

Журнал физической химии, 2021, T. 95, № 8, стр. 1137-1147
Формирование объемной керамики на основе оксида алюминия методом электрофоретического осаждения из суспензий наночастиц
Е. Г. Калинина a, b, *, Д. С. Русакова b, А. С. Кайгородов a, b, А. С. Фарленков b, c, А. П. Сафронов a, b
a Российская академия наук, Уральское отделение,
Институт электрофизики
Екатеринбург, Россия
b Уральский федеральный университет имени первого Президента России Б.Н. Ельцина
Екатеринбург, Россия
c Российская академия наук, Уральское отделение,
Институт высокотемпературной электрохимии
Екатеринбург, Россия
* E-mail: jelen456@yandex.ru
Поступила в редакцию 19.11.2020
После доработки 19.11.2020
Принята к публикации 03.12.2020
Аннотация
Методом электрофоретического осаждения (ЭФО) сформирована высокоплотная керамика из суспензий нанопорошков на основе оксида алюминия. Нанопорошки на основе Al2O3 получены методом электрического взрыва проволоки (ЭВП) из сплава Al–Mg с содержанием Mg 1.3 мас. %. Устойчивые неводные суспензии для ЭФО получены с применением слабоагрегированных наночастиц Al2O3, имеющих сферическую форму с удельной поверхностью 40 м2/г. Показано, что с использованием метода ЭФО и спекания при температуре 1650°С возможно формирование высокоплотной керамики с плотностью 3.93 г/см3 (98.6% от теор.) и микротвердостью 17.7 ± 0.5 ГПа. Установлено, что применение нанопорошка на основе оксида алюминия, полученного из сплава Al–Mg, привело к существенному улучшению спекаемости керамики за счет образования ≈2.5 мас. % шпинели MgAl2O4, что позволило повысить плотность и микротвердость керамических образцов.
Керамика на основе оксида алюминия обладает уникальными свойствами, среди которых можно выделить химическую стойкость, огнеупорность, высокую прочность и низкую электропроводность, что обусловливает ее применение в широком спектре высокотехнологичных отраслей энергетики, машиностроения, электротехнической промышленности и приборостроения [1–3]. На основе оксида алюминия формируют тонкопленочные покрытия, используя методы импульсного лазерного осаждения [4, 5], химического осаждения из газовой фазы [6, 7], золь–гель-метод [8]. Компактирование порошков оксида алюминия осуществляют методами изостатического и магнитно-импульсного прессования [9–11], шликерного литья [12, 13]. Методы одноосного и изостатического прессования имеют существенные недостатки и требуют дорогостоящего оборудования. В силу высокой энергии поверхностного слоя частицы порошка при прессовании активно взаимодействуют между собой и оказывают значительное сопротивление при компактировании, что приводит к формированию неоднородностей плотности внутри компакта, внутренних напряжений, пор внутри керамики при последующем спекании [14].
Для получения высокоплотной керамики перспективно применение нанопорошков, однако, наряду с имеющимися преимуществами нанопорошков, такими как сниженная температура спекания керамики, меньший размер зерен, возникают и значительные сложности при их прессовании ввиду развитой поверхности наночастиц и избыточной поверхностной энергии [15]. Применение коллоидных методов при компактировании порошков исключает напряженность компактов, однако, в суспензии принципиально важна роль взаимодействия частиц в жидкой среде и их агрегация [16].
Перспективным коллоидным методом компактирования частиц из устойчивых суспензий порошков является метод электрофоретического осаждения (ЭФО), который обладает следующими преимуществами: простота и низкая стоимость оборудования, индифферентность к форме покрываемой поверхности, регулирование толщины, морфологии тонкой пленки или компакта путем изменения времени и приложенного напряжения [17–20]. Известно, что методом ЭФО получают не только тонкие пленки на основе оксида алюминия [21–23], но и объемные плотные компакты [24, 25], а также пористые образцы [26]. Наряду с отмеченными выше преимуществами ЭФО, применение этой технологии при формировании объемной керамики позволяет улучшить однородность компактов, дает возможность гибко регулировать форму образцов, проводить локальное осаждение для формирования 3D-структур [27], в том числе при реализации аддитивных технологий [28]. В качестве примера применения ЭФО в аддитивных технологиях изготовления керамики можно привести работу [29], в которой применяли полый электрод для осуществления локального осаждения частиц из суспензии оксида алюминия со средним размером 200 нм на поверхности пористой и проницаемой для ионов фторопластовой мембраны. В результате экспериментов были получены локальные участки осажденного оксида алюминия с высотой и поперечным размером ~ 500 мкм.
Необходимый этап реализации ЭФО – приготовление устойчивой суспензии [20]. Для получения устойчивых суспензий на основе оксида алюминия, например, с использованием коммерческих порошков α-Al2O3 TM-DAR (Boehringer Ingelheim Chemicals, Tokyo, Japan), α-Al2O3 AKP-50 (Sumitomo Chem. Co., Ltd.), многие авторы вынуждены применять различные дисперсанты, такие как монохлоруксусная кислота [30], полиэтилеимин [31], фосфатные эфиры [24]. Авторы [30] применяли коммерческие порошки Al2O3 с добавкой 0.05 мас. % MgO (Malakoff Ind., USA) со средним размером частиц 330 и 470 нм. При проведении ЭФО из суспензии в изопропаноле с добавлением монохлоруксусной кислоты и поливинилбутираля были получены объемные образцы керамики плотностью 98.5–99.2% от теор. и микротвердостью 17.8–18.0 ГПа. В [31] сообщается о применении горячего изостатического прессования (180 МПа, 1250°C) для уплотнения керамики после компактирования методом ЭФО. В результате была достигнута плотность алюмооксидной керамики 95.3% от теор.
В работе [25] были проанализированы потенциальные возможности технологии ЭФО при формировании объемной керамики из различных коммерческих нано- и субмикронных порошков оксида алюминия, было определено, что при применении нанопорошков со средним размером частиц 45 нм (Nanotek, Nanophase Technologies Corporation, USA) и 50 нм (MTI Corporation, USA) плотность неспеченных компактов составляла 27 и 42% соответственно, а при применении субмикронных порошков со средним размером частиц 150 нм (Inframat Advanced Materials, USA) и 700 нм (Alcoa, USA) – 56 и 59% от теоретической соответственно. Для повышения абсолютного значения дзета-потенциала суспензии на основе субмикронных порошков Inframat и Alcoa в этаноле авторы использовали добавку уксусной кислоты 0.2 и 0.3 мас. %, что сдвигало дзета-потенциал в сторону отрицательных значений (–49 и –60 мВ), в то время как суспензии нанопорошков Nanotek и MTI не требовали дополнительной стабилизации и характеризовались высокими положительными значениями дзета-потенциала (+61 и +79 мВ). В работе [32] было показано влияние плазменной обработки применяемого для ЭФО субмикронного оксида алюминия (DBET = 150 нм, TM-DAR, Taimei Chemicals, Japan) на свойства полученных осадков, т.е. плазменная активация поверхности порошка улучшает гладкость поверхности осадков и повышает их плотность до и после спекания.
Использование слабоагрегированных нанопорошков со сферической формой частиц, полученных методом электрического взрыва проволоки (ЭВП) [33, 34] позволит упростить приготовление самостабилизированных суспензий для ЭФО [35]. Отсутствие дополнительной стабилизации суспензии (например, введение дисперсантов, неорганических и органических кислот, оснований и молекулярного йода) позволит избежать отрицательного влияния дисперсантов и зарядовых агентов на процесс спекания керамических образцов. Однако, переход к нанопорошкам порождает ряд физико-химических проблем, связанных с преодолением агрегации наночастиц в суспензиях. Наши более ранние работы посвящены исследованию устойчивых суспензий (дисперсности, фракционного состава, электрокинетического потенциала, влияния ультразвуковой обработки и центрифугирования) [36] и формированию методом ЭФО как индивидуальных тонкопленочных слоев на основе оксидов циркония, церия и церата бария [37, 38], так и композитных покрытий, полученных из смешанных суспензий, содержащих нанопорошки различной природы [39]. Тематика наших работ наиболее полно отражена в обзоре [18].
Как показал анализ литературы, характерная особенность применения ЭФО – сравнительно невысокая плотность неспеченных компактов (~27–60 мас. % от теор.), причем при применении нанопорошков плотность компактов снижается, как было отмечено в [25], поэтому представляет интерес поиск методов интенсификации процесса уплотнения керамики в ходе спекания. Для получения выскопоплотной керамики при применении коллоидных методов, в том числе с использованием ЭФО, возможно применение композитных суспензий, содержащих микроразмерные и наноразмерные частицы α-Al2O3 и δ-Al2O3, что оказывает влияние на последующее спекание и уплотнение керамики [40]. Известно, что добавление MgO к оксиду алюминия оказывает влияние на процесс спекания, а именно, вызывает уплотнение керамики при спекании за счет интенсификации диффузионных процессов на границах зерен, поэтому применение допированного магнием оксида алюминия можно использовать в качестве эффективного метода повышения плотности спеченной керамики [41]. Можно предположить, что состав, морфология и предыстория применяемых порошков, определяемая технологией их получения, играют ключевую роль в свойствах применяемых суспензий на их основе, а также в реализации процесса ЭФО, свойствах полученных компактов и спекании керамики. Таким образом, можно сделать вывод о необходимости индивидуального подхода к применяемой дисперсной системе для достижения необходимых свойств керамики.
В настоящей работе мы исследовали особенности формирования плотной объемной керамики методом ЭФО из неводных самостабилизированных суспензий слабоагрегированных нанопорошков недопированного и допированного Mg оксида алюминия, изготовленных методом ЭВП без использования прессования. Особенностью реализации метода ЭФО выступает применение деагрегированных суспензий без использования дополнительных дисперсантов и связующих. В рамках данной работы исследованы морфология и микроструктура, фазовый состав, микротвердость полученных керамических образцов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Нанопорошки на основе оксида алюминия получали методом электрического взрыва проволоки (ЭВП) [33, 42]. Партии нанопорошков A1 и A2 получали электрическим взрывом Al-проволоки и проволоки, изготовленной из сплава Al–Mg с содержанием Mg 1.3 мас. % соответственно. По данным просвечивающей электронной микроскопии двух партий нанопорошков, частицы имели сферическую форму. Распределения частиц по размерам, полученные посредством графического анализа микрофотографий нанопорошков А1 и А2, имели логнормальный вид и характеризовались функцией следующего вида:
(1)
$f(D) = \frac{1}{{D\sigma \sqrt {2\pi } }}{{e}^{{ - \frac{{{{{(\ln D - \ln \mu )}}^{2}}}}{{2{{\sigma }^{2}}}}}}},$Среднечисловой диаметр наночастиц А1 и А2 составил 18 и 22 нм, соответственно. Удельную поверхность нанопорошков А1 и А2 определяли методом БЭТ (SБЭТ = 42 и 40 м2/г соответственно). Для сферических частиц значение удельной поверхности позволяет рассчитать средний диаметр частиц (dБЭТ) по формуле [43]:
где SБЭТ – удельная поверхность нанопорошка, ρ – плотность материала (${{\rho }_{{\gamma {\text{ - A}}{{{\text{l}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{3}}}}}}}$ = 3.68 г/см3). Средний диаметр частиц A1 и A2, вычисленный по формуле (2), составил 39 и 41 нм соответственно. Полученные значения dБЭТ хорошо согласуются со средними диаметрами частиц по поверхности (dS) (для A1 – 38 нм, для A2 – 41 нм), рассчитанными по данным числового распределения частиц по размерам с использованием уравнения (1):По данным РФА, нанопорошок А1 содержал две кристаллические фазы: γ-Al2O3 (≈15 мас. %), кубическая, пространственная группа Fd-3m, параметр решетки а = 7.919(±0.008) Å, ОКР = 18 (±1) нм и δ-Al2O3 (≈85 мас. %), орторомбическая, пространственная группа P222 с параметрами решетки а = 7.934 Å, b = 7.956 Å, c = 11.711 Å и ОКР = = 18(±2) нм. Нанопорошок A2 содержал четыре кристаллические фазы: γ-Al2O3 (≈31 мас. %), кубическая, пространственная группа Fd-3m, а = = 7.950(±0.010) Å, ОКР = 26(±2) нм; металлический Al (≈0.3 мас. %), кубическая, пространственная группа Fm-3m, a = 4.054(±0.004) Å; α-Al2O3 (≈0.4 мас. %), корунд, ромбоэдрическая, пространственная группа R-3c, a = 4.764(±0.004) Å, c = 12.99(±0.02) Å; δ-Al2O3 (≈69 мас. %), орторомбическая, пространственная группа P222, параметры решетки а = 7.934 Å, b = 7.956 Å, c = = 11.711 Å и ОКР = 18(±2) нм.
Нанопорошки A1 и A2 использовали для приготовления устойчивых суспензий и формирования объемных компактов методом ЭФО. Для получения устойчивых суспензий нанопорошков в качестве дисперсионной среды использовали изопропанол (“ос.ч.”, ООО “Компонент-Реактив”, Москва). Суспензии готовили без введения дисперсантов или других добавок. Исходные суспензии концентрацией 50, 70 и 180 г/л готовили по точной навеске нанопорошка и обрабатывали ультразвуком с помощью ультразвуковой ванны УЗВ-13/150-ТН (“Рэлтек”, Екатеринбург) в течение 125 мин. Удаление неразрушенных крупных агрегатов при ультразвуковой обработке в суспензии проводили методом центрифугирования с помощью центрифуги Hermle Z383 со скоростью 6000 об/мин в течение 3 мин. Измерение электрокинетического дзета-потенциала и рН в суспензиях проводили электроакустическим методом с помощью анализатора DT-300 (Dispersion Technology, США). Распределение частиц по размерам в суспензиях на основе нанопорошков А1 и А2 получали методом динамического рассеяния света (ДРС) с помощью анализатора размера частиц ZetaPlus (Brookhaven Instruments Corporation, NY, USA). Все измерения для суспензий проводили в изотермических условиях на воздухе при 25°C.
Электрофоретическое осаждение выполняли на специализированной компьютеризированной установке, обеспечивающей режимы постоянства напряжения, которая была разработана и изготовлена в ИЭФ УрО РАН. ЭФО выполняли при горизонтальном расположении электродов (вертикальное осаждение). Катодом служил диск из алюминиевой фольги площадью 113 мм2, анодом – диск из нержавеющей стали, расстояние между электродами составляло 10 мм. Для изготовления объемных компактов оксида алюминия с использованием электрофоретического осаждения наночастиц использовали следующий режим осаждения: для А1 – постоянное напряжение составляло 100 В, для А2 – 20 и 100 В. Время осаждения во всех случаях составляло 150 мин. Во время ЭФО применяли перекачку суспензии со дна ячейки для осаждения в ее верхнюю часть. Полученный осадок сушили на электроде в течение нескольких дней с целью исключения возникновения трещин. Для этого использовали эксикатор с небольшим количеством изопропилового спирта для замедления процесса сушки. Образцы, полученные методом ЭФО, спекали на воздухе при температуре 1500 и 1650°С в течение 2–8 ч (Nabertherm LHT-04/18). Толщина образцов керамики составляла 1.2–4.7 мм.
Морфологию и микроструктуру наночастиц оксида алюминия двух партий А1 и А2 исследовали с помощью просвечивающего электронного микроскопа JEOL JEM 2100 (JEOL, Токио, Япония). Микроструктуру керамики исследовали с помощью сканирующего электронного микроскопа Mira 3 LMU (Tescan, Чехия). Напыление проводящих углеродных покрытий (толщина < 10 нм) проводили с использованием системы Q150T ES (Quorum Technologies, Великобритания). Рентгенофазовый анализ проводили на дифрактометре D8 DISCOVER в медном излучении с графитовым монохроматором на дифрагированном луче. Обработку выполняли с использованием программы TOPAS-3. Удельную поверхность определяли объемным вариантом метода БЭТ по низкотемпературной равновесной сорбции паров азота из смеси с гелием на вакуумной сорбционной установке Micromeritics TriStar 3000. Плотность и пористость образцов керамики на основе оксида алюминия определяли методом гидростатического взвешивания. Микротвердость керамики исследовали методом индентирования на приборе Nanotest 600 с использованием алмазной пирамидки Виккерса. Максимальная приложенная нагрузка составила 2 Н. Скорость нагружения/снятия нагрузки выбирали таким образом, чтобы выполнялся следующий временной цикл: нагружение – 20 с, выдержка при максимальной нагрузке – 5 с, снятие нагрузки – 20 с. Значения усредняли по 10 измерениям.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Приготовление стабильной суспензии для ЭФО
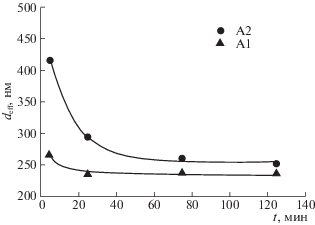
С целью электрофоретического формирования объемных образцов были приготовлены суспензии на основе нанопорошков оксида алюминия двух разных партий А1 и А2 в изопропаноле и проведена оценка дисперсности частиц в этих суспензиях. На рис. 1 приведена зависимость эффективного гидродинамического диаметра агрегатов (deff) для суспензий А1 и А2 в зависимости от времени ультразвуковой обработки (УЗО) с непрерывным охлаждением суспензии. Из рис. 1 видно, что в процессе УЗО размер агрегатов в суспензиях нанопорошков закономерно снижается, особенно эта тенденция выражена для суспензии нанопорошка А2. Зависимость эффективного гидродинамического диаметра агрегатов от времени УЗО для суспензии А2 лежит выше, чем для суспензии А1. Так, исходное значение deff для суспензии А2 составляет 414 нм, в то время как для А1 равно 266 нм. Для суспензии А1 в течение 25 мин УЗО значение deff снижается с 266 до 235 нм, а затем выходит на насыщение, в то время как для А2 – постепенно снижается с 414 до 250 нм. УЗО в течение 125 мин для суспензий А1 и А2 позволяет понизить гидродинамический размер агрегатов. Неразрушенные крупные агрегаты, присутствующие в суспензиях, могут быть отделены центрифугированием.
Рис. 1.
Зависимости эффективного гидродинамического диаметра агрегатов (deff) в суспензиях нанопорошков А1 и А2 от времени непрерывной УЗО.
На рис. 2 представлены унимодальные распределения частиц по интенсивности рассеяния неводных суспензий нанопорошков А1 и А2, полученные методом ДРС, после обработки ультразвуком в течение 5–125 мин и последующего центрифугирования в течение 3 мин со скоростью 6000 об/мин. Из рис. 2 видно, что с увеличением времени УЗО происходит сужение распределений, что подтверждается значениями GSD (геометрическое стандартное отклонение), которое служит параметром ширины распределения и связано со среднеквадратическим отклонением по формуле σ = ln(GSD). Для суспензий А2, обработанных ультразвуком в течение 5, 25, 75 и 125 мин, значение GSD = 1.64 ± 0.07, 1.51 ± 0.07, 1.46 ± 0.07 и 1.44 ± 0.07 соответственно; для суспензий А1, обработанных ультразвуком в течение 5, 25, 75 и 125 мин, значение GSD = 1.60 ± 0.07, 1.51 ± 0.07, 1.52 ± 0.07 и 1.48 ± 0.07 соответственно. Длительность УЗО повлияла на эффективный гидродинамический диаметр частиц в суспензиях А2 и А1, который уменьшился с 414 до 250 нм и с 266 до 236 нм соответственно. После 75 мин УЗО произошло некоторое незначительное уширение распределения для суспензии А1. Для обеих суспензий А1 и А2 после центрифугирования распределение частиц по размерам стало более узким. Эффективный гидродинамический диаметр частиц в суспензиях A2 и A1 и их GSD составили 162 и 158 нм и 1.39 ± 0.06 и 1.41 ± 0.06 соответственно, что связано с удалением крупной фракции частиц в процессе центрифугирования. Таким образом, комбинирование УЗО и центрифугирования позволяет получать узкие распределения частиц по размерам в суспензиях.
Рис. 2.
Унимодальные распределения для суспензий A2 (слева) и А1 (справа); УЗО: a – 5, б – 25, в – 75, г – 125 мин; д –125 мин + центрифугирование 3 мин со скоростью 6000 об/мин; Pint(d) – плотность вероятности унимодального распределения частиц, d – эффективный диаметр частиц.
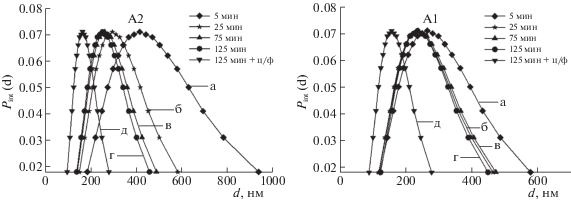
По данным определения фракционного состава мультимодального распределения для суспензий нанопорошков А1 и А2 было получено, что исходная суспензия А2 (после 5 мин УЗО) содержала три фракции частиц и их агрегатов в различном весовом соотношении: 90 нм (5%), 427 нм (87%) и 2030 нм (8%). После 25 мин УЗО из суспензии удалось исключить крупную фракцию (средний размер агрегатов 2030 нм) и повысить долю средней фракции со средним размером агрегатов 441 нм до 97%. После УЗО в течение 125 мин и последующего центрифугирования в суспензии остались только мелкие агрегаты размером 174 нм (44%), и появились частицы размером 46 нм (56%). Исходная суспензия А1 (после 5 мин УЗО) имела бимодальное распределение агрегатов по размерам, а именно, первая фракция – 196 нм (25%) и вторая фракция – 761 нм (75%). После УЗО в течение 25 мин удалось снизить средний размер агрегатов первой и второй фракции до 135 нм (24%) и 412 нм (76%) соответственно. После 125 мин УЗО и последующего центрифугирования средние размеры агрегатов первой и второй фракции составили 115 нм (77%) и 231 нм (23%) соответственно.
Электрофоретическое осаждение компактов и характеристика керамики на основе Al2O3
С целью получения компактов методом ЭФО была проведена серия экспериментов, в которой использовали суспензии на основе нанопорошков А1 и А2, отличающихся способами обработки, концентрацией частиц, значениями дзета-потенциала и рН. В табл. 1 приведены характеристики суспензий, параметры электрофоретического осаждения компактов на основе нанопорошков А1 и А2. Для того чтобы в процессе ЭФО под действием электрического поля наночастицы осаждались на электроде, они должны нести электрический заряд. Поверхность твердых частиц, диспергированных в жидкой полярной среде, несет избыточный электрический заряд, обусловленный либо поверхностной диссоциацией, либо адсорбцией ионов [44]. Количественной характеристикой поверхностного заряда, который не может быть измерен непосредственно, служит величина ζ-потенциала, который имеет тот же знак, что и заряд поверхности частиц, и соответствует электрическому потенциалу на плоскости скольжения, расположенной вблизи плотной части двойного электрического слоя [44]. Значение ζ-потенциала влияет как на устойчивость, так и на электрофоретическую подвижность частиц коллоидной суспензии. Чем больше величина ζ-потенциала, тем больший заряд имеет поверхность частицы, что способствует повышению эффективности ЭФО. Из табл. 1 видно, что до проведения ЭФО исходные суспензии на основе нанопорошков А1 и А2 в изопропаноле характеризовались кислой средой, в отдельных случаях близкой к нейтральной (рН 3.9–7.3) и достаточно высоким начальным значением ζ-потенциала (от +35 до +48 мВ). Согласно теоретическим представлениям и многочисленным экспериментальным данным, для того чтобы суспензия была устойчива, абсолютное значение дзета-потенциала должно быть >26 мВ независимо от его знака [45, 46].
Таблица 1.
Характеристики суспензий нанопорошков А1 и А2 и параметры ЭФО (Образцы А1_1, А1_3, А1_8 получены из суспензии нанопорошка А1; А2_10, А2_13, А2_14 – из суспензии нанопорошка А2)
| Образец | Суспензия (концентрация, условия обработки) | рН до/после осаждения |
ζ-потенциал до/после осаждения, мВ |
Сила тока
от времени (I), среднее значение Iср |
Режим ЭФО/масса сухого компакта |
|---|---|---|---|---|---|
| A1_1 | 50 г/л, УЗО 125 мин, свежеприготовленная суспензия |
5.6/4.2 | +40/+33 | I снижается от 1.223
до 0.090 мA (в 13.6 раза) Iср = 0.255 мА |
100 В, 3 ч/329 мг |
| A1_3 | 50 г/л, УЗО 125 мин, суспензия выдержана 7 суток |
6.7/5.9 | +44/+38 | I снижается от 0.949
до 0.095 мA (в 10 раз) Iср = 0.195 мА |
100 В, 3 ч/262.5 мг |
| A1_8 | 62 г/л, УЗО 125 мин, центрифугирование 6000 об/мин, 3 мин | 3.9/5.6 | +35/+36 | I снижается от 0.762
до 0.044 мA (в 17.3 раза Iср = 0.140 мА |
100 В, 3 ч/276.2 мг |
| A2_10 | 62 г/л, УЗО 125 мин, центрифугирование 6000 об/мин, 3 мин | 7.3/6.8 | +48/+38 | I снижается от 0.302
до 0.064 мA (в 4.7 раза) Iср = 0.113 мА |
100 В, 3 ч/671.2 мг |
| A2_13 | 62 г/л, УЗО 125 мин, центрифугирование 6000 об/мин, 3 мин | 7.3/6.2 | +43/+36 | I снижается от 0.081
до 0.053 мА (в 1.5 раза) Iср= 0.068 мА |
20 В, 3 ч /180.8 мг |
| A2_14 | 180 г/л, УЗО 125 мин | 6.4/5.9 | +37/+38 | I снижается от 0.072
до 0.041 мА (в 1.7 раза) Iср= 0.061 мА |
20 В, 3 ч/1043 мг |
В ряде случаев после проведения ЭФО ζ-потенциал снижался, но оставался положительным и достаточно высоким, в этом случае происходило снижение рН в более кислую сторону. Как было отмечено ранее, основная фракция суспензии А2, доля которой составляла 56%, содержала более мелкие агрегаты (46 нм), в то время как средний диаметр агрегатов основной фракции (77%) суспензии А1 составлял 115 нм. Это предопределяет высокую дисперсность и как следствие более высокий ζ-потенциал суспензии А2 по сравнению с суспензией А1 концентраций 62 г/л. Увеличение концентрации с 62 до 180 г/л в случае суспензии А2 привело к снижению ζ-потенциала. Следует отметить, что выдержка суспензии в течение 7 суток повлияла на начальное значение рН и дзета-потенциал, возможно, вследствие изменения ионного состава суспензии, что вызвало снижение начального значения тока (I0). Так, при ЭФО (U = 100 В) из свежеприготовленной суспензии значение I0 = 1.223 мА, тогда как для выдержанной суспензии I0= 0.949 мА. Массы образовавшихся объемных осадков для образцов А1_1 и А1_3 составили 329 и 262.5 мг, соответственно. При ЭФО из суспензии после центрифугирования (образец А1_8) наблюдалась совершенно иная картина. Так, значение рН после осаждения увеличилось с 3.9 до 5.6, в то время как ζ-потенциал практически не изменился.
При использовании порошка А2 потребовалось снизить напряжение осаждения со 100 до 20 В для обеспечения однородности и гладкости поверхности получаемого осадка. На рис. 3 приведены фотографии осадков после сушки, полученных из суспензии нанопорошка А2 при напряжениях 100 В (образец А2_10, слева) и 20 В (образец А2_13, справа). Из рис. 3 (слева) видно, что поверхность образца существенно неоднородная, имеются наплывы и трещины, тогда как при снижении напряжения до 20 В удалось получить гладкую поверхность осадка без трещин (рис. 3, справа). Массы образовавшихся объемных осадков для образцов А2_10 и А2_13 составили 671.2 и 180.8 мг соответственно. Вероятно, что для более высокодисперсной суспензии А2 во время ЭФО в режиме постоянного напряжения 100 В возникали интенсивные электрогидродинамические и электроосмотические потоки, которые приводили к неоднородному осаждению, что и определяло необходимость снижения напряжения осаждения до 20 В [47, 48].
Рис. 3.
Компакты (после сушки) при ЭФО из суспензии нанопорошка А2 при напряжении 100 В, образец А2_10 (слева) и 20 В, образец А2_13 (справа).

Выдержка суспензии А1 в течение 7 дней и ее центрифугрование не оказали влияния на плотность компакта и керамики (табл. 1 и 2). Однако, как видно из табл. 2 центрифугирование суспензии и повышение температуры спекания с 1500 до 1650°С для образцов А1_1, А1_3, А1_8 позволили увеличить плотность и микротвердость керамики, а также снизить пористость. Применение суспензии на основе нанопорошка А2 оказало значительное влияние на характеристики образцов A2_13, A2_14. Керамика, полученная из суспензии А2, характеризовалась меньшей пористостью и высокой плотностью и микротвердостью. Проведение ЭФО из суспензии А2 с концентрацией 180 г/л привело к увеличению массы компакта до 1043 мг (образец А2_14) по сравнению с осаждением образца А2_13 из суспензии с концентрацией 62 г/л, для которого масса осадка составляла 180.8 мг. Применение центрифугирования для суспензии нанопорошка А2 позволило обеспечить удаление крупных агрегатов, максимальный размер оставшейся фракции в суспензии не превышал 174 нм, при этом формирование компакта А2_13 из полученной суспензии привело к увеличению плотности с 3.87 до 3.93 г/см3 и микротвердости с 12.3 ± 0.8 до 17.7 ± 0.5 ГПа (табл. 2).
Таблица 2.
Плотность компакта до спекания (ρ), плотность (ρк), пористость (ε) и микротвердость (Н) керамики после спекания
| Образец | Режим спекания | ρ, г/см3 | ρк, г/см3 | ε, % | Н, ГПа |
|---|---|---|---|---|---|
| A1_1 | 1500°С, 2 ч | 1.19 | 1.79 | 55 | 0.36 ± 0.01 |
| A1_3 | 1650°С, 4 ч | 1.21 | 2.50 | 37 | 2.20 ± 0.07 |
| A1_8 | 1650°С, 8 ч | 1.23 | 2.58 | 35 | 3.30 ± 0.07 |
| A2_13 | 1650°С, 8 ч | 1.28 | 3.93 | 1 | 17.7 ± 0.5 |
| A2_14 | 1650°С, 8 ч | 1.75 | 3.87 | 3 | 12.3 ± 0.8 |
Как было отмечено ранее в экспериментальной части, применяемые две партии нанопорошков А1 и А2, полученные методом ЭВП, отличались по составу алюминиевой проволоки. Партия нанопорошка А1 получена из алюминиевой проволоки, тогда как партия нанопорошка А2 – из сплава Al–Mg (содержание Mg 1.3 мас. %). По данным РФА, Mg в исходном порошке А2 не образовал отдельную кристаллическую фазу, содержащую Mg, тогда как в ходе длительного спекания полученной керамики А2_13 формируется материал, содержащий две кристаллические фазы: 1) α-Al2O3 – корунд (≈97.5 мас. %), ромбоэдрическая, пространственная группа R-3c с периодами решетки a = 4.763(±0.002) Å, c = 13.011 (±0.005) Å и ОКР > 200 нм; 2) MgAl2O4 – шпинель (≈2.5 мас. %), кубическая, пространственная группа Fd-3m с периодом решетки a = 8.080 (±0.02) Å и ОКР = 110(±40) нм. Кристаллической фазы шпинели MgAl2O4 в составе порошка не было обнаружено, можно предположить, что при формировании частиц методом ЭВП Mg присутствует в виде ионов внедрения или замещения в кристаллической решетке Al2O3. Образец керамики А1_8 однофазный: α-Al2O3, корунд, ромбоэдрическая, пространственная группа R-3c с периодами решетки a = 4.760 (±0.004) Å, c = = 12.998(±0.004) Å и ОКР ≈ 200 нм. Присутствие Mg в составе применяемого нанопорошка А2 привело к существенному улучшению спекаемости керамики за счет образования ≈2.5 мас. % шпинели MgAl2O4, что позволило повысить плотность и микротвердость образцов керамики А2_13 и А2_14.
На рис. 4 представлены электронные микрофотографии для образцов А1_1 (рис. 4 сверху, поверхность слева, скол справа) и А1_3 (рис. 4 снизу, поверхность слева, скол справа). Поверхность и скол образца А1_1 характеризуются отдельными зернами размером ~0.3 мкм и их агломератами до 1 мкм со значительным количеством пор между ними размером до 1 мкм (рис. 4 сверху). Для поверхности и скола образца А1_3 характерны укрупнение отдельных зерен до размера 1–1.5 мкм и их агломератов до 2–3 мкм и присутствие значительного количества пор между зернами размером около 1 мкм (рис. 4, снизу). Изменение микроструктуры образца А1_3 по сравнению с образцом А1_1 обусловлено более высокой температурой спекания и временем выдержки, а именно (1650°С, 4 ч) и (1500°С, 2 ч) соответственно. Изменение в режиме спекания также повлияло на значение плотности, пористости и микротвердости образцов А1_1 и А1_3 (табл. 2), однако, увеличение температуры спекания образца А1_3 до 1650°С не позволило достигнуть плотности, близкой к теоретической (теоретическая плотность керамики на основе α-Al2O3 составляет 3.98 г/см3) [49] и высоких значений микротвердости в зависимости от метода компактирования керамики [50–52].
Рис. 4.
Электронные микрофотографии керамики: образец А1_1 (сверху) – поверхность (слева) и скол (справа); образец А1_3 (снизу) – поверхность (слева) и скол (справа).

На рис. 5 представлены электронные микрофотографии для образцов А2_13 (рис. 5, сверху) и А2_14 (рис. 5, снизу). Для образца А2_13 характерна плотная зеренная структура с размером зерен 1–4 мкм и небольшим количеством закрытых пор размером ~ 1 мкм (рис. 5, сверху). Образец А2_14 также характеризуется плотной зеренной структурой с размером зерен 2–8 мкм и незначительным наличием пор размером ~1 мкм (рис. 5, снизу). Вероятно, что указанное различие в микроструктуре связано с наличием разного дисперсного состава используемых для ЭФО суспензий нанопорошка А2 при осаждении образцов А2_13 и А2_14, а именно, суспензия для ЭФО образца А2_13 была подвергнута центрифугированию с удалением крупной фракции агрегатов. Для образца керамики А2_13 после спекания была получена плотность, составляющая 98.6% от теоретической, и достигнута достаточно высокая микротвердость (17.7 ± 0.5 ГПа), что сравнимо с данными авторов [52], которые, применяя метод прессования с последующим микроволновым спеканием, получили микротвердость 16.19 ± 0.58 ГПа.
Рис. 5.
Электронные микрофотографии керамики: образец А2_13 (сверху) – поверхность (слева) и скол (справа); образец А2_14 (снизу) – поверхность (cлева) и скол (справа).

В работе [53] была исследована зависимость твердости от температуры, и при 20°С получено значение твердости 15 ± 2 ГПа, что также согласуется с нашими результатами, которые подтверждают возможность применения метода ЭФО с последующим спеканием для формирования керамики на основе оксида алюминия с добавкой Mg с плотностью и микротвердостью, близкими к теоретическим значениям. Показана перспективность применения самостабилизированных суспензий на основе ЭВП нанопорошков, полученных из сплава Al–Mg, без добавления дисперсантов, полимерных связующих и зарядовых агентов для формирования высокоплотной керамики методом ЭФО. Полученные нами результаты по формированию объемной керамики методом ЭФО без прессования могут быть использованы в дальнейших работах в области технологии керамики, в том числе при оптимизации процесса спекания с ограничением роста зерен, например, как сообщается в работе [54].
ЗАКЛЮЧЕНИЕ
Электрофоретическим осаждением из устойчивых суспензий нанопорошков на основе оксида алюминия в изопропиловом спирте без добавления дисперсантов, зарядовых агентов и органических связующих была сформирована высокоплотная керамика. Нанопорошки на основе Al2O3 были получены методом электрического взрыва Al-проволоки и проволоки из сплава Al–Mg с содержанием Mg 1.3 мас. %. Было показано, что с использованием метода ЭФО и спекания при температуре 1650°С возможно формирование высокоплотной керамики толщиной до 4.7 мм с плотностью 3.93 г/см3 (98.6% от теор.) и микротвердостью 17.7 ± ± 0.5 ГПа. Установлено, что применение нанопорошка на основе оксида алюминия, полученного из сплава Al–Mg, привело к существенному улучшению спекаемости керамики за счет образования ≈ 2.5 мас. % шпинели MgAl2O4, что позволило повысить плотность и микротвердость керамических образцов. Наилучшие результаты по плотности и микротвердости достигнуты для образца керамики, полученного с использованием центрифугирования суспензии. В этом случае после спекания образца при температуре 1650°С формируется плотная структура с размером зерен 1–4 мкм и небольшим количеством закрытых пор размером ~1 мкм.
Работа была частично выполнена с использованием оборудования коллективных центров ИЭФ УрО РАН и ИВТЭ УрО РАН (“Состав вещества”).
Авторы благодарят заведующего лабораторией импульсных процессов И.В. Бекетова и А.В. Багазеева за развитие метода получения нанопорошков (метод ЭВП), а также К.И. Демидову – за проведение РФА порошков и керамик, А.М. Мурзакаева – за исследование морфологии наночастиц и инженера С.Ю. Ивина – за определение плотности и пористости образцов керамики методом гидростатического взвешивания.
Список литературы
Hirata Y., Shimonosono T., Sameshima S., Tominaga H. // Ceram. Int. 2015. V. 41. № 9. P. 11449. https://doi.org/10.1016/j.ceramint.2015.05.109
Ruys A. Alumina Ceramics: Biomedical and Clinical Applications. United Kingdom: Woodhead Publishing, 2019. 580 p.
Rakshit R., Das A. // Precis. Eng. 2019. V. 59. P. 90. https://doi.org/10.1016/j.precisioneng.2019.05.009
Boidin R., Halenkovič T., Nazabal V. et al. // Ceram. Int. 2016. V. 42. № 1. P. 1177. https://doi.org/10.1016/j.ceramint.2015.09.048
Korhonen H., Syväluoto A., Leskinen J.T.T., Lappalainen R. // Opt. Laser Technol. 2018. V. 98. P. 373. https://doi.org/10.1016/j.optlastec.2017.07.050
Kelekanjeri V., Siva Kumar G., Carter W.B., Hampikian J.M. // Thin Solid Films. 2006. V. 515. № 4. P. 1905. https://doi.org/10.1016/j.tsf.2006.07.033
Ogita Y., Saito N. // Ibid. 2015. V. 575. P. 47. https://doi.org/10.1016/j.tsf.2014.10.022
He J., Avnir D., Zhang L. // Acta Mater. 2019. V. 174. P. 418. https://doi.org/10.1016/j.actamat.2019.05.062
Ivanov V.V., Paranin S.N., Khrustov V.R. // Phys. Metals Metallogr. 2002. V. 94. P. S98.
Kaygorodov A., Rhee C., Kim W. et al. // Mater. Sci. Forum. 2007. V. 534−536. P. 1053. https://doi.org/10.4028/www.scientific.net/MSF.534-536.1053
Promdej C., Pavarajarn V., Wada S. et al. // Curr. Appl. Phys. 2009. V. 9. № 5. P. 960. https://doi.org/10.1016/j.cap.2008.09.011
Makiya A., Tanaka S., Shoji D. et al. // J. Eur. Ceram. Soc. 2007. V. 27. № 12. P. 3339. https://doi.org/10.1016/j.jeurceramsoc.2006.11.041
Takao Y., Hotta T., Naito M. et al. // Ibid. 2002. V. 22. № 4. P. 397. https://doi.org/10.1016/S0955-2219(01)00307-7
Okuma G., Watanabe S., Shinobe K. et al. // Sci. Rep. 2019. V. 9. P. 11595. https://doi.org/10.1038/s41598-019-48127-y
Bowen P., Carry C. // Powder Technol. 2002. V. 128. № 2−3. P. 248. https://doi.org/10.1016/s0032-5910(02)00183-3
Lewis J.A. // J. Am. Ceram. Soc. 2000. V. 83. № 10. P. 2341. https://doi.org/10.1111/j.1151-2916.2000.tb01560.x
Corni I., Ryan M.P., Boccaccini A.R. // J. Eur. Ceram. Soc. 2008. V. 28. № 7. P. 1353. https://doi.org/10.1016/j.jeurceramsoc.2007.12.011
Pikalova E., Kalinina E. // J. of Energy Prod. and Mgm. 2019. V. 4. № 1. P. 1. https://doi.org/10.2495/EQ-V4-N1-1-27
Pikalova E.Yu., Kalinina E.G. // Renew. Sust. Energ. Rev. 2019. V. 116. P. 109440. https://doi.org/10.1016/j.rser.2019.109440
Калинина Е.Г., Пикалова Е.Ю. // Успехи химии. 2019. Т. 88. № 12. С. 1179.
Kishida S., Ju D., He H., Li Y. // J. Environ. Sci. 2009. V. 21. P. S112. https://doi.org/10.1016/S1001-0742(09)60051-6
Novak S., König K. // Ceram. Int. 2009. V. 35. № 7. P. 2823. https://doi.org/10.1016/j.ceramint.2009.03.033
Song G., Xu G., Quan Y. et al. // Surf. Coat. Technol. 2016. V. 286. P. 268. https://doi.org/10.1016/j.surfcoat.2015.12.039
Uchikoshi T., Sakka Y. // J. Am. Ceram. Soc. 2008. V. 91. № 6. P. 1923. https://doi.org/10.1111/j.1551-2916.2008.02379.x
König K., Novak S., Boccaccini A.R., Kobe S. // J. Mater. Process. Technol. 2010. V. 210. № 1. P. 96. https://doi.org/10.1016/j.jmatprotec.2009.08.007
Moritz K., Aneziris C.G. // Key Eng. Mater. 2015. V. 654. P. 101. https://doi.org/10.4028/www.scientific.net/KEM.654.101
Nold A., Assion T., Zeiner J., Clasen R. // Ibid. 2009. V. 412. P. 307. https://doi.org/10.4028/www.scientific.net/kem.412.307
Pascall A.J., Qian F., Wang G. et al. // Adv. Mater. 2014. V. 26. № 14. P. 2252. https://doi.org/10.1002/adma.201304953
Vogt L., Schäfer M., Kurth D., Raether F. // Ceram. Int. 2019. V. 45. № 11. P. 14214. https://doi.org/10.1016/j.ceramint.2019.04.129
Maca K., Hadraba H., Cihlar J. // Ibid. 2004. V. 30. № 6. P. 843. https://doi.org/10.1016/j.ceramint.2003.09.021
Shan M., Mao X., Zhang J., Wang S. // Ibid. 2009. V. 35. № 5. P. 1855. https://doi.org/10.1016/j.ceramint.2008.10.033
Drdlik D., Moravek T., Rahel J. et al. // Ceram. Int. 2018. V. 44. № 8. P. 9787. https://doi.org/10.1016/j.ceramint.2018.02.215
Kotov Y.A. // J. Nanopart. Res. 2003. V. 5. P. 539. https://doi.org/10.1023/B:NANO.0000006069.45073.0b
Kotov Y.A. // Nanotechnol. Russia. 2009. V. 4. P. 415. https://doi.org/10.1134/S1995078009070039
Сафронов А.П., Калинина Е.Г., Смирнова Т.А. и др. // Журн. физ. химии. 2010. Т. 84. № 12. С. 2319.
Калинина Е.Г., Саматов О.М., Сафронов А.П. // Неорган. материалы. 2016. Т. 52. № 8. С. 922.
Kalinina E.G., Efimov A.A., Safronov A.P. // Thin Solid Films. 2016. V. 612. P. 66. https://doi.org/10.1016/j.tsf.2016.05.039
Kalinina E., Pikalova E., Kolchugin A. et al. // Materials. 2019. V. 12. № 16. P. 2545. https://doi.org/10.3390/ma12162545
Калинина Е.Г., Ефимов А.А., Сафронов А.П. // Неорган. материалы. 2016. Т. 52. № 12. С 1379.
Boccaccini A.R., Kaya C. // Ceram. Int. 2002. V. 28. № 8. P. 893. https://doi.org/10.1016/s0272-8842(02)00070-6
Harun Z., Ismail N.F., Badarulzaman N.A. // Adv. Mat. Res. 2012. V. 488−489. P. 335. https://doi.org/10.4028/www.scientific.net/AMR.488-489.335
Kotov Yu.A., Beketov I.V., Azarkevich E.I., Murzakaev A.M. // Proceedings of the Ninth CIMTEC-World Ceramic Congress “Ceramics: Getting into the 2000s”. 1998. P. 277.
Gregg S.J., Sing K.S.W. Adsorption, Surface Area and Porosity. New York: Academic Press, 1982. 303 p.
Dukhin S.S., Derjaguin B.V. Surface and Colloid Sciences. New York: Wiley-Interscience, 1974. 356 p.
Bhattacharjee S. // J. Control. Release. 2016. V. 235. P. 337. https://doi.org/10.1016/j.jconrel.2016.06.017
Aznam I., Mah J.C.W., Muchtar A. et al. // Zhejiang Univ. Sci. A. 2018. V. 19. P. 811. https://doi.org/10.1631/jzus.A1700604
Besra L., Liu M. // Prog. Mater. Sci. 2007. V. 52. № 1. P. 1. https://doi.org/10.1016/j.pmatsci.2006.07.001
Solomentsev Yu., Böhmer M., Anderson J.L. // Langmuir. 1997. V. 13. P. 6058. https://doi.org/10.1021/la970294a
Wefers K., Misra C. Oxides and Hydroxides of Aluminum. Alcoa Research Laboratories, 1987. 92 p.
McColm I.J. Ceramic Hardness. New York: Plenum Press, 1990. 324 p.
Cook R.F., Pharr G.M. // J. Am. Ceram. Soc. 1990. V. 73. № 4. P. 787. https://doi.org/10.1111/j.1151-2916.1990.tb05119.x
Lee K.-Y., Cropsey L.C.G., Tyszka B.R., Case E.D. // Mater. Res. Bull. 1997. V. 32. № 3. P. 287. https://doi.org/10.1016/S0025-5408(96)00196-1
Munro R.G. // J. Am. Ceram. Soc. 1997. V. 80. № 8. P. 1919. https://doi.org/10.1111/j.1151-2916.1997.tb03074.x
Chen I., Wang X. // Nature. 2000. V. 404. P. 168. https://doi.org/10.1038/35004548
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии