

Журнал физической химии, 2021, T. 95, № 9, стр. 1414-1425
Рекуррентная аппроксимация параметров удерживания N-замещенных п-толуолсульфонамидов в обращенно-фазовой высокоэффективной жидкостной хроматографии для выявления образования их гидратов
И. Г. Зенкевич a, *, Д. А. Никитина a, Т. А. Корнилова a
a Санкт-Петербургский государственный университет, Институт химии
Санкт-Петербург, Россия
* E-mail: izenkevich@yandex.ru
Поступила в редакцию 20.01.2021
После доработки 21.02.2021
Принята к публикации 24.02.2021
Аннотация
Рекуррентная аппроксимация времен удерживания в обращенно-фазовой высокоэффективной жидкостной хроматографии, tR(C + ΔC) = atR(C) + b, где С – концентрация ацетонитрила в составе элюента, ΔC – постоянный “шаг” ее варьирования, на примере шести специально синтезированных N-замещенных п-толуолсульфонамидов подтверждает наличие аномалий, выявленных ранее для некоторых сложных полифункциональных органических соединений. Причина таких аномалий – присутствие в молекулах сульфонамидных фрагментов –SO2–NH, что приводит к гидратированию сорбатов в водных растворах, точнее – к изменению соотношения их негидратированных и гидратированных форм вследствие смещения положений равновесия Х + Н2О $ \rightleftarrows $ Х ⋅ Н2О (*) в результате изменения состава элюента. Об этом же эффекте свидетельствует сильная антибатная зависимость индексов удерживания RI(С) всех охарактеризованных сульфонамидов; коэффициенты dRI/dC варьируют в диапазоне от –1.9 до –4.0, что по абсолютным величинам значительно больше, чем у не образующих гидраты соединений. Независимым дополнительным подтверждением трансформации сорбатов при вариациях состава элюента служит зависимость относительных оптических плотностей Аотн = А(254)/А(220) от содержания ацетонитрила в элюенте. Это свидетельствует об изменениях химической природы хромофоров в молекулах сульфонамидов в зависимости от положения равновесия (*).
Особенность высокоэффективной жидкостной хроматографии (ВЭЖХ) состоит в том, что разделяемые компоненты выходят из хроматографической колонки в потоке элюентов, которые в обращенно-фазовом варианте (ОФ) этого метода содержат воду. Это определяет возможность образования сорбатами (Х) их гидратов (чаще всего n = 1):
(1)
${\text{Х}} + n{{{\text{Н}}}_{{\text{2}}}}{\text{О}} \rightleftarrows {\text{Х}} \cdot n{{{\text{Н}}}_{{\text{2}}}}{\text{О}}.$В отличие от гидратов неорганических соединений (прежде всего, солей), образование не менее многочисленных гидратов органических соединений принимают во внимание относительно редко, что, в том числе, обусловлено нестабильностью многих из них. Характеристикой образования гидратов служат константы гидратации (Kравн), однако из-за экспериментальных сложностей, прежде всего, низкой растворимости многих органических соединений в воде и их летучести, они определены лишь для небольшого числа соединений [1]:
(2)
${{K}_{{{\text{равн}}}}} = [{\text{X}} \cdot n{{{\text{H}}}_{{\text{2}}}}{\text{O}}]{\text{/}}\{ [{\text{X}}] \cdot {{[{{{\text{H}}}_{{\text{2}}}}{\text{O}}]}^{n}}\} .$Если значения Kравн ≪ 1, то присутствием гидратов в водных растворах можно пренебречь, а исходные (негидратированные) сорбаты могут быть выделены из таких растворов в неизменном виде, но если Kравн ≫ 1, то пренебречь можно уже наличием безводных форм аналитов. При этом существует некоторая область значений Kравн ≈ 1, когда в ОФ ВЭЖХ соотношение гидратированных и негидратированных форм сорбатов в соответствии с равновесием (1) может в заметной степени изменяться при вариациях содержания воды в элюенте в рабочих диапазонах (от ~90 до 10 об. %). Для некоторых соединений это приводит к аномалиям хроматографического удерживания.
Рассмотрение особенностей хроматографического удерживания в ОФ ВЭЖХ нескольких лекарственных препаратов [2], относящихся к сложным полифункциональным органическим соединениям, позволило выявить структурный фактор, определяющий возможность образования их гидратов: присутствие в молекуле сульфонамидных фрагментов –SO2–NH2 или –SO2–NH–. Образование гидратов хорошо известно для многих препаратов, относящихся к классу сульфонамидов [3–7]. Многие из них образуют устойчивые кристаллогидраты, следовательно, в водных растворах (при избытке воды) такие соединения тем более существуют в форме гидратов.
Нелинейные зависимости параметров удерживания (tR) от концентрации органического компонента элюента (C) аппроксимируют различными уравнениями, в том числе Снайдера ($\lg t_{{\text{R}}}^{'}$ = = a + bС), Снайдера–Сочевинского, Скотта–Кучеры (1/$t_{{\text{R}}}^{'}$ = a + bC) и другими, $t_{{\text{R}}}^{'}$ = tR – t0, t0 – время удерживания несорбируемого компонента. Однако для выявления образования гидратов целесообразнее всего оказалось их представление с использованием линейных рекуррентных соотношений [8–10]:
где ΔС = const – постоянный “шаг” изменения концентрации органического модификатора в составе элюента, a и b – коэффициенты, вычисляемые методом наименьших квадратов. К преимуществам рекуррентной аппроксимации можно отнести отсутствие необходимости экспериментального определения или оценки значений t0.Рекуррентные соотношения применимы к монотонным функциям целочисленных или равноотстоящих значений аргументов и обладают рядом необычных математических свойств. Например, они сочетают свойства арифметических и геометрических прогрессий, а их математическими эквивалентами служат полиномы переменных степеней [8–10]. Применимость к функциям равноотстоящих значений аргументов распространяется на свойства, зависящие от температуры, давления или концентраций компонентов смесей; в нашем случае это концентрации органического компонента в составе элюента (С). Каждая точка на графиках рекуррентных зависимостей соответствует двум последовательным значениям, а значения аргумента на таких графиках вовсе не представлены.
Типичный вид графика, соответствующего рекуррентной аппроксимации параметров удерживания образующего гидрат сорбата приведен на рис. 1 (использованы данные для соединения с тривиальным названием “Пазопаниб” [2]). Две точки, соответствующие наибольшим значениям tR и, следовательно, максимальному содержанию воды в элюенте, отклоняются “вниз” от линии регрессии, причем при увеличении содержания воды в элюенте отклонения увеличиваются (крайняя правая точка на графике).
Рис. 1.
(а) Типичный вид графика, соответствующего рекуррентной аппроксимации времен удерживания образующего гидрат сорбата: точки, соответствующие наибольшим значениям tR и, следовательно, максимальному содержанию воды в элюенте, отклоняются “вниз” от линии регрессии; использованы данные для соединения с тривиальным названием “Пазопаниб”, содержащего фрагмент –SO2–NH2 [2]; (б) структурная формула пазопаниба.

Подобные аномалии на графиках рекуррентных зависимостей (3) наблюдались только для соединений, содержащих фрагменты –SO2–NH2 или –SO2–NH–, т.е. для двух из пяти охарактеризованных в работе [2] сорбатов. Иные аномалии, например, у соединения с тривиальным названием “Иматиниб” [2] в области высоких содержаний ацетонитрила, обусловлены другими причинами. Для подтверждения того, что наличие сульфонамидных фрагментов в молекуле – главная причина образования гидратов в условиях ОФ ВЭЖХ (что и приводит к аномалиям рекуррентной аппроксимации параметров удерживания), были специально синтезированы и охарактеризованы в [11] три соединения, относящиеся к ряду N-замещенных п-толуолсульфонамидов (не содержат никаких функциональных групп, кроме сульфонамидных). Однако для их разделения был использован элюент, содержащий не ацетонитрил, а метанол, способный к образованию водородных связей с сорбатами, что усложняет проявление эффектов гидратации. Возможно, что в результате этого только для одного из таких сульфонамидов (N,N-диэтил-) была выявлена зависимость (3), подобная приведенной на рис. 1.
Настоящая работа посвящена подробной характеристике особенностей рекуррентной аппроксимации параметров удерживания в режиме ОФ ВЭЖХ шести N-замещенных сульфонамидов с целью выявления возможного образования их гидратных форм в системе вода/ацетонитрил.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы, получение реакционных смесей и их подготовка к анализу
п-Толуолсульфонамиды синтезировали из п-толуолсульфохлорида (99%, ACROS ORGANICS, Бельгия) как самого доступного реагента класса сульфонилхлоридов, и следующих аминов: аллил- (I, “ч.д.а.”, Берлин-Адлерхоф, ГДР), диэтил- (II, “х.ч.”, Ангарский химический завод), трет-бутил- (III, 99%, ACROS ORGANICS, Бельгия), анилин (IV, ХЧ, Березниковский химический завод), бензиламин (V, Merck, Дармштадт, Германия) и н-гексиламин (VI, доступен в виде гидрохлорида, Реахим, Москва):
Приготовление образцов для хроматографического анализа
Для приготовления подвижных фаз использовали деионизованную воду (удельное сопротивление 18.2 мОм м), полученную на установке Milli-Q (Millipore, США), и ацетонитрил (HPLC-gradient grade, Panreac, Испания). Дегазирование элюента осуществляли фильтрованием под вакуумом и обработкой приготовленных водно-ацетонитрильных смесей ультразвуком с использованием установки Сапфир ТТЦ, 420 Вт (Сапфир, Россия). Перед анализом 10 мкл реакционных смесей разбавляли 10 мл смеси вода–ацетонитрил в соотношении 50 : 50 (объемн.), т.е. в 103 раз. Смесь трех реперных н-алкилфенилкетонов разбавляли в 105 раз.
Условия хроматографического анализа
Хроматографические эксперименты выполняли на жидкостном хроматографе высокого давления Agilent 1260 Infinity с диодно-матричным детектором при сканировании в диапазоне длин волн 220–340 нм; обработку хроматограмм проводили для длин волн 220 и 254 нм. Для характеристики хроматографического поведения N-замещенных п-толуолсульфонамидов в различных изократических режимах использовали колонку Agilent Poroshell 120 EC-CN, 100 × 3.0 мм (размер частиц сорбента 2.7 мкм). Последовательно увеличивали концентрацию ацетонитрила с шагом в 5%. Расход элюента 0.5 мл/мин. Температура колонки 40°C, объем дозируемых проб 5 мкл. Для дозирования использовали автосамплер SN G1329A. Для каждого образца в каждом из режимов проводили по три параллельных определения; разброс времен удерживания в большинстве случаев не превышал 0.01–0.02 мин. Времена удерживания несорбируемого компонента в каждом из режимов вычисляли по временам удерживания трех гомологов реперных н-алкилфенилкетонов; полученные значения отличаются от экспериментальной оценки, определенной по 0.1 М водному раствору бромида калия (“х.ч.”, Sigma-Aldrich), равной 0.94 мин для содержания ацетонитрила в составе элюента 50 об. %.
Обработка результатов
Хроматограммы получали, обрабатывали и хранили в программе Mass Hunter (Agilent Technologies); для статистической обработки данных использовали ПО Excel (Microsoft Office, 2010). Вычисление параметров рекуррентных зависимостей проводили с использованием ПО ORIGIN (версии 4.1 и 8.1) с шагом варьирования концентраций ацетонитрила в элюенте ΔС = 5%. Времена удерживания N-замещенных п-толуолсульфонамидов и реперных алкилфенилкетонов, а также вычисленные по ним значения t0 для различных режимов изократического элюирования приведены в табл. 1.
Таблица 1.
Абсолютные времена удерживания (t0, мин) N-замещенных п-толуолсульфонамидов и реперных алкилфенилкетонов в различных режимах изократического элюирования (С – содержание CH3CN в элюенте)
| C, об. % | Заместители при атоме азота | C6H5COCH3 | C6H5COC2H5 | C6H5COC3H7 | t0 (мин) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| –CH2CH=CH2 (I) |
(C2H5)2 (II) |
–C(CH3)3 (III) |
–C6H5 (IV) |
–CH2C6H5 (V) |
–C6H13 (VI) |
|||||
| 85 | 2.31 | 2.79 | 2.58 | 2.43 | 2.51 | 3.20 | 2.38 | 2.62 | 2.89 | 0.35 |
| 80 | 2.43 | 3.08 | 2.78 | 2.62 | 2.73 | 3.72 | 2.50 | 2.84 | 3.24 | 0.57 |
| 75 | 2.60 | 3.46 | 3.05 | 2.86 | 3.03 | 4.42 | 2.63 | 3.08 | 3.62 | 0.38 |
| 70 | 2.82 | 3.97 | 3.41 | 3.20 | 3.45 | 5.42 | 2.79 | 3.38 | 4.11 | 0.27 |
| 65 | 3.13 | 4.70 | 3.92 | 3.72 | 4.06 | 6.92 | 3.01 | 3.79 | 4.80 | 0.37 |
| 60 | 3.55 | 5.73 | 4.62 | 4.43 | 4.96 | 9.21 | 3.28 | 4.33 | 5.73 | 0.13 |
| 55 | 4.17 | 7.31 | 5.70 | 5.56 | 6.37 | 12.96 | 3.65 | 5.08 | 7.08 | 0.08 |
| 50 | 5.11 | 9.87 | 7.39 | 7.42 | 8.76 | 19.75 | 4.10 | 6.12 | 9.12 | -0.06 |
Примечание. Значения t0 вычислены по временам удерживания трех гомологов н-алкилфенилкетонов по формуле (4).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Характеристика не образующего гидрат сорбата
Обсуждение результатов целесообразно начать со сравнения различных способов аппроксимации параметров удерживания соединений, не образующих гидратов в водных растворах. При этом нет необходимости выбирать дополнительные сорбаты, так как для этого вполне подойдет один из реперных н-алкилфенилкетонов, используемых для определения индексов удерживания, например, бутирофенон. Если значение Kравн (2) гидратации ацетофенона составляет ∼6.6 × 10–6 [1], то для бутирофенона оно меньше из-за электронодонорных свойств алкильных групп, следовательно, образованием его гидрата в водных растворах можно пренебречь.
В уравнениях Снайдера–Сочевинского и Скотта–Кучеры, описывающих зависимости tR(C) в ОФ ВЭЖХ, фигурируют значения исправленных времен удерживания ($t_{{\text{R}}}^{'}$), что требует предварительного экспериментального определения или оценки времени удерживания несорбируемого компонента (“мертвое” время, t0). Простейший способ вычисления таких оценок основан на временах удерживания трех последовательных гомологов одного и того же ряда (метод Петерсона и Хирша [8, 12]); в газовой хроматографии чаще всего используют н-алканы, а в ОФ ВЭЖХ то же можно сделать при выборе н-алкилфенилкетонов:
(4)
${{t}_{0}} \approx ({{t}_{n}}{{t}_{{n + 2}}}--t_{{n - 1}}^{2}){\text{/}}{{t}_{n}} + {{t}_{{n + 2}}}--2{{t}_{{n + 1}}}).$Рис. 2.
(а) Зависимости логарифмов времен удерживания ($\lg t_{{\text{R}}}^{'}$, мин) бутирофенона от содержания ацетонитрила в элюенте (С, об. %), значения $t_{{\text{R}}}^{'}$ вычислены при оценке величин t0 по соотношению (4); (б) то же при использовании одинакового для всех режимов экспериментально определенного значения t0 = 0.94 мин; (в) не имеющий аномалий во всем диапазоне 50 ≤ С ≤ 85% график рекуррентной зависимости (3) для бутирофенона, R = 0.9993.

Особенность зависимости $\lg t_{{\text{R}}}^{'}$(C) при высоком содержании ацетонитрила (85%) можно было бы объяснить проявлением так называемого гидрофильного режима хроматографического разделения [13], если бы не ее исчезновение при использовании фиксированного экспериментально определенного значения t0 = 0.94 мин (рис. 2б). Аномалия точки $\lg t_{{\text{R}}}^{'}$ при С = 85% при этом исчезает, однако сохраняется перегиб в области С ≈ ≈ 65%. Рассмотрение причин появления такого перегиба выходит за рамки настоящего обсуждения, но важно отметить, что рекуррентное представление (3) параметров удерживания бутирофенона (рис. 3в) демонстрирует отсутствие каких-либо аномалий, а коэффициент корреляции зависимости (3) при этом составляет 0.9993. Таким образом, пример соединения, не образующего гидратных форм в водных растворах, подтверждает отсутствие аномалий рекуррентных зависимостей (3).
Рис. 3.
(а) Зависимости логарифмов времен удерживания ($\lg t_{{\text{R}}}^{'}$, мин) N-аллил-п-толуолсульфонамида (I) от содержания ацетонитрила в элюенте (С, об. %), параметры линейной регрессии вычислены по четырем наибольшим значениям $\lg t_{{\text{R}}}^{'}$: a = –0.0179 ± 0.0006, b = 1.60 ± 0.03, R = –0.998, S0 = 0.007; (б) линейная рекуррентная зависимость tR(C + 5%) = = atR(C) + b, параметры линейной регрессии: a = 0.733 ± 0.004, b = 0.53 ± 0.01, R = 0.99995, S0 = 0.004. Отклонения от линии регрессии двух точек, отвечающих минимальным концентрациям CH3CN 55–60 и 50–55%, составляют –0.05 и –0.11, соответственно.

Выявление корреляций между параметрами удерживания аналитов различной химической природы в ОФ ВЭЖХ и составом элюента – наиболее распространенная форма характеристики хроматографических свойств как аналитов, так и сорбентов хроматографических колонок [14–19]. При этом интерпретация линейных зависимостей значительно предпочтительнее, чем нелинейных. В таких случаях особый интерес представляют отклонения от линейности, так как чаще всего они связаны с проявлениями каких-либо свойств органических соединений. Так, наиболее подробно рассмотренным примером является использование ОФ ВЭЖХ для определения констант диссоциации органических кислот [20–23]:
В случае рассматриваемых нами N-замещенных п-толуолсульфонамидов такие кислотно-основные равновесия можно не принимать во внимание, так как значения (как экспериментальные, так и расчетные) pKa сульфонамидов CH3–C6Н4–SO2NHR значительно превышают рН элюента: 10.3 ± 0.2 (R = H), 11.7 ± 0.3 (R = CH3), 10.9 (R = C2H5), 11.8 (R = C3H7) и т.д.
Рекуррентная аппроксимация времен удерживания как способ выявления образования гидратов N-замещенных п-толуолсульфонамидов
Особенность следующего фрагмента обсуждения состоит в том, что для иллюстрации образования гидратов, так же как и в работах [2, 11], нагляднее всего графическое представление рекуррентных зависимостей tR(C + 5%) = atR(C ) + b вида (3). Поэтому на рис. 3–8 представлены такие графики для всех шести рассматриваемых N-замещенных п-толуолсульфонамидов (I–VI). На рисунках (а) приведены “традиционные” зависимости $\lg t_{{\text{R}}}^{'}$(C), а на рисунках (б) – соответствующие им рекуррентные “эквиваленты” вида (3).
Рис. 4.
(а) Зависимости логарифмов времен удерживания ($\lg t_{{\text{R}}}^{'}$, мин) N,N-диэтил-п-толуолсульфонамида (II) от содержания ацетонитрила в элюенте (С, об. %), параметры линейной регрессии вычислены по трем значениям $\lg t_{{\text{R}}}^{'}$: a = = –0.0223 ± 0.0001, b = 2.086 ± 0.003, R = –1.000, S0 = 0.0004; (б) линейная рекуррентная зависимость tR(C + 5%) = = atR(C) + b, параметры линейной регрессии: a = 0.719 ± 0.002, b = 0.59 ± 0.01, R = 0.99999, S0 = 0.01. Отклонения от линии регрессии двух точек, отвечающих минимальным концентрациям CH3CN 55–60 и 50–55%, составляют –0.16 и –0.52, соответственно.

Рис. 5.
(а) Зависимости логарифмов времен удерживания ($\lg t_{{\text{R}}}^{'}$, мин) N-трет-бутил-п-толуолсульфонамида (III) от содержания CH3CN в элюенте (С, об. %), параметры линейной регрессии вычислены по четырем наибольшим значениям $\lg t_{{\text{R}}}^{'}$: a = –0.0213 ± 0.0007, b = 1.93 ± 0.04, R = –0.998, S0 = 0.008; (б) линейная рекуррентная зависимость tR(C + 5%) = atR(C) + b, параметры линейной регрессии: a = 0.56 ± 0.01, b = 0.727 ± 0.004, R = 0.99996, S0 = 0.005. Отклонения от линии регрессии двух точек, отвечающих минимальным концентрациям CH3CN 55–60 и 50–55%, составляют –0.12 и –0.32, соответственно.
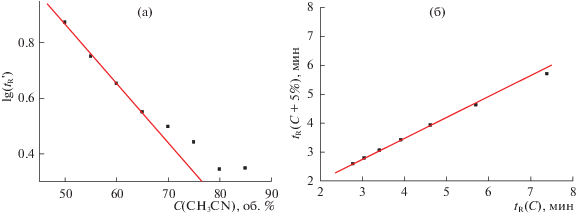
Рис. 6.
(а) Зависимости логарифмов времен удерживания ($\lg t_{{\text{R}}}^{'}$, мин) N-фенил-п-толуолсульфонамида (IV) от содержания CH3CN в элюенте (С, об. %), параметры линейной регрессии вычислены по трем значениям $\lg t_{{\text{R}}}^{'}$: a = –0.0214 ± ± 0.0001, b = 1.916 ± 0.007, R = –0.99999, S0 = 0.0008; (б) линейная рекуррентная зависимость tR(C + 5%) = atR(C) + b, параметры линейной регрессии: a = 0.70 ± 0.01, b = 0.59 ± 0.01, R = 0.9997, S0 = 0.01. Отклонения от линии регрессии двух точек, отвечающих минимальным концентрациям CH3CN 55–60 и 50–55%, составляют –0.11 и –0.37, соответственно.

Рис. 7.
(а) Зависимости логарифмов времен удерживания ($\lg t_{{\text{R}}}^{'}$, мин) N-бензил-п-толуолсульфонамида (V) от содержания CH3CN в элюенте (С, об. %), параметры линейной регрессии вычислены по трем значениям $\lg t_{{\text{R}}}^{'}$: a = –0.0244 ± 0.0008, b = 2.14 ± 0.05, R = –0.9994, S0 = 0.006; (б) линейная рекуррентная зависимость tR(C + 5%) = atR(C) + b, параметры линейной регрессии: a = 0.694 ± 0.006, b = 0.63 ± 0.02, R = 0.9999, S0 = 0.01. Отклонения от линии регрессии двух точек, отвечающих минимальным концентрациям CH3CN 55–60 и 50–55%, составляют –0.13 и –0.49, соответственно.
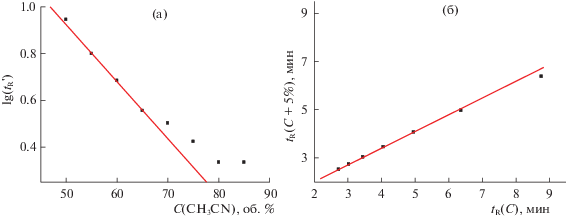
Рис. 8.
(а) Зависимости логарифмов времен удерживания ($\lg t_{{\text{R}}}^{'}$, мин) N-гексил-п-толуолсульфонамида (VI) от содержания CH3CN в элюенте (С, об. %), параметры линейной регрессии вычислены по трем значениям $\lg t_{{\text{R}}}^{'}$: a = –0.0294 ± ± 0.0006, b = =2.72 ± 0.03, R = –0.9998, S0 = 0.004; c(б) линейная рекуррентная зависимость tR(C + 5%) = atR(C) + b, параметры линейной регрессии: a = 0.675 ± 0.008, b = 0.73 ± 0.05, R = 0.9997, S0 = 0.04. Отклонения от линии регрессии двух точек, отвечающих минимальным концентрациям CH3CN 55–60 и 50–55%, составляют –0.40 и –1.62, соответственно.

Сравнение рис. 3–8 показывает, что зависимости (а) для всех шести сульфонамидов имеют идентичный вид, полностью соответствующий этой зависимости для бутирофенона (рис. 2а). На всех графиках проявляется перегиб в области С ≈ 65% и аномалии точек, отвечающих концентрации ацетонитрила 85%, обусловленные, как было показано выше, вариациями параметра t0, в наибольшей степени влияющими на минимальные значения времен удерживания, т.е. при высоких содержаниях ацетонитрила в элюенте. На рис. 3б–8б для всех п-толуолсульфонамидов (I–VI) приведены графики рекуррентных зависимостей (3), которые также однотипны. Важно, что для всех сорбатов не наблюдается никаких аномалий в области высоких содержаний ацетонитрила в элюенте (от ~85 до 60%), так что коэффициенты корреляции линейных регрессий вида (3) для этих участков составляют от 0.9997 до 0.99999. Что же касается точек, соответствующих сочетаниям концентраций ацетонитрила 60–55 и 55–50%, то они, хоть и незначительно, но (различимо даже визуально) отклоняются “вниз” от линии регрессии. Для первой из них такие отклонения по вертикали составляют от –0.05 до –0.40 мин, а для второй – от –0.11 до –1.62 мин. Величины отклонений симбатны абсолютным временам удерживания сорбатов, т.е. минимальны для N-аллилсульфонамида, а максимальны для N-гексилсульфонамида.
Подобные аномалии полностью аналогичны наблюдаемым для двух синтетических лекарственных препаратов, содержащих фрагмент структуры –SO2–NH– (Бозентан и Пазопаниб) [2], а также для N,N-диэтил-п-толуолсульфонамида в системе вода/метанол [11]. Подобные отклонения точек (занижение параметров удерживания по сравнению с зависимостью tR(C + 5%) = = atR(C) + b для более высоких содержаний ацетонитрила), соответствующих минимальным содержаниям ацетонитрила и, следовательно, максимальным содержаниям воды, означают, что формы сорбатов в элюенте в этой области более гидрофильны, чем при высоких содержаниях ацетонитрила. Увеличение гидрофильности согласуется со смещением равновесия (1) вправо, т.е. с возрастанием концентраций гидратов анализируемых соединений в элюенте. Таким образом, данные для модельных соединений ряда N-замещенных п-толуолсульфонамидов подтверждают эффект, впервые выявленный для более сложных объектов [2].
В целом же рассматриваемый эффект образования гидратов сорбатов в элюенте проявляется в достаточно слабой степени. Так, отклонения точки, соответствующей сочетанию концентраций ацетонитрила 60–55%, для пяти из шести сульфонамидов составляют всего (–0.05)–(–0.16) мин и только для N-гексилсульфонамида достигают ‒0.40 мин. Это означает, что они могут быть выявлены только при достаточно высокой точности измерения параметров удерживания (в нашем случае на уровне ±0.01–0.02 мин). При худшей воспроизводимости экспериментальных данных рассматриваемый эффект может остаться незамеченным.
Особенности индексов удерживания N-замещенных п-толуолсульфонамидов
Образование гидратов анализируемых соединений может быть выявлено только в результате рекуррентного представления времен удерживания. В сравнении с этим небезынтересно проверить, как образование гидратов сказывается на таких характеристиках как индексы удерживания (RI)11:
(5)
$\begin{gathered} {\text{RI}} = {\text{R}}{{{\text{I}}}_{n}} + ({\text{R}}{{{\text{I}}}_{{n + z}}}--{\text{R}}{{{\text{I}}}_{n}})[f({{t}_{{R,x}}})-- \\ - \;f({{t}_{{R,n}}})]{\text{/}}[f({{t}_{{R,n + z}}})--f({{t}_{{R,n}}})], \\ \end{gathered} $Концепцию индексов широко применяют в ГХ [16, 17], но в ОФ ВЭЖХ – в меньшей степени [26, 27]. Это обусловлено влиянием значительно бóльшего числа экспериментальных условий на значения RI в ВЭЖХ, что приводит в результате к их худшей межлабораторной воспроизводимости и, следовательно, меньшей информативности для хроматографической идентификации. Однако в отдельных случаях их рассмотрение полезно.
Интерполяционная формула (5) в изократических условиях элюирования эквивалентна линейному соотношению RI = alg($t_{{\text{R}}}^{'}$) + b. В нашем случае в качестве реперных компонентов мы использовали ацетофенон (RIn = 800) и бутирофенон (${\text{R}}{{{\text{I}}}_{{n + z}}}$ = 1000), что дает возможность вычислять RI во всем диапазоне от t0 (экстраполяцией) до ~1200. Полученные значения для всех N-замещенных п-толуолсульфонамидов во всех режимах анализа приведены в табл. 2.
Таблица 2.
Индексы удерживания (RI) N-замещенных п-толуолсульфонамидов в различных режимах изократического элюирования и параметры уравнений RI = aС + b
| C, об. % | Заместители при атоме азота | |||||
|---|---|---|---|---|---|---|
| –CH2CH=CH2 (I) |
(C2H5)2 (II) |
–C(CH3)3 (III) |
–C6H5 (IV) |
–CH2C6H5 (V) |
–C6H13 (VI) |
|
| 85 | 769 | 964* | 884* | 822 | 864* | 1103 |
| 80 | 778 | 961 | 884 | 836 | 867 | 1102 |
| 75 | 793 | 972 | 894 | 854 | 892 | 1121 |
| 70 | 806 | 982 | 904 | 872 | 910 | 1140 |
| 65 | 817 | 991 | 914 | 892 | 930 | 1151 |
| 60 | 829 | 1000 | 924 | 908 | 949 | 1168 |
| 55 | 840 | 1010 | 935 | 927 | 969 | 1181 |
| 50 | 855 | 1020 | 948 | 948 | 990 | 1194 |
| a = dRI/dC | –2.45 ± 0.04 | –1.94 ± 0.02 | –2.10 ± 0.04 | –3.62 ± 0.06 | –4.01 ± 0.06 | –3.03 ± 0.11 |
| b | 977 ± 3 | 1117 ± 2 | 1051 ± 3 | 1126 ± 4 | 1190 ± 4 | 1348 ± 7 |
| R | –0.9990 | –0.9996 | –0.9998 | –0.9992 | –0.9994 | –0.997 |
| S0 | 1.4 | 0.6 | 1.1 | 1.9 | 1.6 | 2.8 |
Главной особенностью RI сульфонамидов оказывается их сильно выраженная зависимость от состава элюента, которую с высокой точностью можно аппроксимировать линейным уравнением:
Коэффициенты корреляции зависимости (6) для пяти сульфонамидов превышают по абсолютной величине 0.999 и только для наиболее гидрофобного N-гексильного производного несколько меньше (все значения RI вычислены экстраполяцией). Значения так называемой генеральной дисперсии S0 (в интервале от 0.6 до 2.8 ед. индекса) подтверждают высокую среднюю точность аппроксимации RI по уравнению (6). Коэффициенты а в соотношении (6), a = dRI/dC, фактически отражающие зависимость RI от содержания органической составляющей элюента, варьируют от ‒1.9 до –4.0 (табл. 2). В газовой хроматографии подобные отрицательные величины dRI/dT, где Т – температура принципиально невозможны, а столь большие по абсолютной величине положительные значения dRI/dT присущи только полициклическим ароматическим углеводородам, содержащим не менее 5–6 циклов в молекулах. Скорее всего, именно существенные отрицательные значения a = dRI/dC и служат характерным признаком сорбатов, образующих гидраты в условиях ОФ ВЭЖХ и полностью согласуются с аномалиями времен удерживания, выявляемыми в результате рассмотрения рекуррентных зависимостей. Действительно, увеличение концентрации воды в элюенте приводит к образованию гидратов и увеличению полярности сорбатов относительно реперных компонентов системы RI (менее полярных н-алкилфенилкетонов) и, следовательно, к увеличению их индексов удерживания.Подобные существенные изменения параметров удерживания образующих гидраты соединений (по сравнению с менее склонными к этому сорбатами) при вариациях состава элюента отмечали и ранее. Так, например, увеличение концентрации ацетонитрила в элюенте с 10 до 30% уменьшает время удерживания фенола с 23.8 до 6.7 мин (т.е. в 3.6 раза), тогда как время удерживания дигидрокверцетина (практически нацело гидратированного в водных растворах) – с 68.6 до 4.5 мин (т.е. в 15.2 раза) [24]. Более отчетливо выраженные вариации индексов удерживания по сравнению с временами удерживания означают, что они предъявляют меньшие требования к точности измерений, что, следовательно, делает возможным использование более простых хроматографов.
Дополнительное подтверждение образования гидратов. Зависимость относительных оптических плотностей от состава элюента
Таким образом, изменение соотношения негидратированных и гидратированных форм некоторых органических соединений в результате смещения равновесия (1) при изменении содержания воды в элюентах в ОФ ВЭЖХ проявляется как в аномалиях времен удерживания (выявляется в результате их рекуррентной аппроксимации), так и в больших отрицательных значениях коэффициентов зависимости индексов удерживания от состава элюента, dRI/dC. Несмотря на это представляется весьма желательным подтвердить изменение соотношения негидратированных и гидратированных форм N-замещенных п-толуолсульфонамидов дополнительным независимым способом.
Образование гидратов Х ⋅ nН2О может изменять химическую природу хромофоров в молекулах, хотя детали таких процессов принимают во внимание сравнительно редко. Так, например, в известных публикациях, посвященных гидратации сульфонамидов [3–7], характер связывания воды остается неопределенным. Тем не менее, можно полагать, что, по крайней мере, в отдельных случаях вариации УФ-спектральных характеристик сорбатов в элюентах разного состава заметны настолько, что их можно проследить экспериментально. Для таких целей пригоден УФ-спектральный параметр, предложенный для повышения надежности идентификации сорбатов в ВЭЖХ – относительные оптические плотности Аотн [28]. Основанная на таких характеристиках концепция идентификации была реализована в некоторых отечественных хроматографах серии “Милихром”:
Таблица 3.
Относительные оптические плотности Аотн = А(254)/А(220) N-замещенных п-толуолсульфонамидов в различных режимах изократического элюирования
| C, об. % | Заместители при атоме азота | |||||
|---|---|---|---|---|---|---|
| –CH2CH=CH2 (I) |
(C2H5)2 (II) |
–C(CH3)3 (III) |
–C6H5 (IV) |
–CH2C6H5 (V) |
–C6H13 (VI) |
|
| 85 | 0.062 | 0.283 | 0.108* | – | 0.200* | 0.069 |
| 80 | 0.072* | 0.282 | 0.102 | 0.337* | 0.199* | 0.069 |
| 75 | 0.064 | 0.283 | 0.100 | 0.337* | 0.139 | 0.072 |
| 70 | 0.062 | 0.283 | 0.099 | 0.239* | 0.146 | 0.071 |
| 65 | 0.065 | 0.284 | 0.101 | 0.296 | 0.158 | 0.073 |
| 60 | 0.068 | 0.284 | 0.104 | 0.299 | 0.159 | 0.074 |
| 55 | 0.068 | 0.283 | 0.104 | 0.296 | 0.165 | 0.074 |
| 50 | 0.075 | 0.284 | 0.106 | 0.294 | 0.166 | 0.075 |
| a = dАотн/dC | (–3.3 ± 0.5) × 10–4 | – | (1.8 ± 0.7) × 10–4 | – | (–11 ± 2) × 10–4 | (–1.8 ± 0.2) × 10–4 |
| R | –0.866 | – | –0.772 | – | –0.957 | –0.953 |
| Среднее значение Аотн | – | 0.283 ± 0.001 | 0.102 ± 0.003 | 0.296 ± 0.002 | – | – |
Стандартные отклонения площадей хроматографических пиков (S) при разных длинах волн составляют менее 1%, что объясняет хорошую воспроизводимость отношений (7). “Естественным” ограничением применимости параметров Аотн для идентификации является перекрывание пиков целевых компонентов с примесями, что наиболее вероятно при минимальных временах удерживания, т.е. при высоком содержании ацетонитрила в элюенте. Именно по этой причине несколько значений Аотн в табл. 3 исключены из рассмотрения. Характер последующей обработки значений Аотн зависит от диапазона их вариаций при изменении содержания ацетонитрила в элюенте. Если эти отношения практически не зависят от С (для соединений II, III и IV), то наиболее рационально вычисление их средних арифметических значений и стандартных отклонений. Однако для трех других сульфонамидов (I, V и VI) вариации Аотн(C) более заметны, что позволяет обработку данных методом наименьших квадратов с вычислением параметров регрессионного уравнения Аотн= = аС + b. Наибольшие диапазоны вариаций и максимальные абсолютные значения коэффициентов корреляции выявлены у наиболее гидрофобных сульфонамидов – N-бензил и N-гексил. Зависимости Аотн = аС + b для N-гексил-п-толуолсульфонамида приведены на рис. 9. Важно заметить, именно для этого соединения отмечены наибольшие аномалии рекуррентной аппроксимации времен удерживания. Следовательно, образование гидратов (точнее, изменение соотношения негидратированных и гидратированных форм в результате вариаций состава элюента) для гидрофобных сульфонамидов выражено сильнее, чем у гидрофильных представителей ряда. Эта особенность объясняет, почему рассматриваемые эффекты оказались выраженными в наибольшей степени у сложных полифункциональных лекарственных препаратов, для которых они и были выявлены впервые [2].
Рис. 9.
Зависимость относительной оптической плотности N-гексил-п-толуолсульфонамида (VI) от содержания CH3CN в элюенте (С, об. %), из параметров линейной регрессии Аотн = аС + b в табл. 3 приведены только значения а = dAотн/dC и коэффициенты корреляции R.

Таким образом, рассмотрение зависимостей параметров удерживания шести специально синтезированных N-замещенных п-толуолсульфонамидов от содержания ацетонитрила в элюенте подтверждает, что причиной аномалий их рекуррентной аппроксимации действительно является изменение соотношения негидратированных и гидратированных форм сорбатов при изменении состава элюента. Этот вывод дополнительно подтвержден существенной антибатной зависимостью индексов удерживания сульфонамидов от концентрации ацетонитрила в элюенте; коэффициенты dRI/dC варьируют в интервале от –1.9 до –4.0. Кроме того, для трех из шести рассмотренных сульфонамидов выявлено влияние содержания ацетонитрила в элюенте на относительные оптические плотности Аотн = А(254)/А(220), что свидетельствует об изменениях химической природы хромофоров в молекулах в зависимости от положения равновесия Х + Н2О $ \rightleftarrows $ Х ⋅ Н2О.
Список литературы
Rawn J.D., Quellette R. Organic Chemistry. 2nd Edn. New York: Acad. Press. 2019. 1056 p.
Зенкевич И.Г., Никитина Д.А. // Журн. физ. химии. 2021. Т. 95. № 2. С. 285. https://doi.org/10.31857/S004445372102028X
Guo M.-L. // Crystallogr. Commun. 2004. V. 60. Part 4. P. 574. https://doi.org/10.1107/S1600536804005446
Suchetan S.F., Foro S., Gowda B.T., Prakash M.S. // Acta Crystallogr. Sect. E Struct. Rep. Online. 2012. V. E68. № 1. P. o46. https://doi.org/10.1107/S1600536811051932
Kompella A., Kasa S., Balina V.S., et al. // Science J. Chem. 2014. V. 2. № 6–1. P. 9. https://doi.org/10.11648/j.sjc.s.2014020601.12
Jatezak M., Sidoryk K., Kossykowska M. et al. // Chromatographia. 2016. V. 79. P. 1131–1141. https://doi.org/10.1007/s10337-016-3124y
Jurczak E., Mazurek A.H., Szeleszczuk L. et al. // Pharmaceuticals. 2020. V. 12. 25 p. https://doi.org/10.3390/pharmaceuticals12100959
Zenkevich I.G. // J. Chemometr. 2009. V. 23. P. 179. https://doi.org/10.1002/cem.1214
Zenkevich I.G. Recurrent Relationships in Separation Science. Ch. 24 in Chemometrics in Chromatography. / Eds. L. Komsta, Y.V. Heyden, J. Sherma. London: CRC Press. 2018. P. 449.
Zenkevich I.G. // J. Chemometr. 2010. V. 24. P. 158. https://doi.org/10.1002/cem.1297
Корнилова Т.А., Деруиш А., Зенкевич И.Г. // Аналитика и контроль. 2020. Т. 24. № 4. С. 315. https://doi.org/10.15826/analitika.2020.24.4.006
Peterson M.L., Hirsch J. // J. Lipid Res. 1959. V. 1. P. 132.
Патрушев Ю.В., Сотникова Ю.С., Сидельников В.Н. // Физикохимия поверхности и защита материалов. 2020. Т. 56. № 1. С. 53.
Lanin S.N., Ledenkova M.Y., Nikitin Y.S. // J. Chromatogr. A. 1998. V. 797. P. 3. https://doi.org10.1016/S0021-9873(97)01206-5
Zapala W. // J. Chromatogr. Sci. 2003. V. 41. № 7. P. 289.
Чиркин В.А., Карпов С.И., Селеменев В.Ф. // Журн. анал. химии. 2013. Т. 68. № 4. С. 369. https://doi.org/10.1134/S10619348113020056
Сайфутдинов Б.Р., Даванков В.А., Ильин М.М. // Журн. физ. химии. 2014. Т. 88. № 3. С. 373. https://doi.org/10.1134/S0036024414030224
Сайфутдинов Б.Р., Даванков В.А., Петухова Г.А. и др. // Докл. РАН. 2015. Т. 462. № 6. С. 675. https://doi.org/10.1134/S0012501615060056.
Савченкова А.С., Буряк А.К., Курбатова С.В. // Журн. физ. химии. 2015. Т. 89. № 9. С. 1450. https://doi.org/10.1134/S0036024415090277
Canals I., Portal J.A., Roses M., Bosch E. // Chromatographia. 2022. V. 55. № 9/10. P. 565–571.
Sanli N., Sanli S., Ozkan G., Denizli A. // J. Braz. Chem. Soc. V. 21. № 10. P. 1952.
Remko M. // J. Molec. Struct. THEOCHEM. 2010. V. 941. P. 34. doi: . theochem.2009.12.017.https://doi.org/10.1016/j
Caine B.A., Bronzato M.B., Popelier L.A. // J. Chem. Sci. 2019. V. 10. P. 6368. https://doi.org/10.1039/c9sc01818b
Zenkevich I.G., Kochetova M.V., Larionov O.G. et al. // J. Liquid Chromatogr. & Related Technol. 2005. V. 28. P. 2141. https://doi.org/10.108/JLC-200064000
Kovats’ Retention Index System. In Encyclopedia of Chromatography. Ed. J. Cazes. 3rd Edn. New York: Taylor & Francis. 2010. 2850 p. V. 2. P. 1304-1310.
Retention and Selectivity in Liquid Chromatography. J. Chromatogr. Libr., V. 57. Ed. R.M. Smith. Amsterdam: Elsevier. 1995. 462 p. P. 93–144.
Руководство по газовой хроматографии. Под ред. Э. Лейбница и Х.Г. Штруппе. Пер. с нем. М.: Мир, 1988. В 2-х тт. (Handbuch der Gaschromatographie. Herausgegeben von E. Leibnitz, H.G. Struppe. Leipzig, Akademische Verlag, 1984.).
Зенкевич И.Г., Косман В.М. // Журн. прикл. химии. 1997. Т. 70. № 11. С. 1861.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии