111), демонстрируют разную атомную структуру, морфологию и электронные свойства [16]. На основе исследований методами СТМ, ДМЭ, РФЭС, РД и расчетов методом функционала плотности показано формирование нескольких нестехиометрических фаз TiOx (x < 2), обусловленных различным взаимным расположением ионов титана и кислорода и наличием упорядоченных массивов дефектов. Существенную роль при этом играет симметрия подложки. Расположение ионов Ti, находящихся в непосредственном контакте с Pt(111), повторяет симметрию подложки, в то время как за счет различий в величине постоянной решетки Pt и оксида титана в процессе дальнейшего роста пленки образуется так называемая бороздчатая морфология пленки. При этом размеры и направления бороздок определяются характером координационного окружения титана, при котором четырехкратно координированные ионы Ti обладают наибольшей величиной заряда. Контролируемое формирование той или иной структуры пленки оксида титана осуществляется посредством изменения режимов ее формирования – скорости потока напыляемых атомов титана, парциального давления кислорода в вакуумной камере, температуры подложки, длительности последующей термической обработки сформированной пленки. В свою очередь, состояние пленки определяет состояние формируемых на ее поверхности кластеров металлов, что позволяет настраивать свойства металлооксидной системы для целенаправленной реализации тех или иных адсорбционно-реакционных процессов на ее поверхности.
По аналогии с TiO2, одной из наиболее подходящих подложек для формирования пленки FeO является Pt(111). Один из распространенных способов формирования данной оксидной пленки в условиях сверхвысокого вакуума – напыление атомов Fe на подложку, поддерживаемую при комнатной температуре с последующим отжигом сформированной пленки при 1000 К в атмосфере кислорода при парциальном давлении 10-5 Торр в течение нескольких минут [17]. По данным СТМ, образующаяся пленка представляют собой двойной слой FeO с постоянной решетки 0.309 нм, слегка большей, чем у массивного кристалла (0.304 нм). При этом результаты рентгенодифракционных исследований указывают на то, что анионы кислорода занимают дырочные центры относительно нижележащего подслоя катионов, образующих двумерную решетку гексагональной симметрии [18]. Несоответствие постоянных решеток FeO (0.31 нм) и Pt(111) (0.277 нм) приводит к формированию достаточно большой элементарной ячейки пленки (2.6 нм), повернутой на 0.6 град относительно элементарной ячейки подложки. При этом в зависимости от режима роста пленки возможно формирование пленок, в которых ионы Fe могут находиться в разных адсорбционных центрах на поверхности Pt(111) – непосредственно над атомом Pt, в мостиковом центре, в тетраэдрических и октаэдрических пустотах [19, 20]. Это, в свою очередь, приводит к различию поверхностного потенциала оксидной пленки, определяя разный характер адсорбции и взаимодействия молекул на ее поверхности, что позволяет контролируемым образом настраивать реакционные свойства поверхности оксида [21]. При этом, в отличие от пленки TiO2, формируемая на поверхности Pt(111) пленка FeO обладает более “гладкой” морфологией с латеральной неоднородностью, не превышающей 0.01 нм.
В отличие от TiO2 и FeO, Al2O3 является оксидом простого (непереходного) металла, что определяет свои особенности пленки оксида алюминия. Один из наиболее распространенных способов получения сверхтонкой пленки оксида алюминия – окисление поверхности сплава Ni3Al(111) [22, 23]. Для получения сплошной упорядоченной пленки оксида проводят окисление поверхности сплава при экспозиции кислорода ~40 L при температуре подложки 1000 К с последующей термической обработкой в течение 5 мин. Хотя образующаяся при этом пленка оксида алюминия согласована с подложкой, ее структура довольно сложна. Элементарная ячейка пленки повернута относительно подложки на 23 град и представляет собой чередующееся сочетание анионов и катионов Al–O–Al–O, причем непосредственно на поверхности пленки их расположение практически сопланарно. Особенностью пленки является образование анионных вакансий в трехкратно-координированных центрах решетки, а также неупорядоченных точечных дефектов, которые играют ключевую роль в процессах формирования на поверхности оксида наноразмерных кластеров металлов и в адсорбционно-реакционных процессах.
Другой способ формирования пленки оксида алюминия – реактивное напыление металлического алюминия в атмосфере кислорода на поверхность тугоплавких подложек (W, Mo, Re), поддерживаемых при повышенной температуре [24]. Преимущество этого метода в том, что в зависимости от структуры используемой подложки, можно формировать соответствующую структуру пленки оксида алюминия α-Al2O3(1000), (01-10), γ‑Al2O3(111). Кроме того, в зависимости от соотношения парциального давления кислорода в процессе роста пленки и плотности потока напыляемых атомов алюминия, можно варьировать стехиометрию, природу и плотность дефектов оксида, что определяет ее реакционную эффективность как модельного катализатора. Недостаток данного способа формирования сверхтонкой пленки оксида алюминия – происходит окисление поверхности подложки кислородом, находящимся в вакуумной камере. Это кардинальным образом меняет термодинамику процесса роста пленки, поскольку удельная поверхностная энергия оксида существенно меньше, чем для чистого металла.
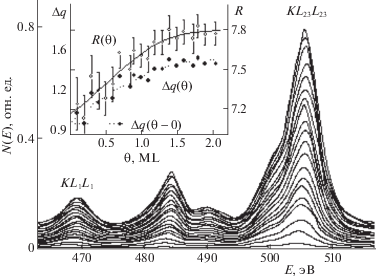
Для преодоления этого недостатка используют способ термического испарения массивного оксида алюминия, когда на поверхность металлической подложки доставляются уже частицы оксида AlxOy [25, 26]. По мере роста поверхностной концентрации частиц оксида алюминия на поверхности Мо(110) происходит плавное снижение работы выхода с 5.0 до 4.2 эВ при покрытии, соответствующем двум монослоям. Такое изменение работы выхода, в соответствии с дипольной моделью, можно приписать тому факту, что адсорбируемые частицы ориентируются на поверхности таким образом, что анион находится ближе к поверхности, а катион смещен в сторону вакуума. Предпочтительность такой ориентации может быть обусловлена тем, что, согласно расчетам, наиболее энергетически выгодные связи при взаимодействии оксида алюминия с переходным металлом, формируются между ионами кислорода и атомами металла [27]. Причем следует предположить, что эти связи являются достаточно прочными: десорбция оксида алюминия с поверхности Мо(110) начинается при температуре ∼1700 К. Формирование таких связей приводит к тому, что величина переноса заряда от катиона к аниону в частицах, непосредственно взаимодействующих с поверхностью подложки, отличается от таковой в объеме оксида. Для оценки абсолютной величины ослабления связи проведено исследование характера трансформации оже-линии кислорода в процессе роста покрытия оксида алюминия (Θ), начиная с субмонослойных значений (рис. 1). Для этого строилась концентрационная зависимость отношения интенсивностей оже-компонент KL23L23 и KL1L1, R(Θ), приведенная на вставке. С использованием соотношения, предложенного в работах [28], построены концентрационные зависимости величины связывающего заряда (Δq(Θ)) между Al и О, также приведенные на вставке. Видно, что с ростом покрытия величина Δq растет, стабилизируясь при покрытии, близком к 2 монослоям. Экстраполяция этой зависимости к нулевому покрытию (Θ → 0) позволяет оценить величину внутримолекулярной связи в одиночной частице оксида алюминия, адсорбированной на поверхности Мо(110), которая составляет ~0.9 (в единицах заряда электрона). Это означает, что эффективность межионной связи Alx+ – Oy– в такой частице на ~30% меньше, чем в объемном оксиде алюминия. По-видимому, подобное ослабление межионной связи, происходит вследствие того, что при адсорбции частицы оксида на поверхности формируются новые хемосорбционные связи за счет перераспределения электронной плотности с внутримолекулярных орбиталей на связи между адсорбатом и подложкой [27]. Другой возможностью, объясняющей ослабление внутримолекулярной связи при адсорбции полярных частиц, по мнению авторов [29], может быть попарное отталкивание ионов от своих зеркальных изображений в металле.
Рис. 1.
Последовательность оже-линий кислорода KLL в процессе роста пленки оксида алюминия термическим напылением на поверхности Мо(110), начиная с субмонослойных покрытий и до покрытия Θ = 2 ML. На вставке – зависимость отношения интенсивностей (R = I(KL23L23/I(KL1L1)) оже-переходов KL23L23 и KL1L1 от покрытия Θ, а также зависимость величины связывающего заряда Al-O от покрытия Δq(Θ) оксида алюминия на поверхности Мо(110).
Приведенные выше пленки оксидов являются неполярными, т.е. их поверхность не обладает макроскопическим дипольным моментом. В то же время именно полярные пленки оксидов могут представлять повышенный интерес как модельные катализаторы высокой эффективности. Их формирование вызывает, однако, серьезные затруднения, связанные со стремлением таких полярных поверхностей к релаксации. Вместе с тем, такие полярные поверхности могут быть сформированы в виде тонких пленок на поверхности ориентирующих металлических подложек соответствующей симметрии. Один из характерных примеров – пленка MgO(111) на поверхности Мо(110) [30, 31]. Данная поверхность кристалла MgO нестабильна вследствие некомпенсированного дипольного момента и релаксирует с образованием фасеток стабильной структуры MgO(100) [31]. В виде же тонкой пленки структура MgO(111) может быть сформирована на ориентирующей подложке с близкими параметрами решетки. В качестве такой подложки может быть использована поверхность кристалла Мо(110) [30–32]. В отличие от свойств неполярных граней MgO – (100), (110), адсорбционные свойства поверхности MgO(111) остаются практически неизученными, за исключением нескольких последних исследований [30, 33].
Другой оксид с довольно легко настраиваемой стехиометрией, который может быть хорошим кандидатом в качестве модельного катализатора, – оксид церия [7, 34]. Как и в предыдущих случаях, формирование пленок этого оксида в условиях сверхвысокого вакуума происходит в результате реактивного испарения металлического церия в атмосфере кислорода. На поверхности таких изоструктурных материалов как Ni(111), Pd(111), Pt(111), Rh(111) происходит формирование пленки СеО2 со структурой поверхности (111). Однако, в отличие от TiO2, FeO, Al2O3, MgO, пленка СеО2 менее стабильна. Это обусловлено большей реакционной способностью Се к образованию сплавов с подложкой, в результате чего происходит разложение СеО2, сопровождаемое диффузией кислорода в объем подложки или его десорбцией в газовую фазу. Восстановить исходную структуру СеО2 в ряде случаев можно за счет последующей экспозиции нестехиометрического оксида в атмосфере кислорода при повышенной температуре подложки. Помимо металлов, другой перспективной подложкой для формирования структурно совершенных пленок оксида церия, например, СеО2(100), служит SrTiO3. Это обусловлено близостью параметров решетки двух материалов с расхождением не более 2% при развороте подложки на 45 град, так что кристаллографические направления [011] и [001] становятся параллельными. Другая оксидная подложка для эпитаксиального роста пленки СеО2(111) – стабилизированный иттрием оксид циркония ZrO2(111) [34]. Несмотря на то, что расхождение параметров решетки в данном случае составляет 5.6%, формирование структурных несовершенств на границе раздела – дислокаций, вакансий, сдвигов, замещений – приводит к уменьшению этой величины до 1%. Довольно легкое изменение стехиометрии, структуры, морфологии пленки оксида церия делает этот материал перспективным в качестве катализатора высокой активности и селективности.
МОДИФИЦИРОВАНИЕ СОСТОЯНИЯ МЕТАЛЛООКСИДНЫХ СТРУКТУР КАК СПОСОБ УПРАВЛЕНИЯ ХАРАКТЕРОМ АДСОРБЦИОННО-РЕАКЦИОННЫХ ПРОЦЕССОВ НА ИХ ПОВЕРХНОСТИ
Как видно из изложенного выше, состояние поверхности пленок оксидов может быть изменено в широких пределах в зависимости от режимов их формирования. Кроме того, состояние уже сформированных оксидных пленок может быть далее контролируемым образом модифицировано за счет внешних воздействий – облучения электронами, ионами, светового воздействия различных длин волн, интенсивности и длительности, термической обработки. Это, в свою очередь, влияет на состояние нанесенных кластеров металлов и характер адсорбции и превращения молекул на их поверхности. Одной из наиболее характерных в этом плане систем – Au/TiO2 [35]. Андерсон и др., изучая рост кластеров Au на поверхности TiO2(110) до и после облучения ее ионам Не+, обнаружили, что в последнем случае кластеры в равновесном состоянии более сферически симметричны, и размер кластеров больше в 2–2.5 раза [36]. Это связывается с преимущественной нуклеацией кластеров вблизи анионных вакансий, образующихся в результате ионной бомбардировки оксида титана. На поверхности другой структурной модификации оксида титана TiO2(101), по данным СТМ, наблюдается различное поведение кластеров Au и Pt, осаждаемых на ее поверхность термическим напылением: золото образует кластеры больших размеров на границах ступенек, в то время как Pt формирует мономеры на террасах с довольно большой энергией связи – 2.2 эВ [37]. Модифицирование поверхности оксида электронной бомбардировкой существенно трансформирует состояние кластеров Au, сместив их в область образованных анионных вакансий, в то время как состояние частиц Pt не претерпевает заметных изменений.
На поверхности пленки СеО2(111) поведение Au и Pd близко к наблюдаемому для TiO2. Фройнт и др. показали, что Au и Pd при субмонослойных концентрациях преимущественно конденсируются на границах ступеней, и только после их заполнения происходит адсорбция на террасах [38]. После ионной бомбардировки, когда формируется довольно высокая плотность анионных вакансий, преимущественными центрами нуклеации частиц Au и Pd на поверхности СеОх (x < 2) служат F-центры, причем по мере роста поверхностной концентрации адатомов происходит рост размера связанных с этим центром кластеров. При этом особенностью для кластеров как Au, так и Pd, является формирование атомного упорядочения – структур (111) грани кластера, параллельной поверхности подложки, и (100) и (110) боковых фасеток. Сочетание таких атомных структур и границ раздела между ними в пределах одного наноразмерного кластера может иметь особое влияние на адсорбционно-реакционные характеристики молекул на его поверхности [7, 34].
В отличие от СеО2, энергия связи атомов Au, Pt, Pd, а также других металлов на поверхности MgO(100) и MgO(110) довольно незначительна, как правило, меньше 1 эВ [39]. Это приводит к тому, что осаждаемые атомы благородных металлов обладают повышенной латеральной подвижностью, вследствие чего, как правило, формируются эпитаксиальные покрытия Au(111)//MgO(001) и Au(110)//MgO(110). Модифицирование поверхности оксида электронной бомбардировкой приводит к кардинальному изменению механизмов роста благородных металлов: последние преимущественно формируют трехмерные кластеры вокруг анионных вакансий [40].
С точки зрения каталитической эффективности, наноразмерные частицы золота и других благородных металлов (Pt, Pd) на поверхности легко восстанавливаемых оксидов (TiO2, FeO, Cr2O3, CeO2) проявляют большую активность по сравнению со случаем трудно восстанавливаемых оксидов (MgO, SiO2, Al2O3) [41, 42]. В целом, сформировалась точка зрения, согласно которой, в первом случае реакции происходит по механизму Ленгмюра–Хинселвуда, во втором – по механизму Элея–Ридела. В обоих случаях ключевым остается вопрос, происходит ли, например, в реакции окисления СО, диссоциация кислорода на поверхности подложки, или же реакция происходит при взаимодействии молекул СО непосредственно с О2 [43]. Большинство расчетов сходится на том, что диссоциативная адсорбция О2 требует преодоления довольно существенного энергетического барьера, в то время как прямое взаимодействие соадсорбированных СО и О2 – более предпочтительный процесс [44]. Такое поведение связывается с инертным характером частиц Au (Pt, Pd), которые способны связывать адсорбаты, когда находятся в низкокоординированных состояниях. В этих состояниях возможна адсорбция молекул кислорода, сопровождаемая ослаблением внутримолекулярной связи О=О и последующим ее разрушением, инициированным дефектами подложки восстанавливаемого оксида. Один из параметров каталитической эффективности нанесенной на оксид частицы благородного металла (Au, Pd, Pt, Rh) – низкая энергия связи с адсорбатом, что способствует десорбции молекул с поверхности в газовую фазу, тем самым ослабляя эффект “отравления” катализатора. Другой ключевой фактор высокой каталитической эффективности рассматриваемых металлооксидных систем – наноразмерные частицы благородного металла, несмотря на свою инертность, могут приобретать ненулевой электронный заряд, величина которого регулирует активность и селективность катализатора. Кроме того, существенную роль в процессах адсорбции и превращения молекул играет и граница раздела между оксидом и нанесенным кластером металла [44–47]. Для изучения межфазной границы раздела металл/оксид и управления ее адсорбционно-реакционными свойствами часто используют так называемый подход инверсных металлооксидных катализаторов [48]. Его суть заключается в том, что металл и оксид “меняются местами”: сначала формируется пленка металла, на поверхность которой осаждаются кластеры соответствующего оксида. Это позволяет в наиболее непосредственном виде установить роль границы раздела металл/оксид в процессах адсорбции и превращения молекул. Такой подход позволил, например, установить, что именно граница раздела между оксидом и металлом (Au/TiO2, Pd/TiO2, Au/CeO2, Pt/Cr2O3, Pd/Fe2O3) играет ключевую роль в процессах окисления молекул СО, NO, H2 [44–48]. В частности, на основе измерений методами ИКС, РФЭС, СОРИНЭ, ЭОС, ДМЭ показано, что кластеры оксида титана со стехиометрией TiO2 могут быть сформированы на поверхности пленки золота с ориентацией поверхности (111) на подложке Мо(110) [45]. При этом, несмотря на то, что молекулы СО не адсорбируются как на Au(111), так и на TiO2 при 200 К, на поверхности сформированной системы TiO2/Au(111) происходит достаточно активная адсорбция молекул СО. Это указывает на то, что адсорбционные центры данной инверсной металлооксидной системы расположены на границе раздела металл/оксид. При этом концентрация адсорбированных молекул СО, как видно из спада интенсивности ИК-полосы поглощения СО, довольно интенсивно уменьшается при экспозиции системы CO/TiO2/Au(111) в атмосфере молекулярного кислорода (рис. 2). Активность этого процесса тем выше, чем меньше первоначальная концентрация молекул оксида углерода, что иллюстрируется на рис. 2. Это обстоятельство указывает на ключевую роль свободных реакционных центров на границе раздела металл/оксид и предлагает способ управления активностью рассматриваемого каталитического процесса.
Рис. 2.
Зависимости интенсивности ИК-сигнала СО от времени выдержки инверсной металлооксидной системы CO/TiO2/Au в атмосфере кислорода при различных первоначальных экспозициях СО (L): 1 – 100, 2 – 80, 3 – 60, 4 – 40. Угол наклона (α) этих кривых при начальных экспозициях О2 характеризует активность окисления СО σ = $\operatorname{tg} a$. На вставке – зависимость σ от первоначальной экспозиции СО.

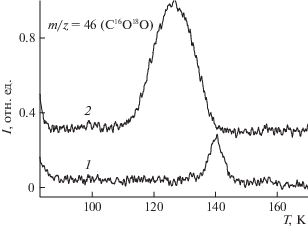
Помимо межфазной границы раздела, настройка зарядового состояния катионов металла оксида позволяет управлять характером адсорбционно-реакционных процессов. Показано, например, что система, образованная формированием нанокластеров Au на поверхности нестехиометрической пленки TiOx (x < 2), обладает существенно большей каталитической эффективностью в процессе окисления молекул СО, чем система Au/TiO2 [49]. Более того, как показано с использованием “меченых атомов” (изотопа 18О), в процессе окисления молекул СО участвует не только кислород, находящийся в газовой фазе, но и кислород, входящий в состав оксида титана, причем в большей степени в случае нестехиометрического оксида. Это иллюстрируется сопоставлением соответствующих термодесорбционных спектров, приведенных на рис. 3. Факт регистрации десорбционного пика C16O18О довольно однозначно свидетельствует в пользу прямого участия кислорода, входящего в состав оксида титана, полученного с использованием 18О, в процессе каталитического окисления СО на поверхности систем Au/TiO2 и Au/TiOx . Существенно большая его интенсивность в последнем случае указывает на большую эффективность системы Au/TiOx в данном процессе по сравнению с Au/TiO2, хотя и следовало бы предположить обратное, учитывая большую концентрацию кислорода в случае TiO2. Подобная закономерность имеет место и в случае непосредственного взаимодействия оксида углерода с системами Au/TiO2 и Au/TiOx; в последнем случае эффективность преобразования СО в CO2 выше [50]. По-видимому, в данном процессе решающую роль играет не столько величина абсолютной концентрации кислорода, сколько его электронное состояние как непосредственно в оксиде, так и на межфазовой границе металл/оксид. Кроме того, следует предположить, что взаимообмен кислородом между газовой фазой и оксидом титана играет существенную роль в процессе транспорта атомов кислорода на поверхности адсорбента к молекулам СО. В пользу такой возможности свидетельствует тот факт, что спектры РФЭС в области Ti 2p, весьма чувствительные к степени окисления Ti, идентичны до и после многократных циклов адсорбции–десорбции оксида и диоксида углерода. С учетом наблюдающегося при этом “ухода” кислорода оксида титана (18O) в виде C16O18O следовало бы ожидать изменения степени окисления оксида титана и соответственного изменения формы фотоэлектронной линии Ti 2p. Данное обстоятельство свидетельствует о том, что меньшая энергия связи и большая степень делокализации кислорода в случае TiOx по сравнению с TiO2 способствуют большей эффективности процесса окисления оксида углерода на поверхности системы Au/TiOx.
Рис. 3.
Термодесорбционные спектры частиц с массовым числом 46 (C16O18O), соответствующие системам CO/Au/TiO2 (спектр 1) и CO/Au/TiOx (x < 2) (спектр 2).
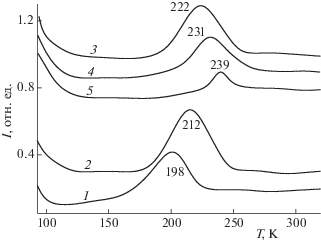
Настроить каталитическое поведение металлооксидной системы можно, придав оксидной подложке особую структуру. В качестве последних примеров можно привести результаты сравнительного исследования взаимодействия молекул СО, NO, Н2, О2 на поверхности пленок MgO(100) и MgO(111) [33, 45, 47]. С использованием методов УФЭС, ИКС, ДМЭ, ТДС показано, что интенсивность внутримолекулярной колебательной моды СО на поверхности MgO(111) существенно выше, чем на поверхности MgO(100), что связано с усилением динамического дипольного момента молекулы некомпенсированным электростатическим полем поверхности MgO(111). Это поле обусловливает взаимодействие СО и Н2 с образованием прекурсоров С–О–Н, приводящее к кардинальному ослаблению первоначальной колебательной интенсивности С=О. Существенно отличается и активность взаимодействия молекул СО, NO, O2 на поверхности Au/MgO(111) и Au/MgO(100). Причина такого различия в том, что частицы золота размером 2–3 нм, нанесенные на поверхность пленки MgO(111), приобретают эффективный положительный заряд в результате переноса электронной плотности в сторону подложки для стабилизации некомпенсированного дипольного момента полярной поверхности MgO(111). Такое состояние данной металлооксидной системы проявляется в более эффективном процессе окисления молекул СО, как кислородом, так и соадсорбированными молекулами NO, по сравнению с системой на основе неполярной поверхности MgO(100). Особенности, отличающие наноразмерные кластеры Au на поверхности MgO(111) от массивных металлических адсорбентов, проявляются, например, при совместной адсорбции молекул CO и NO. Это иллюстрируется данными рис. 4, на котором приведен инфракрасный фурье-спектр, соответствующий насыщающей экспозиции кислорода 100 L на поверхности системы CO/Au/MgO(111), поддерживаемой при температуре 90 K (спектр 3). Очевидная особенность при этом – сдвиг колебательного волнового числа СО от 2133 до 2140 см–1. Такой фиолетовый сдвиг свидетельствует о том, что соадсорбция кислорода приводит к понижению электронной плотности на разрыхляющей орбитали 2π* CO. Для объяснения такого сдвига волнового числа можно предположить, что происходит диссоциативная адсорбция кислорода, так что электронная плотность с орбитали 2π* смещается в сторону более электроотрицательного уровня О 2p. ИК-спектр, соответствующий системе, образованной адсорбцией СО на предварительно выдержанной в атмосфере NO до экспозиции 100 L системе Au/MgO(111), приведен на рис. 4 (вставка, спектр 1'). Наблюдаются две основные линии поглощения в спектральных областях, соответствующих внутримолекулярным колебаниям NO (1520 см–1) и СО (2040 см–1), причем для СО наблюдается фиолетовый сдвиг по сравнению с величиной, характерной для адсорбции на поверхности кристаллов золота различной атомной структуры [51, 52]. Последнее может быть связано с эффективным положительным зарядом нанесенных на MgO(111) наноразмерных кластеров золота, снижающим эффект заполнения разрыхляющей молекулярной 2π*-орбитали. Для NO, наоборот, частота молекулярных колебаний, хотя и в меньшей степени, но смещена в красную область. Такое различие может быть связано с различием кратности связи в молекулах СО и NO и ее трансформацией при хемосорбции [53]. Прогрев системы усиливает данную тенденцию, приводя к дальнейшему красному сдвигу частоты колебаний NO, сопровождающему процесс ослабления внутримолекулярной связи и, в конечном итоге, к диссоциации молекулы. Выделяющийся при этом кислород, как и в случае (СО + + О2), приводит к окислению соадсорбированных молекул СО. Об этом свидетельствует соответствующий термодесорбционный спектр, основной сигнал которого соответствует m/z = 44 (СО2) (рис. 5, спектр 3). Такая же ситуация имеет место и для кластеров Au близкой морфологии на поверхности MgO(100), однако в этом случае, как видно из соответствующего термодесорбционного спектра (рис. 5, спектр 4), эффективность окисления заметно меньше. Такое различие может быть связано с разным состоянием частиц Au на поверхности MgO(111) и MgO(100), а также с более эффективным процессом диссоциации NO за счет более активной аккомодации образующихся атомов N как реализации механизма стабилизации полярной поверхности MgO(111). Последнее, в свою очередь, может приводить к изменению состояния частиц Au на поверхности и, как следствие, к снижению эффективности процесса окисления СО за счет молекул NO. Об этом свидетельствуют, в частности, довольно заметные сигналы СО в термодесорбционном (рис. 5, спектр 5) и инфракрасном (рис. 4, вставка, спектр 2') спектрах, сохраняющиеся после реализации процесса взаимодействия (NO + CO).
Рис. 4.
Инфракрасные фурье-спектры в области внутримолекулярных колебаний молекул СО, адсорбированных на поверхности систем Au/MgO(100) (1), Au/MgO(111) (2) и после адсорбции кислорода на поверхность системы CO/Au/MgO(111) (3). Во всех случаях адсорбция молекул газов осуществлялась при температуре подложки 90 К. Экспозиция СО и О2 составляет 50 и 100 L соответственно. На вставке – ИК-спектры, соответствующие экспозиции СО на предварительно выдержанной в атмосфере NO до экспозиции 100 L системе Au/MgO(111) (спектр 1') при 90 К. Спектр 2' соответствует этой же системе после ее отжига до температуры 300 К.

Рис. 5.
Термодесорбционные спектры СО2 (m/z = 44), соответствующие системам (О2 + СО) на поверхности MgO(100) (1) и MgO(111) (2), зарегистрированные при поддержании парциального давления кислорода на уровне 10–6 Торр и скорости роста температуры образца 2 К/с (3–5); термодесорбционные спектры СО2, соответствующие системам, образованным адсорбцией 100 L СО на предварительно выдержанных в атмосфере NO до экспозиции 100 L системам Au/MgO(111) (спектр 3) и Au/MgO(100) (спектр 4), поддерживаемым при температуре 90 К. Спектр 5 соответствует m/z = 28 (СО), сопровождающему сигнал СО2 для системы Au/MgO(111) (спектр 3).
Другой важный молекулярный адсорбат, участвующий в каталитических процессах, имеющих как фундаментальное, так и прикладное значение, – водород Н2. Особенность этой молекулы как адсорбата для оксидных материалов в том, что она вызывает довольно активные процессы восстановления подложек, в свою очередь, влияя на их каталитическую активность и селективность. В наибольшей степени это относится к легко восстанавливаемым оксидам – оксидам переходных и редкоземельных металлов. При этом следует отметить, что в случае “идеальных” структурно упорядоченных и стехиометрических оксидов, например, СеО2(111), СеО2(110), существует достаточно большой активационный барьер адсорбции [54]. На поверхности с пониженной стехиометрией и наличием структурных дефектов, водород адсорбируется с образованием гидроксильных групп ОН, которые, соединяясь с адсорбируемым водородом, образуют молекулы воды, десорбируемые в газовую фазу при температуре >200 К [55]. В отличие от Н2 адсорбция воды (Н2О) на поверхности оксида церия приводит как к молекулярной, так и диссоциативной адсорбции, а также к сочетанию обоих типов адсорбции одновременно [56, 57]. Впрочем, на данный момент в этом вопросе нет единой точки зрения: с одной стороны, считается, что наиболее предпочтительна молекулярная адсорбция воды на поверхности структурно упорядоченных стехиометрических оксидов СеО2(111), СеО2(110), с другой – диссоциативная [7]. Более однозначная позиция существует в отношении нестехиометрических оксидов церия: практически для всех исследованных структур поверхности преимущественно реализуется диссоциативная адсорбция (Н2О → Н + ОН). Данное обстоятельство определяет высокую эффективность нестехиометрических пленок оксида церия в качестве катализаторов реакции водяного газа.
Оксидом, каталитические свойства поверхности которого также могут быть настроены в широких пределах, является оксид молибдена. Существует несколько подходов формировать оксиды молибдена в виде тонких пленок на поверхности подложек в условиях сверхвысокого вакуума и контролируемым образом вводить в них дефекты. В одном из них пленка МоО3 формируется на поверхности Au(111) последовательным нанесением Мо(СО)6 и NO2 при температуре подложки 450 К [58]. При этом, согласно результатов ДМЭ, СТМ, РФЭС, СЭПВР, образуется стехиометрическая упорядоченная пленка МоО3. Показано, что термическая обработка такой пленки при 650 К приводит к появлению ионов Мо5+ и кислородных вакансий. При этом концентрация указанных ионов повышается с увеличением длительности отжига до достижения равного соотношения концентрации катионов Мо5+ и Мо6+. Формирования катионов Мо с меньшей степенью окисления, например, Мо4+ (МоО2), не наблюдалось. Восстановленная пленка МоОх сохраняет двумерный характер, как и исходная стехиометрическая пленка МоО3, что связывается с достаточно существенным барьером структурной реконструкции оксида, объясняющим отсутствие ионов Мо с меньшей, чем +5 степенью окисления. На основе данных СТМ и теоретических расчетов делается вывод, что формирование ионов Мо5+ в процессе термической обработки связано не с появлением анионных точечных вакансий, а с образованием кристаллографического сдвига. В отличие от приведенных результатов, Chang и др. при изучении системы МоО3/Au(111), полученной аналогичным способом, наблюдали появление ионов Мо4+ при термической обработке пленки МоО3 [59]. Авторы также продемонстрировали пассивность упорядоченной стехиометрической пленки МоО3 в качестве возможного модельного катализатора диссоциации/окисления этилена.
Другой способ формирования пленки оксида молибдена – окисление кристалла Мо(110) в атмосфере кислорода при парциальном давлении 10–6 Торр и температуре подложки 1300 К [60]. При этом образуется структура, соответствующая МоО2(010), и от пленки МоО3 на поверхности Au(111) ее отличает высокая стабильность, обусловленная сильным взаимодействием на межфазной границе раздела МоО2/Мо(110), характеризуемой энергией адгезии ~7 Дж/м2. Согласно данным СТМ и ДМЭ, упорядоченная пленка МоО2 ориентирована на поверхности Мо(110) таким образом, что направление МоО2[201] параллельно направлению Мо[1–1–3], а направление МоО2[100 ] – направлению Мо[00–1]. Пленка оксида искажена на 4.9% относительно структуры подложки. Результаты расчетов свидетельствуют о том, что ион кислорода на межфазной границе находится в трехкоординированном положении. Пленка МоО2 может быть сформирована до достаточно больших толщин, хотя наиболее стабильна тонкая пленка, находящаяся в непосредственном контакте с подложкой в силу указанной заметной величины энергии адгезии. Последняя обусловлена существенным перераспределением заряда в приповерхностной области металл/оксид. Особенности формирования кислородных вакансий пленки МоО2 и их влияние на процесс восстановления молекул диоксида углерода исследованы с использованием бомбардировки ионами аргона и фотоэлектронных измерений [28]. Показано, что бомбардировка ионами аргона довольно эффективно формирует анионные вакансии, концентрацию которых можно контролируемым образом менять длительностью, дозой облучения и энергией ионов. В отличие от стехиометрической пленки МоО2, пленка с анионными вакансиями, как видно из серии фотоэлектронных спектров (рис. 6), служит довольно эффективной подложкой для фотоиндуцированного преобразования молекул углекислого газа и воды в метанол последовательностью циклов CHOOH–CH2O–CH3O–CH3OH. При этом данный процесс наиболее эффективен только при определенной поверхностной концентрации анионных вакансий (~25%), необходимой для того, чтобы происходила аккомодация образующихся в процессе реакции H2O → H+ + OH– гидроксид-ионов стехиометрической подложкой, что возможно за счет формирования оксигидроксид-ионов металла MoO(OH)–. Возможность настройки стехиометрии, структуры, зарядового состояния катионов Мох+ позволяет настраивать селективность катализатора. Несмотря на то, что оксид молибдена преимущественно является катализатором окисления за счет низкой энергии его восстановления, при определенной настройке он может выступать в роли не менее активного катализатора гидрирования ненасыщенных углеводородов. Высокий потенциал использования оксида молибдена в качестве катализатора гидрирования алкенов продемонстрирован недавно с использованием оксида пористой структуры в качестве модельного катализатора [61]. Это связывается с возможностью достижения высокой концентрации атомарного водорода в приповерхностной области оксида. Предполагается достижение еще большей эффективности гидрирования алкенов в случае нестехиометрического оксида за счет повышения плотности растворенного водорода и достижение особого состояния атомов водорода, расположенных в анионных вакансиях.
Рис. 6.
Ультрафиолетовые фотоэлектронные спектры Мо (110) (кривая 1), Мо(110) после экспозиции в атмосфере СО2 и Н2О при 95 К при суммарном парциальном давлении 10–6 Торр и воздействии лазерных импульсов с энергией 6.4 эВ в течение 5 мин (кривая 2). Для наглядности приведено сопоставление разностного спектра (2–1) (кривая 3) со спектром конденсированной на поверхности МоОх пленки метанола (кривая 4).

На основе теоретических расчетов адсорбции и гидрирования этилена на поверхности оксида молибдена показано, что наиболее предпочтительна наклонная геометрия адсорбции молекулы на поверхности с предадсорбированными атомами водорода в результате взаимодействия π‑электрона этилена и протона [62]. Довольно слабое взаимодействие этилена с подложкой не приводит к существенной активации внутримолекулярных связей, что обеспечивает одностадийный процесс гидрирования, в отличие от случая адсорбции и реакции на металлических подложках, на которых возмущение молекулы за счет образования сильных хемосорбционных связей с подложкой приводит к появлению сторонних реакций. Рассчитанные энергетические барьеры гидрирования на поверхности оксида сопоставимы или меньше наблюдаемых для Pt(111), когда процесс гидрирования этилена является двухстадийным.
ЗАКЛЮЧЕНИЕ
Оксиды металлов и системы, образованные нанесением на поверхность таких оксидов наноразмерных кластеров металлов, – широко и подробно исследуемые материалы, представляющие повышенный интерес как с фундаментальной, так и с прикладной точки зрения, в частности, в качестве систем, моделирующих каталитические процессы на поверхности металлооксидных систем. Эффективность таких систем как катализаторов обусловлена сочетанием ряда параметров – свойствами нанесенного нанокластера металла, часто кардинально отличающимися от свойств соответствующих “свободных” кластеров, вследствие взаимодействия с оксидной подложкой, структурным, стехиометрическим, электронным состоянием оксида, характером взаимодействия на границе раздела металл/оксид, изменением состояния системы при адсорбции и взаимодействии на ее поверхности атомов и молекул. Наличие такого широкого набора параметров позволяет подбирать их таким образом, чтобы контролируемым образом настраивать активность и селективность катализатора. Наибольшей гибкостью в этом плане обладают системы на основе оксидов с легко изменяемой стехиометрией – оксидов переходных и редкоземельных металлов. Варьируя стехиометрию за счет подбора режимов формирования таких оксидов, их обработки потоками заряженных частиц и фотонов, можно контролируемым образом менять их атомную структуру, электронное состояние катионов и анионов, вносить дефекты и, таким образом, управлять характером адсорбции и взаимодействия молекул на поверхности соответствующих металлооксидных систем. В основе такой взаимосвязи лежит высокая чувствительность адсорбционно-реакционных свойств поверхности к деталям ее электронного и атомного строения. Современные тенденции развития исследований в этом направлении таковы, что в дальнейшем следует ожидать переход к более тонкой настройке состояния поверхности адсорбентов. В качестве таковых наиболее многообещающими являются именно металлооксидные системы, вследствие возможности подбора большего сочетания контролируемо подстраиваемых параметров, в частности, особых структурных состояний тонких пленок оксидов, формирования точно выверенных одно-, двух- и трехмерных сверхструктур металлов, межфазных границ раздела.
Тенденции развития исследований в данной области таковы, что в дальнейшем следует ожидать реализации более тонкой подстройки параметров металлооксидных систем на уровне отдельных атомов для управления характером адсорбции и взаимодействия молекул на их поверхности.
Список литературы
Henrich V.E. // Progr. Surf. Sci. 1995. V. 50. P. 77. https://doi.org/10.1016/0079-6816(95)00046-1
Campbell C.T. // Surf. Sci. Rep. 1997. V. 27. P. 1. https://doi.org/10.1016/S0167-5729(96)00011-8
Henry C.R. // Surf. Sci. Rep. 1998. V. 31. P. 231. https://doi.org/10.1016/S0167-5729(98)00002-8
Bonnel D.A. // Progr. Surf. Sci. 1998. V. 57. P. 187. https://doi.org/10.1016/S0079-6816(98)00016-1
Bauemer M., Freund H.-J. // Progr. Surf. Sci. 1999. V. 61. P. 127.https://doi.org/10.1016/S0079-6816(99)00012-X
Chambers S.A. // Surf. Sci. Rep. 2000. V. 39. № 1. P. 105. https://doi.org/10.1016/S0167-5729(00)00005-4
Ma Y., Gao W., Zhang Z. et al. // Surf. Sci. Rep. 2018. V. 73. № 1. P. 1−36.https://doi.org/10.1016/j.surfrep.2018.02.001
Chen S., Xiong F., Huang W. // Surf. Sci. Rep. 2019. V. 74. № 4. P. 100471. https://doi.org/10.1016/j.surfrep.2019.100471
Picone A., Riva M., Brambilla A. et al. // Surf. Sci. Rep. 2016. V. 71. P. 32.https://doi.org/10.1016/j.surfrep.2016.01.003
Parkinson G.S. // Surf. Sci. Rep. 2016. V. 71. P. 272. https://doi.org/10.1016/j.surfrep.2016.02.001
Yates J.T., Jr. Experimental Innovations in Surface Science: a Guide to Practical Laboratory Methods and Instruments. New York: AIP, 1998. 904 p. (ISBN 978-3-319-17668-0).
Wilde M., Fukutani K. // Surf. Sci. Rep. 2014. V. 69. P. 196.https://doi.org/10.1016/j.surfrep.2014.08.002
Honkala K. // Surf. Sci. Rep. 2014. V. 69. P. 366. https://doi.org/10.1016/j.surfrep.2014.09.002
Fu Q., Wagner T. // Surf. Sci. Rep. 2007. V. 62. P. 431. https://doi.org/10.1016/j.surfrep.2007.07.001
Магкоев Т.Т., Григоркина Г.С., Заалишвили В.Б. и др. // Поверхность. Рентгеновские, синхротронные и нейтронные исследования. 2020. № 6. С. 1. https://doi.org/10.31857/S1028096020060138
Wu Q.H., Fortunelli A., Granozzi G. // Int. Rev. Phys. Chem. 2009. V. 28. P. 517. https://doi.org/10.1080/01442350903172453
Galloway H.C., Sautet P., Salmeron M. // Phys. Rev. B. 1996. V. 54. P. R11145.https://doi.org/10.1103/physrevb.54.r11145
Shaikhutdinov S.K., Meyer R., Lahav D. et al. // Phys. Rev. Lett. 2003. V. 91. P. 076102.https://doi.org/10.1103/PhysRevLett.91.076102
Ritter M., Ranke W., Weiss W. // Phys. Rev. B. 1998. V. 57. P. 7240. https://doi.org/10.1103/PhysRevB.57.7240
Kim Y.J., Westphal C., Ynzunza R.X. et al. // Phys. Rev. B. 1997. V. 55. P. R13448. https://doi.org/10.1103/PhysRevB.55.R13448
Rienks E., Nilius N., Rust H.P., Freund H.-J. // Phys. Rev. B. 2005. V. 71. P. 241404. https://doi.org/10.1103/PhysRevB.71.241404
Bardi U., Atrei A., Rovida G. // Surf. Sci. 1990. V. 239. P. L511. https://doi.org/10.1016/0039-6028(90)90607-A
Degen S., Krupski A., Kralj M. et al. // Surf. Sci. 2005. V. 576. P. L57. https://doi.org/10.1016/j.susc.2004.12.020
Chen P.J., Goodman D.W. // Surf. Sci. 1994. V. 312. № 3. P. L767. https://doi.org/10.1016/0039-6028(94)90719-6
Magkoev T.T., Christmann K., Moutinho A.M.C., Murata Y. // Surf. Sci. – 2002. V. 515. P. 538. https://doi.org/10.1016/S0039-6028(02)01972-6
Magkoev T.T., Vladimirov G.G. // J. Phys.: Condens. Matter. 2001. V. 13. P. L655. https://doi.org/10.1088/0953-8984/13/28/101
Kelber J.A., Niu C., Shepherd K., Jennison D.R., Bogicevic A. // Surf. Sci. 2000. V. 446. № 1/2. P. 76. https://doi.org/10.1016/S0039-6028(99)01089-4
Weissmann R. // Solid State Commun. 1979. V. 31. № 5. P. 347. https://doi.org/10.1016/0038-1098(79)90553-2
Efrima S., Metiu H. // Surf. Sci. 1981. V. 108. № 1. P. 329. https://doi.org/10.1016/0039-6028(81)90453-2
Grigorkina G.S., Ramonova A.G., Kibizov D.D. et al. // Solid State Commun. 2017. V. 257. № 1. P. 16. https://doi.org/10.1016/j.ssc.2017.03.014
Xue M., Guo Q. // J. Chem. Phys. 2007. V. 127. № 8. P. 054705. https://doi.org/10.1063/1.2756831
He J.-W., Estrada C.A., Corneille J.S. et al. // Surf. Sci. 1992. V. 261 № 1. P. 164. https://doi.org/10.1016/0039-6028(92)90228-X
Магкоев Т.Т., Григоркина Г.С., Заалишвили В.Б. и др. // Журн. физ. химии. 2020. Т. 94. № 2. С. 276. (Magkoev T.T., Grigorkina G.S., Zaalishvili V.B. et al. // Russ. J. Phys.Chem. A. 2020. V. 94. № 2. P. 401). https://doi.org/10.1134/S0036024420010203).https://doi.org/10.1134/S0044453720010203
Mullins D.R. // Surf. Sci. Rep. 2015. V. 70. P. 42. https://doi.org/10.1016/j.surfrep.2014.12.001
Panayotov D.A., Morris J.R. // Surf. Sci. Rep. 2016. V. 71. P. 77.https://doi.org/10.1016/j.surfrep.2016.01.002
Lee C., Fan T., Wu S., Anderson L. // Surf. Sci. 2005. V. 578. P. 5. https://doi.org/10.1016/j.susc.2005.01.017
Gong X.-Q., Selloni A., Dulub O. et al. // J. Am. Chem. Soc. 2008. V. 130. P. 370. https://doi.org/10.1021/ja0773148
Lu J.L., Gao H.J., Shaikhutdinov S., Freund H.-J. // Catal. Lett. 2007. V. 114. P. 8. https://doi.org/10.1007/s10562-007-9039-3
Hojrup-Hansen K., Ferrero S., Henry C.R. // Appl. Surf. Sci. 2004. V. 226. P. 167. https://doi.org/10.1016/j.apsusc.2003.11.017
Benia H.M., Lin X., Gao H.J. et al. // J. Phys. Chem. C. 2007. V. 111. P. 10528. https://doi.org/10.1021/jp0726891
Liu Z.-P., Gong X.-Q., Kohanoff J. et al. // Phys. Rev. Lett. 2003. V. 91. P. 266102. https://doi.org/10.1103/PhysRevLett.91.266102
Molina L.M., Hammer B. // Phys. Rev. Lett. 2003. V. 90. P. 206102. https://doi.org/10.1103/PhysRevLett.90.206102
Chen Y., Crawford P., Hu P. // Catal. Lett. 2007. V. 119. P. 21. https://doi.org/10.1007/s10562-007-9200-z
Molina L.M., Hammer B. // Appl. Catal. A: Gen. 2005. V. 291. P. 21. https://doi.org/10.1016/j.apcata.2005.01.050
Magkoev T.T. // Surf, Sci. 2007. V. 601. P. 3143. https://doi.org/10.1016/j.susc.2007.05.015
Григоркина Г.С., Рамонова А.Г., Кибизов Д.Д. и др. // Письма ЖТФ. 2017. Т. 43. № 13. С. 43. https://doi.org/10.21883/PJTF.2017.13.44810.16757
Grigorkina G.S., Zaalishvili V.B., Burdzieva O.G. et al. // Solid State Commun. 2018. V. 276. P. 28. https://doi.org/10.1016/j.ssc.2018.04.001
Zhang J., Medlin J.W. // Surf. Sci. Rep. 2018. V. 73. P. 117.https://doi.org/10.1016/j.surfrep.2018.06.002
Тваури И.В., Ремар Д.Ф., Туриев А.М. и др. // Письма ЖТФ. 2010. Т. 36. № 10. С. 59.https://doi.org/10.1134/S106378501005024X
Магкоев Т.Т., Розенталь Д., Шредер С.Л.М., Кристман К. // Письма ЖТФ. 2000. Т. 26. № 20. С. 1. (1063-7850/00/2610-0894$20.00).
Carabineiro S.A.C., Nieuwenhuys B.E. // Gold Bulletin. 2009. V. 42. № 4. P. 288. https://doi.org/10.1007/BF03214951
Hoffmann F.M. // Surf. Sci. Rep. 1983. V. 3. № 2−3. P. 107. https://doi.org/10.1016/0167-5729(83)90001-8
Zhdanov V.P., Kasemo B. // Surf. Sci. Rep. 1997. V. 29. № 2. P. 35. https://doi.org/10.1016/S0167-5729(97)00009-5
Watkins M.B., Foster A.S., Shluger A.L. // J. Phys. Chem. C. 2007. V. 111. P. 15337. https://doi.org/10.1021/jp071715s
Chen B.H., Ma Y.S., Ding L.B. et al. // J. Phys. Chem. C. 2013. V. 117. P. 5800. https://doi.org/10.1021/jp312406f
Lykhach Y., Johánek V., Aleksandrov H.A. et al. // J. Phys. Chem. C. 2012. V. 116. P. 12103. https://doi.org/10.1021/jp302229x
Jiang S., Zhang R., Liu H. et al. // J. Am. Chem. Soc. 2020. https://doi.org/10.1021/jacs.9b13915
Deng X., Quek S.Y., Biener M.M. et al. // Surf. Sci. 2008. V. 60. P. 1166. https://doi.org/10.1016/j.susc.2008.01.014
Chang Z., Song Z., Liu G. et al. // Surf. Sci. 2002. V. 512. P. L353. https://doi.org/10.1016/S0039-6028(02)01495-4
Radican K., Berdunov N., Manai G., Shvets I.V. // Phys. Rev. B. 2007. V. 75. P. 155434. https://doi.org/10.1103/PhysRevB.75.155434
Fei S., Xia K., Tian X. et al. // Int. Journ. Hydrogen Energy. 2016. V. 41. P. 5652. https://doi.org/10.1016/j.ijhydene.2016.02.079
Yang M., Han B., Cheng H. // J. Phys. Chem. C. 2012. V. 116. P. 24630. https://doi.org/10.1021/jp308255a
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии